

Abstract
Oxidative stress and inflammation play an important role in the pathogenesis of early brain injury (EBI) following subarachnoid hemorrhage (SAH). The present study aimed to evaluate the effect of salvianolic acid A (SalA) on EBI after SAH via its antioxidative, anti-inflammatory, and anti-apoptotic effects. The intraperitoneal administration of SalA (10 and 50 mg/kg/day) significantly alleviated EBI (including neurobehavioral deficits, brain edema, blood-brain barrier permeability, and cortical neuron apoptosis) after SAH in rats. SalA treatment also reduced the post-SAH elevated levels of reactive oxygen species level and malondialdehyde. Further, SalA increased glutathione peroxidase enzymatic activity and the concentrations of glutathione and brain-derived neurotrophic factor in brain cortex, at 24 h after SAH. In addition, SalA also decreased the release of inflammation cytokines (i.e., TNF-α, IL-1β, IL-6, and IL-8) in SAH rats. Expressions of cell apoptosis-related proteins were also regulated by SalA treatment in SAH rats. Meanwhile, SalA also modulated Nrf2 signaling, and the phosphorylation of ERK and P38 MAPK signaling in SAH rats. These results indicated that the administration of SalA may ameliorate EBI and provide neuroprotection after SAH in rat models.
Keywords: Salvianolic acid A, subarachnoid hemorrhage, apoptosis, inflammatory cytokine, oxidative damage
Introduction
Subarachnoid hemorrhage (SAH) is a disease with high disability and mortality rate. The most common cause of SAH is ruptured cerebral aneurysm [1,2]. Approximately 30% of patients die within the first few days, while 10% die in the following days due to various complications. The overall mortality rate is more than 50% [3]. SAH consists of 5-7% of total strokes and affects 10 per 100,000 adults each year. Most deaths occur in the early phase of initial bleeding, which results from the increased ICP or acute hydrocephalus [4]. SAH may induce early brain injury (EBI) and delayed brain injury, which can manifest as cerebral edema, intracranial hypertension, cerebral infarction, neurological disorders, and disturbances of consciousness. These symptoms may result from inflammation, cerebral vasospasm, apoptosis, etc. [5-7]. Currently, however, the specific pathogenesis and treatment strategy for SAH remains poorly understood and thus, warrants further study.
Salvianolic acid A (SalA) is one of the major active components of Salviae Miltiorrhizae Bunge (Danshen in Chinese). Extensive studies have demonstrated the potent protective effects of SalA against ischemia-induced injury both in vitro and in vivo [8,9]. SalA reportedly attenuates inflammation, apoptosis, and oxidative stress, and decreases the expressions of AKT and NF-κB at the cellular level [10,11]. However, there are only a few previous studies on the effects and molecular mechanisms of SalA on the blood-brain-barrier (BBB) and endogenous neurogenesis in ischemic stroke. The objective of the current study was to investigate the effects of SalA in EBI after SAH in rats. In particular, we aimed to elucidate whether treatment with SalA after SAH would protect rats against EBI and neuronal apoptosis, and elucidate possible underlying mechanism(s) of any actions.
Materials and methods
Animal model
The protocols involving animal use in the current study were approved by the Institutional Animal Care and Use Committee of Mindong Hospital, which is affiliated with Fujian Medical University. Clean, adult male Sprague-Dawley rats, weighing 250-300 g, were obtained from Shanghai Laboratory Animal Center, Chinese Academy of Sciences. Animals were housed in a colony room under controlled temperature (22°C), and a 12:12 light-dark cycle, with food and water available ad libitum. A rat model of SAH was induced by endovascular perforation, as described in a previous study [12], with minor modifications. Briefly, rats were anesthetized with an intraperitoneal injection of chloral hydrate (300 mg/kg). Then, the right common carotid artery, external carotid artery, and internal carotid artery (ICA) were carefully exposed through a ventral midline neck incision, and a blunted 3-0 monofilament nylon suture was inserted into the ICA, until resistance was felt (approximately 18-20 mm from the common carotid bifurcation). The suture was carefully pushed approximately 3 mm further to perforate the artery wall to create a SAH. Sham-operation rats were manipulated in the same manner without the perforation.
