

Abstract
Oxidative stress has been considered a major contributing factor to traumatic brain injury (TBI). Formononetin, a phytoestrogen that belongs to the flavonoid family, is extracted from plants and herbs such as the red clover. Growing evidence demonstrates that formononetin has antioxidant properties. Therefore, formononetin has potential use in treating oxidative stress injuries in TBI. In this study, the neuroprotective and antioxidant effects of formononetin against TBI, as well as the related probable mechanisms, were investigated. The TBI model was produced in male Wistar rats through Feeney’s weight-drop model. At 1 day after TBI, the neurological function score and brain water content were assessed. TUNEL assay was used to determine neuronal apoptosis. The expression levels of miR-155, HO-1, and BACH1 were measured by RT-PCR and western blotting. Consequently, our findings showed that formononetin pretreatment for 5 days significantly improved the neurological scores, reduced brain edema and inhibited neuronal apoptosis in rats after TBI. MiR-155 was substantially decreased and BACH1 expression was significantly increased in the TBI model, while pretreatment with formononetin dramatically up-regulated the expression levels of miR-155 and HO-1 and down-regulated the protein expression of BACH1 in rats after TBI. In summary, formononetin has been shown to have neuroprotective effects, and the mechanisms of this effect may be associated with its inhibition of oxidative stress and activation of Nrf2-dependent antioxidant pathways in TBI.
Keywords: Formononetin, phytoestrogen, traumatic brain injury (TBI), miR-155, heme oxygenase-1 (HO-1), BACH1
Introduction
Traumatic brain injury (TBI), a major cause of death and disability around the globe, occurs when an external mechanical force damages the brain structure and can lead to the temporary or permanent impairment of cognitive, physical and psychosocial functions. The most common causes of TBI include violence, traffic accidents, sports and building work. In addition to the damage caused at the time of injury (primary brain injury), a variety of events that occur during the minutes to days following the injury may result in secondary brain injury. The mechanisms of secondary injuries include initiation of the inflammatory cascade, the production of free radicals, an overload of excitatory neurotransmitters, apoptosis, necrosis, destruction of the blood-brain barrier, and an altered energy metabolism [1,2]. Therefore, the pathophysiology of TBI is inseparably linked to numerous detrimental neurobiological consequences.
In recent decades, a considerable amount of research has found that heme oxygenase plays a protective role in the oxidative stress and inflammatory response of TBI [3-5]. Heme oxygenase is induced by a wide variety of stimuli, from oxidative stress to proinflammatory cytokines and endotoxins. Heme oxygenase-1 (HO-1) is an enzyme that catalyzes the degradation of heme to biliverdin, free iron, and carbon monoxide. The metabolites of heme can mediate the cytoprotective and anti-inflammatory properties of HO-1 by serving as antioxidants, thus alleviating the damage caused by reactive oxygen species (ROS).
MicroRNAs, a large family of small (18-24 nucleotide) non-coding RNAs, have been recently considered new players in regulating diverse aspects of cell biology, including oxidative stress and inflammation. Numerous studies have indicated that microRNAs are able to “fine-tune” the nuclear factor erythroid 2-related factor 2 (Nrf2) pathway and to maintain redox homeostasis by regulating the generation of reactive oxygen species [6,7]. For instance, it has been shown that miR-155 is associated with fluoro-octane sulfonate-induced oxidative hepatic damage via a Nrf2-dependent pathway [8]. However, the signaling events regulating cellular microRNA activity and/or function in the oxidative brain damage of TBI remain largely unknown.
Formononetin (FN) is an O-methylated isoflavone phytoestrogen extracted from plants and herbs, such as the red clover, that has been proven to improve blood microcirculation, induce apoptosis in cancer cells, regulate antioxidant effects, etc. [9-11]. In our previous study, it was suggested that formononetin protects TBI rats against neurological lesions and that the underlying mechanisms of this protection are associated with the inhibition of both the intracephalic inflammatory response and oxidative stress [12]. Specifically, formononetin has been shown to up-regulate the protein expression of Nrf2 in TBI rats. However, the function of formononetin and the mechanisms potentially underlying its activity remain unclear. Thus, the current study investigated the involvement of miR-155 in formononetin-induced neuroprotection against TBI and its potential interactions with the Nrf2/ARE/HO-1 signaling pathway.