Experimental groups
Rats were divided into four groups as follows: (i) sham operation group, which underwent sham operation and received vehicle; (ii) SAH group, which was subjected to SAH and received vehicle; (iii) SalA 10 mg/kg group (SalA 10), which was subjected to SAH and treated with SalA 10 mg/kg; and (iv) SalA 50 mg/kg group (SalA 50), which was subjected to SAH and treated with SalA 50 mg/kg. For the vehicle control, the rats were treated with normal saline at the same volume as SalA. SalA (purity > 98%;) was purchased from Shanghai Winherb Medical S & T Development Co., Ltd. (Shanghai, China).
Neurological scoring
Like in our previous study [13], behavioral activity was examined using three scoring systems (Table 1), at 48 h after SAH. Scoring was performed to record appetite, activity, and neurological deficits by two “blinded” investigators; the sequence of testing for the given tasks was randomized. Neurological deficits of the experimental animals were graded as follows: (i) no neurologic deficit (score = 0); (ii) suspicious or minimum neurologic deficit (score = 1); (iii) mild neurologic deficit (score = 2-3); and (iv), severe neurologic deficit (score = 4-6).
Table 1.
Behavior scores
| Category | Behavior | Score |
|---|---|---|
| Appetite | Finished meal | 0 |
| Left meal unfinished | 1 | |
| Scarcely ate | 2 | |
| Activity | Active, walking, barking, or standing | 0 |
| Lying down, walk and stand with some stimulations | 1 | |
| Almost always lying down | 2 | |
| Deficits | No deficits | 0 |
| Unstable walk | 1 | |
| Impossible to walk and stand | 2 |
Assessment of brain edema
Rats were killed 24 h after SAH and the brains were extracted. Each rat brain was gently blotted with filter paper and weighed on an electronic balance to obtain the wet weight (WW). The brain tissue was then dried for 24 h, at 100 °C, in a vacuum oven, to obtain the dry weight (DW). The cerebral water content was calculated according to the following formulation: H2O (%) = (WW-DW)/WW × 100.
Measurement of BBB permeability
The Evans blue (EB) extravasation was measured to assess BBB permeability. EB dye (2% in saline) was injected into the left jugular vein 23 h after SAH. Thereafter, the rats were transcardially perfused with phosphate buffered saline (PBS) to remove the intravascular dye. The rat brains were then homogenized in a 10-fold volume of 50% trichloroacetic acid solution to precipitate protein and centrifuged for 10 min at 3000 r/min. The supernatant was diluted with ethanol (1:3) and its fluorescence was measured at 610 nm to measure the absorbance of EB. The results were expressed as micrograms per gram tissue.
TUNEL staining
The terminal deoxynucleotidyl transferase (TdT) dUTP Nick-End Labeling (TUNEL) assay was performed according to a previous study [14]. Rats were anesthetized 48 h after SAH and perfused transcardially with 0.9% saline, followed by 4% paraformaldehyde. Brains were removed and kept in 4% paraformaldehyde for 6 h, before being immersed in 30% sucrose for 3 days at 4°C. Brain sections (18 lm) were rinsed three times in PBS and blocked in 10% goat serum/PBS/0.1% Triton X-100 at room temperature for 2 h.
Biochemical analysis
Brain tissues were prepared in phosphate buffer (pH 7.4) to make 1:10 (w/v) homogenates. After centrifugation at 12,000 g for 20 min at 4°C, the supernatants were collected for biochemical analysis. The levels of malondialdehyde (MDA), glutathione (GSH), and glutathione peroxidase (GSH-Px) were determined using assay kits, which were purchased from Nanjing Jiancheng Biotech Ltd. (China). The values were normalized by the protein concentration of the sample, which was measured using a bicinchoninic acid (BCA) assay. The reactive oxygen species (ROS) level in the brain homogenates was measured using 2’,7’-dichlorodihydrofluorescein diacetate (Sigma, USA), as outlined in a previous study [14].
ELISA
Frozen brain tissue (200 mg) was homogenized, at 24 h after the induction of SAH, with a plastic homogenizer in cell lysis buffer containing 1 mM phenylmethanesulfonylfluoride (PMSF), 20 mM Tris pH 7.5, 150 mM NaCl, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM EDTA, 1% Na3VO4, and 0.5 μg/ml leupeptin. Each sample was then centrifuged at 14,000 g for 20 min at 4°C. The cytokine contents (i.e., TNF-α, IL-1β, IL-6, and IL-8) in the brain tissue were measured using rat immunoassay enzyme-linked immunosorbent assay (ELISA) kits (Boster Biological Technology, LTD, Wuhan, China), according to the manufacturer’s instructions. The results were expressed as picograms per milliliter homogenate.
Western blot
Total protein was extracted from the brain samples using a protein extraction kit (Beyotime Biotech. Co., China) according to the manufacturer’s instructions. Protein samples (50 μg) were separated using 10% SDS polyacrylamide gels and transferred to nitrocellulose membranes. The membranes were respectively incubated at 4°C for 2 h with a mouse monoclonal antibody against Nuclear factor E2 related factor 2 (Nrf2; 1:500, Abcam), HO-1 (1:200, Santa Cruz), NQO-1 (1:200, Santa Cruz), caspase-3 (1:500, Abcam), caspase-9 (1:500, Abcam), Bax (1:500, Abcam), Bcl-2 (1:500, Abcam), extracellular signal regulated kinases (ERK) 1/2 (1:500, Abcam), c-Jun N-terminal kinase (JNK) 1/2 (1:500, Abcam), p38 (1:500, Abcam), or GAPDH (1:500, Abcam). The nitrocellulose membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (1:3000, Zhongshan Biotechnology Co. LTD, Beijing) for 2 h at 25°C and developed with an enhanced chemiluminescence detection system. GAPDH was used as a loading control. The optical densities of protein bands were analyzed using Quantity One software (Bio-Rad).
Statistical analyses
Experimental data are presented as mean ± standard deviation (SD). Statistical analysis was performed using analysis of variance, followed by a Bonferroni test, for the individual comparisons between group means (SPSS 13.0 for Windows, USA). Statistical significance was set at P<0.05.
Results
Effect of SalA on neurological scores, brain edema, and BBB Permeability
When the rat brains were quickly removed 24 h after the SAH, subarachnoid blood clots were found on the ipsilateral side, around the circle of Willis, and within the ventral surface of the brain stem (Figure 1A). The neurological scores, which were significantly reduced at 24 h post-SAH, were significantly improved by the SalA (at 10 and 50 mg/kg) administration (P<0.01, Figure 1B-1). The elevation in the water content of the brain was elevated at 24 h post-SAH; this was attenuated by SalA (10 and 50 mg/kg; P<0.01, Figure 1B-2). Further, the obvious increase in EB content at 24 h post-SAH, was also significantly attenuated by the administration of SalA (10 and 50 mg/kg; P<0.01, Figure 1B-3).
Figure 1.

(A) Schematic representation of the cortex sample area taken for the assay. Effect of salvianolic acid A (SalA) administration on (B-1) neurological scores, (B-2) brain water content of cerebral cortex, (B-3) Evans blue (EB) content as indices for BBB permeability, in the control group, subarachnoid hemorrhage (SAH), and SalA (10 and 50 mg/kg)-treated SAH groups. Values represented as mean ± standard deviation (n = 6, each group). ##P<0.01 compared to control group; **P<0.01 compared to SAH group.
Effect of SalA on oxidative stress and expression of inflammatory cytokines
At 24 h post-SAH, compared to the control group, the ROS level of brain cortex homogenate was significantly increased in the vehicle-treated SAH group. However, compared with the vehicle-treated SAH group, the administration of SalA (10 and 50 mg/kg) significantly decreased the production of ROS in the brain cortex (P<0.01, Figure 2A-1). Further, the SAH significantly decreased the GSH concentration and GSH-Px enzyme activity in brain cortex, compared to that in the control group at 24 h. The GSH concentration and GSH-Px enzyme activity in the SalA (10 and 50 mg/kg)-treated SAH groups, however, were significantly increased compared with the vehicle-treated SAH group, at 24 h (P<0.05, Figure 2A-2 and 2A-3). Further, SAH significantly increased the concentration of MDA, i.e., an oxidative damage marker of lipids, in brain cortex, compared to the control group at 24 h. SalA (10 and 50 mg/kg) treatment, however, significantly reversed this increase (P<0.05, Figure 2A-4).
Figure 2.

(A-1) Reactive oxygen species (ROS) production of cerebral cortex homogenate, (A-2) glutathione (GSH) concentration, (A-3) glutathione peroxidase (GSH-Px) activities, (A-4) malondialdehyde (MDA) concentration of cerebral cortex in the sham-operated group, vehicle-treated, or salvianolic acid A (SalA)-treated subarachnoid hemorrhage (SAH) group. (B) Inflammation cytokines (TNF-α, IL-1β, IL-6, and IL-8) were identified by ELISA. Values are represented as mean ± standard deviation (n = 6, each group). #P<0.05, ##P<0.01 compared to sham-operated group; *P<0.05, **P<0.01 compared to SAH group.