Materials and methods
Materials
Formononetin (molecular weight = 268.3, purity ≥ 98.0%, verified by high-performance liquid chromatography) was purchased from Sigma-Aldrich Trading Company Limited Shanghai, China and was dissolved in dimethyl sulfoxide (DMSO). The formononetin stock (200 mM) was stored at 4°C until use.
Experimental animals and formononetin treatment
Young adult male Wistar rats (Grade II, License No. 20160203) weighing 220-280 g were obtained from the Experimental Animal Center of Guilin Medical University. The animals were housed in a temperature-controlled (22-28°C) environment under a 12 h light/dark cycle with free access to food and water and were acclimatized for at least 4 days prior to experimentation.
The TBI rat model used in this study was based on Feeney’s weight-drop model as previously reported with some modifications [13,14]. Briefly, the tested rats were anesthetized with an intraperitoneal injection of 2% sodium pentobarbital solution (3 mL·kg-1, 60 mg·kg-1) and then were placed on a stereotaxic frame. Rectal temperature was maintained at 37-38°C with a heating pad. A 5-mm-diameter bone window was made in the skull with the dura intact by a dental drill (cerebral location: dextral coronal section at 1 mm, midline scalp at 2 mm). Subsequently, traumatic injury was induced by striking the dura with an impactor; briefly, a 40-g steel weight with a flat end was dropped from 30-cm high onto a small pillar (4 mm diameter and 5 mm long) to bump the dura. The weight impacted the intact dura and caused a cortical contusion. After impact, the scalp was sutured closed, and the animals were allowed to recover from the anesthesia. All of the operations were carried out with aseptic techniques. TBI was confirmed in the rat model by the observation of limb convulsions, transitory apnea, and unconsciousness post-injury. In this study, the experimental rats were randomized into different groups: 10 control rats in the sham group (subjected to identical surgical procedures but without the impact), 10 TBI rats in the TBI group, and 20 TBI rats in the formononetin-treated groups (intraperitoneal injection with formononetin 10 mg·kg-1·d-1 or 30 mg·kg-1·d-1 for 5 days before TBI, with 10 rats in each treatment group).
Assessment of neurological conditions
Neurological function of the tested rats was investigated using the modified neurological severity score (mNSS) as previously reported. As a composite of the motor, sensory and reflex tests, the injury severity scores were graded on a scale of 0 to 18, and the higher the score was, the more severe the injury. Neurological function was assessed 1 day after the TBI was incurred. This evaluation was performed by an observer who was blinded to the treatment groups.
Brain tissue sampling preparation
The rats were deeply anesthetized by intraperitoneal injection of ethyl carbamate (200 mg·kg-1). Intracephalic perfusion was completed with 250 mL cold saline to remove the remnant blood. Brain tissue from the lesioned zone of each group was sampled. Some tissue samples were used for PCR, and others were used for western blotting analyses.
Evaluation of brain edema
The injured brain tissues of the TBI rats were immediately harvested and weighed on an electronic balance to obtain the wet weight; then, the tissues were dried for 24 h in an oven at 85°C to constant weight and reweighed to obtain the dry weight. The moisture content of the brain was calculated using the following equation: cerebral moisture content (%) = (wet weight-dry weight)/wet weight × 100%.
Hematoxylin and eosin (H&E) staining and terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) assay
Rat brains were collected, post-fixed, dehydrated, and embedded in paraffin. The paraffin sections were subjected to dewaxing and hydration and then stained with H&E for 5 min. The slices were differentiated with 0.6% hydrochloric acid in alcohol for 30 s, counterstained in acidified eosin alcohol (pH 4.2) for 3 min, dehydrated and cleared. The pathological lesion zones of the brain were examined under a light microscope (Leica, Wetzlar, Germany).