Moreover, compared with the rats in the sham-operation group, the SAH increased the expression of inflammatory cytokines (i.e., TNF-α, IL-1β, IL-6, and IL-8) in the rat brain (P<0.01). SalA (10 and 50 mg/kg) significantly downregulated the release of TNF-α, IL-1β, IL-6 and IL-8 in rats with a SAH (P<0.05, Figure 2B).
Inhibition of cortical apoptosis after SAH by SalA
We assessed the brain cortex in each group using TUNEL to determine whether SalA prevents cortical apoptosis. Compared with the sham-operated group, numerous TUNEL-positive cells were observed in brain cortex of rats in the vehicle-treated SAH group (Figure 3). However, SalA treatment (10 and 50 mg/kg) significantly reduced increase in TUNEL-positive cells in the rats in the SalA-treated SAH groups, compared with that in the vehicle-treated SAH group (Figure 3).
Figure 3.
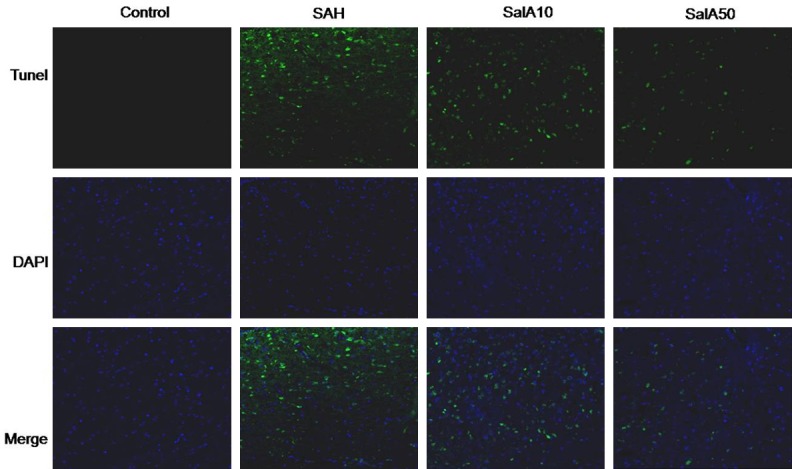
Representative confocal images show TUNEL staining of cerebral cortex in the control group, subarachnoid hemorrhage (SAH) group, and salvianolic acid A (SalA; 10 and 50 mg/kg)-treated SAH groups. The control group shows almost no TUNEL-positive cells. There are numerous TUNEL-positive cells in the SAH group and fewer TUNEL-positive cells in the SalA-treated (10 and 50 mg/kg) SAH group. Scale bar is 100 lm.
Effect of SalA on the expression of apoptosis-related proteins and Nrf2 signaling
The protein expression of caspase-3, caspase-9, and Bax/Bcl-2 was measured using western blot analysis. As shown in Figure 4A, a steep increase in the activities of caspase-3 and caspase-9 was observed in the brain cortex of rats that underwent a SAH. The increased activation of caspase-9 indicated that a SAH can initiate apoptosis, mediated by mitochondria and/or death receptors. However, the SalA (10 and 50 μM) treatment dramatically suppressed the activation of caspase-3 and caspase-9 induced by SAH, in a dose-dependent manner. In addition, compared to the control, the SAH also notably increased the expression of Bax/Bcl-2 (Figure 4A). The SalA (10 and 50 μM), however, suppressed this increase. In conclusion, the results demonstrated that SalA was an effective inhibitor of SAH-induced apoptosis in the brain cortex of rats. The decreased expression of Nrf2 and its downstream targets (HO-1 and NQO-1) in rats that underwent a SAH, was significantly mitigated with SalA (10 and 50 μM) treatment (Figure 4B).
Figure 4.
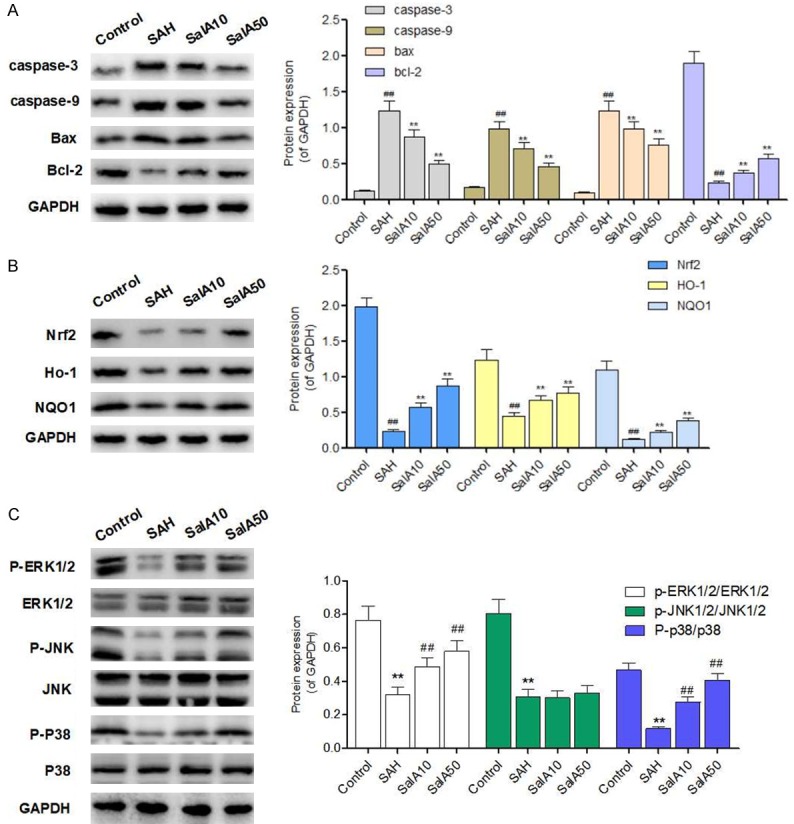
Salvianolic acid A (SalA) regulates the protein expression of apoptosis-related proteins, Nrf2 signaling, and MAPK signaling. A: Representative protein bands of Bax, Bcl-2, caspase-3 and caspase-9 expression in control, subarachnoid hemorrhage (SAH), and SalA-treated (10 and 50 mg/kg) groups were detected by western blot. B: Representative protein bands of Nrf2, HO-1 and NQO-1 expression in control, SAH, and SalA-treated (10 and 50 mg/kg) groups were detected by western blot. C: Representative protein bands of the proteins and the phosphorylation of the proteins in MAPK signaling in control, SAH, and SalA-treated (10 and 50 mg/kg) groups were detected by western blot. Values are represented as mean ± standard deviation (n = 5, each group). #P<0.05, ##P<0.01 compared to sham-operated group; *P<0.05, **P<0.01 compared to SAH group.
Effect of SalA on mitogen-activated protein kinase (MAPK) signaling
Next, we used a western blot to detect the protective effect of SalA on the phosphorylation of ERK1/2, P38, and JNK1/2, to determine the protective effect of SalA against oxidative damage via MAPK signaling. As shown in Figure 4C, the SAH increased the phosphorylation of p38 MAPK and decreased the phosphorylation of ERK; these changes were reversed by the SalA treatment. Although the phosphorylation of JNK was also increased in the SAH group, the SalA treatment had no effect on the phosphorylation of JNK.
Discussion
The current study demonstrated that the intraperitoneal administration of SalA (10 and 50 mg/kg/day) was protective for EBI following SAH, at least partly through its antioxidative, anti-inflammatory, and antiapoptotic effects. The evidence that led to this conclusion is as follows: SalA significantly improved neurological deficits, ameliorated brain edema and BBB permeability, inhibited inflammation factors, and suppressed oxidative stress and cortical neuron apoptosis, at 48 h after a SAH in a rat model. Further, SalA significantly decreased the SAH-induced formation of MDA and ROS, and increased the GSH concentration and GSH-Px activities. These findings suggested that treatment with SalA may provide an effective method for protecting the brain against EBI following SAH in rats.
The key pathological processes that underlie EBI after SAH include neuron cell necrosis, BBB damage, and brain edema [15]. These pathological processes significantly contribute to the inflammatory reaction and oxidative stress of the pressure, which forms after a SAH. The increase in ROS in EBI after SAH can cause neuronal necrosis and destroy the BBB. The destruction of the BBB and inflammation tends to cause vasogenic cerebral edema, which ultimately leads to neurological deterioration. The increased pressure due to brain edema is one of the primary reasons for neurological deterioration, while neurological changes after SAH are the secondary pathological process. In the current study, the brain water content was detected using the wet method. The results showed a significant increase in brain tissue water content in 24 h after SAH. SalA, however, significantly reduced cerebral edema. Further, the SAH increased ROS and MDA content and decreased GSH and GSH-Px; these changes were reversed by SalA treatment. These findings indicated that SalA reduced EBI after SAH, by inhibiting the inflammatory response, lipid peroxidation, and BBB damage.