To visualize DNA fragmentation, TUNEL staining was performed using an In Situ Cell Death Detection Kit® (Roche, Mannheim, Germany) according to the manufacturer’s protocol. The sections were post-fixed in ethanol-acetic acid (2:1), rinsed and then incubated with proteinase K (100 μg/ml). The sections were then rinsed and incubated in 3% H2O2, permeabilized with 0.5% Triton X-100, rinsed again, and incubated in the TUNEL reaction mixture. Finally, the sections were rinsed and visualized using a Converter-POD with 0.03% 3,3’-diaminobenzidine (DAB). Mayer’s hematoxylin (DAKO, Glostrup, Denmark) was used as a counterstain, and the sections were mounted onto gelatin-coated slides. The slides were air-dried overnight at room temperature, and cover slips were mounted using Permount®. The sections were analyzed under a light microscope.
Quantitative real-time PCR (qRT-PCR) detection for miR-155, HO-1 and BACH1
Frozen samples from injured zones of the brain were homogenized, and total RNA was extracted with TRIzol reagent (Beyotime Institute of Biotechnology, Shanghai, China). Total RNA was reverse transcribed with a Revert Aid™ First Strand cDNA Synthesis Kit (Fermentas, Burlington, Ontario, USA) following the manufacturer’s instructions. miR-155, HO-1 and BACH1 expression levels were measured by quantitative real-time PCR using the ABI PRISM 7500 sequence detection system (Applied Biosystems, Carlsbad, CA). The threshold cycle (Ct) data were recorded by the software provided, and the relative expression levels of miR-155 and HO-1 were calculated with the 2-ΔΔCT method. Transcript levels were normalized to the expression level of GAPDH in all cases.
Western blotting analysis for HO-1 and BACH1 expression levels
To assess the protein expression levels of HO-1 and BACH1 after treatments, western blotting analysis was performed. In brief, the cerebral tissue from the lesion zone was separated and homogenized with RIPA lysis buffer (Beyotime Institute of Biotechnology, Shanghai, China). The lysates were incubated on ice for 15 min and centrifuged at 10,000 rpm for 10 min at 4°C. Supernatant protein content was determined using a bicinchoninic acid assay protocol (Sigma-Aldrich, USA). Then, sodium dodecyl sulfate (SDS) sample loading buffer was added to the supernatant, and the mixture was boiled at 100°C for 5 min. Samples containing 50 µg of protein were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and electrotransferred onto polyvinylidene difluoride (PVDF) membranes (Millipore, Bedford, MA, USA). After blocking, the membranes were incubated overnight at 4°C with the following primary antibodies: rabbit anti-rat HO-1, rabbit anti-rat BACH1 antibody (both from Santa Cruz, CA, USA; 1:500), or mouse anti-rat β-actin antibody (Santa Cruz, CA, USA; 1:4000). After three washes in TBST, the membranes were incubated with the appropriate horseradish peroxidase-coupled secondary antibody at room temperature for 1 h. Immunoreactive bands were scanned and visualized using an enhanced chemiluminescence detection system (Life Technologies, USA) and X-ray film. The optical absorbance of bands was quantified with a computer-assisted image analysis system (Bio-Rad, USA) and normalized to the β-actin protein expression level.
Statistical analysis
The results are expressed as the means ± standard deviation. Data were examined using one-way analysis of variance, followed by Tukey’s post hoc test. Statistical analysis was conducted with SPSS 19.0 software (IBM, Chicago, IL, USA). The threshold of significance was defined as P < 0.05.
Results
Formononetin protected against traumatic brain injury in rats
To explore the overall effects of formononetin on functional recovery after traumatic brain injury, we assessed the mNSS 1 day post-injury (Figure 1A). The TBI group showed significant functional deficits compared with the function of the sham group. In addition, the formononetin groups had a lower mNSS than the TBI group (P < 0.05, n = 10), and the decrease in neurological scores was more significant in the group receiving 30 mg·kg-1 formononetin, indicating that formononetin improved brain function.
Figure 1.