The MDA content, which reflects lipid peroxidation [16,17], was significantly increased by the SAH in rats. This was indicative of the presence of oxidative stress damage due to SAH. Our results demonstrated that SalA reduced the content of MDA in rat model of SAH. Thus, the potential neuroprotective effect of SalA may be, at least partially, related to its inhibition of oxidative stress. Nrf2 plays a role in regulating antioxidants during EBI after a SAH [18,19]. The activation of Nrf2 signaling pathways activate the antioxidant signaling pathway to reduce the oxidative stress effect of brain tissue and BBB during EBI [20]. SalA activates the Nrf2 signaling pathway to reduce the pressure of liver oxidative stress [21-23]. In the current study, the expression of Nrf2 protein was significantly decreased at 24 h after SAH. The SalA treatment, however, increased the expression of Nrf2. This indicated that SalA relieved lipid peroxidation and possibly restored the BBB function, by activating Nrf2 antioxidant-associated signaling pathways. A previous study also reported that SalA alleviated ischemic brain injury through the inhibition of inflammation of neurogenesis in mice [24-26]. In our study, we found that SalA also showed an anti-inflammatory effect against EBI after SAH in rats. The SAH-induced increase of pro-inflammation cytokines, such as TNF-α, IL-1β, IL-6, and IL-8, was significantly reversed by SalA.
The MAPK signaling system, which is composed of ERK, JNK, and p38 MAPK [25,26], up-regulates Nrf2 signaling. Generally, p38 MAPK and JNK pathways can be activated by genotoxic agents and cytokine-mediating stress response, causing growth inhibition and apoptosis in cells. Cell apoptosis is closely associated with the activation of JNK and p38 MAPK signaling pathways and inactivation of the ERK signaling pathway, which is characterized by increased protein phosphorylation of p38 MAPK and/or JNK, and/or decreased protein phosphorylation of ERK [26,27]. Therefore, we detected their phosphorylation levels using western blot analysis. SAH increased the phosphorylation of p38 MAPK and decreased the phosphorylation of ERK. These changes were reversed by SalA treatment. JNK phosphorylation was also increased in SAH group. However, the SalA treatment had no effect on the phosphorylation of JNK. This indicated that SalA attenuated EBI following SAH by regulating MAPK signaling (except the JNK signaling pathway).
In conclusion, this study indicated that intraperitoneal administration of SalA (10 and 50 mg/kg/day) confers protection against SAH-induced EBI in rats through the inhibition of oxidative stress and inflammation, and the promotion of BDNF production in vivo. The SAH treatment improved the neurobehavioral deficit, attenuated brain edema and BBB permeability, and reduced oxidative stress and cortical apoptosis. These finding suggested that SAH could be a potential candidate treatment for EBI after SAH.
Acknowledgements
This work is supported by Fujian medical university qihang fund (No. 2016HQ097).
Disclosure of conflict of interest
None.