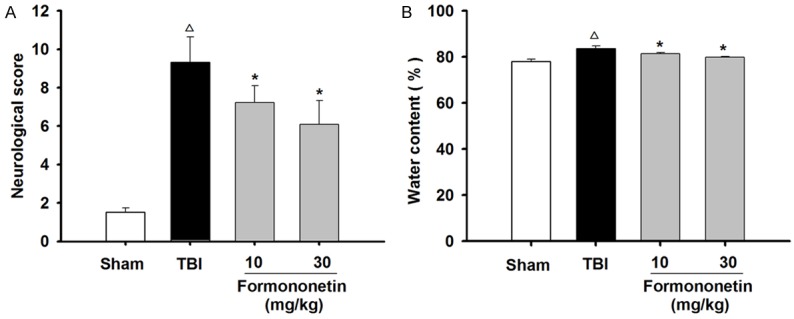
Formononetin decreased the modified neurological severity score (mNSS) and brain water content of TBI rats. A. Neurological severity score was evaluated at 1 day after TBI. B. Water content was calculated by the following equation: (wet weight - dry weight)/wet weight × 100%. The results are expressed as mean ± SD. Δ P < 0.05 TBI vs. Sham group; *P < 0.05 vs. TBI group; n = 10.
To analyze TBI-induced cerebral hemorrhaging in the experimental rats, cerebral water content was evaluated. As shown in Figure 1B, TBI caused a significant increase in the tissue water percentage. Compared with the TBI rats, the cerebral water content was gradually lowered following formononetin treatments (P < 0.05, n = 10).
Formononetin mitigated the pathological lesions and inhibited neuronal apoptosis in the brain tissue of TBI rats
Histological changes in the brain tissues of each group were observed using the H&E staining assay. In the sham group, the nerve cells of brain tissue were arranged uniformly, accompanied by abundant cytoplasm and numerous neurons. In addition, the extracellular space was normally distributed. In contrast, a great number of degenerative and necrotized nerve cells were observed in the pathological lesions of the TBI group. After the formononetin treatments, the edema and necrosis in the lesioned zones of the brain were significantly attenuated, and the number of nerve cells was effectively increased compared with that of the TBI group (Figure 2A).
Figure 2.

Formononetin ameliorated the morphological lesions and reduced the neuronal apoptosis rate in the damaged brains of TBI rats. A. Representative images of H&E staining in the pathological lesion zones. B. The percentage of apoptosis was analyzed by TUNEL staining. The results are expressed as mean ± SD. Δ P < 0.05 TBI vs. Sham group; *P < 0.05 vs. TBI group; n = 10.
We next detected the effect of formononetin on neuronal apoptosis in TBI rats. The TUNEL staining assay showed that there was a significant increase in the apoptotic rate of the TBI group (28.19 ± 3.14%) compared with that of the sham group (2.32 ± 0.13%). Compared with the TBI group, treatment with formononetin showed clear signs of a dose-dependent mitigation of the apoptotic rate (10 mg·kg-1, 23.12 ± 3.19%; 30 mg·kg-1, 14.21 ± 2.45%) (P < 0.05) (Figure 2B). These results demonstrated that formononetin ameliorated the morphological lesions of brain tissue and inhibited neuronal apoptosis in TBI rats.
Formononetin up-regulated the expression levels of miR-155 and HO-1 in TBI rats
To further analyze the formononetin-induced neuroprotective and antioxidative effects, we examined the expression levels of miR-155 and HO-1 (Figure 3). The results from the real-time PCR analysis revealed that miR-155 expression in the injured zone of TBI rats was significantly decreased compared with that in the sham group. Strikingly, formononetin treatment in TBI rats resulted in concentration-dependent up-regulation of miR-155 expression in the lesioned brain (P < 0.05).
Figure 3.

The effects of formononetin on miR-155 and HO-1 levels in TBI rats. The RNA and protein extracted from the injured zone of TBI rats were analyzed by qRT-PCR and western blotting, respectively. A. Quantitative analysis using qRT-PCR of the expression of miR-155, normalized to GAPDH expression. B. Quantitative analysis using qRT-PCR of the expression of OH-1, normalized to GAPDH expression. C. Quantitative analysis using western blot of the expression of miR-155, normalized to β-actin expression. The results are expressed as mean ± SD. Δ P < 0.05 TBI vs. Sham group; *P < 0.05 vs. TBI group; n = 3.
In addition, the relative mRNA and protein expression levels of HO-1 were both up-regulated after TBI. Treatment with formononetin further enhanced the expression of HO-1 in a dose-dependent manner (P < 0.05), indicating that formononetin exhibited antioxidant effects that were cytoprotective against TBI.