References
- 1.Al-Mufti F, Merkler AE, Boehme AK, Dancour E, May T, Schmidt JM, Park S, Connolly ES, Lavine SD, Meyers PM, Claassen J, Agarwal S. Functional outcomes and delayed cerebral ischemia following nonperimesencephalic angiogram-negative subarachnoid hemorrhage similar to aneurysmal subarachnoid hemorrhage. Neurosurgery. 2017 doi: 10.1093/neuros/nyx188. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 2.Madias JE. What could be the mechanism of the delayed normalization of the electrocardiogram in patients with aneurysmal subarachnoid hemorrhage-triggered takotsubo syndrome? World Neurosurg. 2017;102:682. doi: 10.1016/j.wneu.2017.02.055. [DOI] [PubMed] [Google Scholar]
- 3.Harmsen WJ, Ribbers GM, Zegers B, Sneekes EM, Praet SF, Heijenbrok-Kal MH, Khajeh L, van Kooten F, Neggers SJ, van den Berg-Emons RJ. Impaired muscle strength may contribute to fatigue in patients with aneurysmal subarachnoid hemorrhage. Int J Rehabil Res. 2017;40:29–36. doi: 10.1097/MRR.0000000000000197. [DOI] [PubMed] [Google Scholar]
- 4.Filipce V, Caparoski A. The effects of vasospasm and re-bleeding on the outcome of patients with subarachnoid hemorrhage from ruptured intracranial aneurysm. Pril (Makedon Akad Nauk Umet Odd Med Nauki) 2015;36:77–82. doi: 10.1515/prilozi-2015-0081. [DOI] [PubMed] [Google Scholar]
- 5.Lu J, Chen F, Cai B, Chen F, Kang D. A rabbit model of aneurysmal subarachnoid hemorrhage by ear central artery-suprasellar cistern shunt. J Clin Neurosci. 2017;44:300–305. doi: 10.1016/j.jocn.2017.05.031. [DOI] [PubMed] [Google Scholar]
- 6.Zhang H, He X, Wang Y, Sun X, Zhu L, Lei C, Yin J, Li X, Hou F, He W, Zhao D. Neuritin attenuates early brain injury in rats after experimental subarachnoid hemorrhage. Int J Neurosci. 2017;127:1087–1095. doi: 10.1080/00207454.2017.1337013. [DOI] [PubMed] [Google Scholar]
- 7.Suzuki H, Nakano F. To improve translational research in subarachnoid hemorrhage. Transl Stroke Res. 2017 doi: 10.1007/s12975-017-0546-2. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 8.Pan H, Li D, Fang F, Chen D, Qi L, Zhang R, Xu T, Sun H. Salvianolic acid A demonstrates cardioprotective effects in rat hearts and cardiomyocytes after ischemia/reperfusion injury. J Cardiovasc Pharmacol. 2011;58:535–542. doi: 10.1097/FJC.0b013e31822de355. [DOI] [PubMed] [Google Scholar]
- 9.Mahmood Q, Wang GF, Wu G, Wang H, Zhou CX, Yang HY, Liu ZR, Han F, Zhao K. Salvianolic acid A inhibits calpain activation and eNOS uncoupling during focal cerebral ischemia in mice. Phytomedicine. 2017;25:8–14. doi: 10.1016/j.phymed.2016.12.004. [DOI] [PubMed] [Google Scholar]
- 10.Fan H, Yang L, Fu F, Xu H, Meng Q, Zhu H, Teng L, Yang M, Zhang L, Zhang Z, Liu K. Cardioprotective effects of salvianolic Acid a on myocardial ischemia-reperfusion injury in vivo and in vitro. Evid Based Complement Alternat Med. 2012;2012:508938. doi: 10.1155/2012/508938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen Q, Xu T, Li D, Pan D, Wu P, Luo Y, Ma Y, Liu Y. JNK/PI3K/Akt signaling pathway is involved in myocardial ischemia/reperfusion injury in diabetic rats: effects of salvianolic acid A intervention. Am J Transl Res. 2016;8:2534–2548. [PMC free article] [PubMed] [Google Scholar]
- 12.Park S, Yamaguchi M, Zhou C, Calvert JW, Tang J, Zhang JH. Neurovascular protection reduces early brain injury after subarachnoid hemorrhage. Stroke. 2004;35:2412–2417. doi: 10.1161/01.STR.0000141162.29864.e9. [DOI] [PubMed] [Google Scholar]
- 13.Zhang ZY, Sun BL, Yang MF, Li DW, Fang J, Zhang S. Carnosine attenuates early brain injury through its antioxidative and anti-apoptotic effects in a rat experimental subarachnoid hemorrhage model. Cell Mol Neurobiol. 2015;35:147–157. doi: 10.1007/s10571-014-0106-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moroni F, Carpenedo R, Cozzi A, Meli E, Chiarugi A, Pellegrini-Giampietro DE. Studies on the neuroprotective action of kynurenine mono-oxygenase inhibitors in postischemic brain damage. Adv Exp Med Biol. 2003;527:127–136. doi: 10.1007/978-1-4615-0135-0_15. [DOI] [PubMed] [Google Scholar]
- 15.Hu Q, Li T, Wang L, Xie Y, Liu S, Bai X, Zhang T, Bo S, Xin D, Xue H, Li G, Wang Z. Neuroprotective effects of a smoothened receptor agonist against early brain injury after experimental subarachnoid hemorrhage in rats. Front Cell Neurosci. 2016;10:306. doi: 10.3389/fncel.2016.00306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu W, Parakramaweera R, Teng S, Gowda M, Sharad Y, Thakker-Varia S, Alder J, Sesti F. Oxidation of KCNB1 potassium channels causes neurotoxicity and cognitive impairment in a mouse model of traumatic brain injury. J Neurosci. 2016;36:11084–11096. doi: 10.1523/JNEUROSCI.2273-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Manevich Y, Hutchens S, Halushka PV, Tew KD, Townsend DM, Jauch EC, Borg K. Peroxiredoxin VI oxidation in cerebrospinal fluid correlates with traumatic brain injury outcome. Free Radic Biol Med. 2014;72:210–221. doi: 10.1016/j.freeradbiomed.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu X, Zhang X, Ma K, Zhang R, Hou P, Sun B, Yuan S, Wang Z, Liu Z. Matrine alleviates early brain injury after experimental subarachnoid hemorrhage in rats: possible involvement of PI3K/Akt-mediated NF-kappaB inhibition and Keap1/Nrf2-dependent HO-1 inductionn. Cell Mol Biol (Noisy-le-grand) 2016;62:38–44. [PubMed] [Google Scholar]
- 19.Zhao X, Wen L, Dong M, Lu X. Sulforaphane activates the cerebral vascular Nrf2-ARE pathway and suppresses inflammation to attenuate cerebral vasospasm in rat with subarachnoid hemorrhage. Brain Res. 2016;1653:1–7. doi: 10.1016/j.brainres.2016.09.035. [DOI] [PubMed] [Google Scholar]
- 20.Wang Z, Chen G, Zhu WW, Zhou D. Activation of nuclear factor-erythroid 2-related factor 2 (Nrf2) in the basilar artery after subarachnoid hemorrhage in rats. Ann Clin Lab Sci. 2010;40:233–239. [PubMed] [Google Scholar]
- 21.Wu P, Yan Y, Ma LL, Hou BY, He YY, Zhang L, Niu ZR, Song JK, Pang XC, Yang XY, Du GH. Effects of the Nrf2 protein modulator salvianolic acid A alone or combined with metformin on diabetes-associated macrovascular and renal injury. J Biol Chem. 2016;291:22288–22301. doi: 10.1074/jbc.M115.712703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu DS, Wang YS, Bi YL, Guo ZP, Yuan YJ, Tong SM, Su RC, Ge LH, Wang J, Pan YL, Guan TT, Cao Y. Salvianolic acid A ameliorates the integrity of blood-spinal cord barrier via miR-101/Cul3/Nrf2/HO-1 signaling pathway. Brain Res. 2017;1657:279–287. doi: 10.1016/j.brainres.2016.12.007. [DOI] [PubMed] [Google Scholar]
- 23.Zhang H, Liu YY, Jiang Q, Li KR, Zhao YX, Cao C, Yao J. Salvianolic acid A protects RPE cells against oxidative stress through activation of Nrf2/HO-1 signaling. Free Radic Biol Med. 2014;69:219–228. doi: 10.1016/j.freeradbiomed.2014.01.025. [DOI] [PubMed] [Google Scholar]
- 24.Chien MY, Chuang CH, Chern CM, Liou KT, Liu DZ, Hou YC, Shen YC. Salvianolic acid A alleviates ischemic brain injury through the inhibition of inflammation and apoptosis and the promotion of neurogenesis in mice. Free Radic Biol Med. 2016;99:508–519. doi: 10.1016/j.freeradbiomed.2016.09.006. [DOI] [PubMed] [Google Scholar]
- 25.Armstead WM, Riley J, Vavilala MS. Norepinephrine protects cerebral autoregulation and reduces hippocampal necrosis after traumatic brain injury via blockade of ERK MAPK and IL-6 in juvenile pigs. J Neurotrauma. 2016;33:1761–1767. doi: 10.1089/neu.2015.4290. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26.Armstead WM, Bohman LE, Riley J, Yarovoi S, Higazi AA, Cines DB. tPA-S(481)A prevents impairment of cerebrovascular autoregulation by endogenous tPA after traumatic brain injury by upregulating p38 MAPK and inhibiting ET-1. J Neurotrauma. 2013;30:1898–1907. doi: 10.1089/neu.2013.2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bachstetter AD, Rowe RK, Kaneko M, Goulding D, Lifshitz J, Van Eldik LJ. The p38alpha MAPK regulates microglial responsiveness to diffuse traumatic brain injury. J Neurosci. 2013;33:6143–6153. doi: 10.1523/JNEUROSCI.5399-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]