Formononetin decreased the mRNA and protein expression of BACH1 in TBI rats
As one of the key transcriptional repressor proteins in the Nrf2/ARE/HO-1 signaling pathway, BACH1 plays important roles in the oxidative stress and inflammatory response of TBI. Therefore, we examined the changes in BACH1 expression in the injured zone of TBI rat brains. As shown in Figure 4, the mRNA and protein expression levels of BACH1 were markedly increased in the TBI model vs. those in the sham-operated rats, whereas formononetin significantly attenuated the increase in BACH1 levels in the brain tissue of TBI rats (P < 0.05). This indicated that formononetin affected the expression of BACH1 after TBI.
Figure 4.
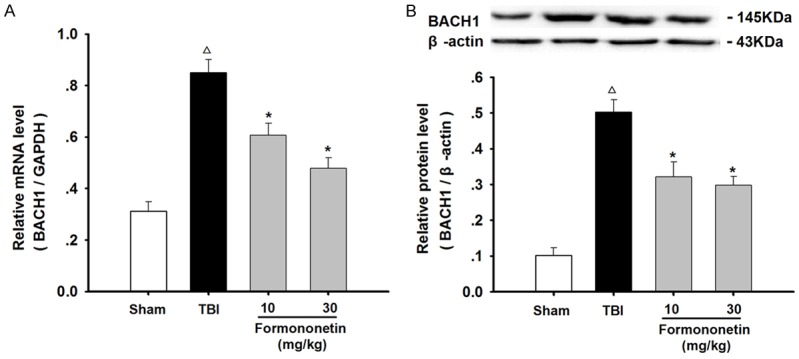
Formononetin down-regulated the mRNA and protein expression levels of BACH1 in the damaged brain of TBI rats. The RNA and protein extracted from the injured zone in rats 24 h after TBI were analyzed for BACH1 expression by qRT-PCR and western blot. A. The mRNA expression of BACH1 was normalized to GAPDH expression. B. The protein expression of BACH1 was normalized to β-actin. The results are expressed as mean ± SD. Δ P < 0.05 TBI vs. Sham group; *P < 0.05 vs. TBI group; n = 3.
Discussion
There is abundant evidence that oxidative stress is implicated in the pathology of TBI damage. However, the evidence is too limited to effectively treat clinical patients suffering from moderate to severe TBI. Reassuringly, the neuroprotective effects of estradiol and phytoestrogen have gained attention in TBI research [15-17]. However, the mechanisms underlying these protective effects are unclear. In the present study, formononetin, a typical isoflavone phytoestrogen, ameliorated brain dysfunction, alleviated cerebral edema and inhibited neuronal apoptosis. In addition, the administration of formononetin increased the expression of miR-155 and attenuated TBI-induced oxidative stress by up-regulating HO-1 expression and down-regulating BACH1 expression. These results suggest that formononetin provides neuroprotection in TBI rats by regulating redox homeostasis via the modulation of key molecules associated with the Nrf2/ARE/HO-1 antioxidant pathway.
Accumulating evidence shows that the Nrf2/ARE/HO-1 pathway participates in the oxidative stress of secondary brain injuries after TBI [18,19]. As a pivotal regulator of the antioxidant response element (ARE), nuclear factor E2-related factor 2 (Nrf2) is responsible for activating transcription in response to oxidative stress. Under various stress conditions, Nrf2 translocates from the cytoplasm to the nucleus where it binds to ARE and transactivates the expression of a series of enzymes, such as heme oxygenase-1 (HO-1), NQO1, and BACH1. These Nrf2-dependent gene products play key roles in protecting cells from oxidative or xenobiotic damage [20]. The expression of HO-1, a Nrf2-regulated cytoprotective enzyme, has been proven to be altered after TBI and cerebral ischemia [21,22]. BACH1 is a transcriptional repressor of the HO-1 gene and plays a critical role in protecting tissue from oxidative stress [23,24]. Our results indicated that the oxidative stress that occurred in TBI rats was accompanied by neuronal apoptosis, as shown with the TUNEL assay. Interestingly, these abnormal conditions were reversed after formononetin treatment and were accompanied by an increase in HO-1 expression and a decrease in BACH1.
During the past decade, miRNAs have been considered new players in regulating the Nrf2 antioxidant pathway and in maintaining redox homeostasis. Pulkkinen, K.H. et al. demonstrated that miR-155 inhibited BACH1 translation in human umbilical vein endothelial cells (HUVECs), resulting in enhanced HO-1 expression and providing cytoprotection against oxidative stress [25]. Recently, emerging studies have also reported changes in the expression of several miRNAs in the brain tissue following TBI [26-28]. Yang T. et al. observed that the serum expression levels of miR-93, miR-191, and miR-499 were significantly increased in TBI patients compared with the levels of the controls at all examined time points [29]. Our present study shows that elevated miR-155 expression is beneficial in formononetin-mediated neuroprotection after TBI. This finding suggests that the protective effect of formononetin in TBI may be associated with the inhibition of BACH1 translation and enhancement of HO-1 expression. Our previous study also demonstrated that formononetin up-regulated Nrf2 and antioxidant enzymes expression to exert neuroprotective effects in TBI rats [12]. Considering these results, we speculated that TBI results in the insufficient antioxidant capacity of the brain, thereby inducing neural impairments. The formononetin-mediated activation of the Nrf2 pathway contributes to reducing oxidative damage and alleviating brain injury in TBI.
Although our studies have provided some information on the pathophysiology during the acute stage after TBI, notable differences exist between animal models and clinical treatments. To verify the relationship between Nrf2/ARE/HO-1 pathway activation and the curative effects of formononetin in TBI, further studies evaluating the injury response and the mechanisms of the functional deficits are warranted.
Conclusion
In conclusion, we demonstrated that formononetin protects the brain against injury after TBI via Nrf2-dependent antioxidant pathways. The mechanisms underlying the protection exhibited by formononetin may involve the suppression of BACH1 and the increased expression of miR-155 and HO-1. Overall, our results suggest that formononetin might be a promising drug candidate for the treatment of TBI.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81460211, 81560378) and the Guangxi Emergency and Medical Rescue Talent Highland (GXJZ201506).
Disclosure of conflict of interest
None.
References
- 1.Abdul-Muneer PM, Chandra N, Haorah J. Interactions of oxidative stress and neurovascular inflammation in the pathogenesis of traumatic brain injury. Mol Neurobiol. 2015;51:966–979. doi: 10.1007/s12035-014-8752-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Simon DW, McGeachy MJ, Bayir H, Clark RS, Loane DJ, Kochanek PM. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat Rev Neurol. 2017;13:171–191. doi: 10.1038/nrneurol.2017.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang Y, Wang H, Li L, Li X, Wang Q, Ding H, Wang X, Ye Z, Wu L, Zhang X, Zhou M, Pan H. Sinomenine provides neuroprotection in model of traumatic brain injury via the Nrf2-ARE pathway. Front Neurosci. 2016;10:580. doi: 10.3389/fnins.2016.00580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ding K, Wang H, Xu J, Li T, Zhang L, Ding Y, Zhu L, He J, Zhou M. Melatonin stimulates antioxidant enzymes and reduces oxidative stress in experimental traumatic brain injury: the Nrf2-ARE signaling pathway as a potential mechanism. Free Radic Biol Med. 2014;73:1–11. doi: 10.1016/j.freeradbiomed.2014.04.031. [DOI] [PubMed] [Google Scholar]
- 5.Zhang L, Wang H. Targeting the NF-E2-Related factor 2 pathway: a novel strategy for traumatic brain injury. Mol Neurobiol. 2017 doi: 10.1007/s12035-017-0456-z. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 6.Cheng X, Ku CH, Siow RC. Regulation of the Nrf2 antioxidant pathway by microRNAs: new players in micromanaging redox homeostasis. Free Radic Biol Med. 2013;64:4–11. doi: 10.1016/j.freeradbiomed.2013.07.025. [DOI] [PubMed] [Google Scholar]
- 7.Wang P, Liang X, Lu Y, Zhao X, Liang J. MicroRNA-93 downregulation ameliorates cerebral ischemic injury through the Nrf2/HO-1 defense pathway. Neurochem Res. 2016;41:2627–2635. doi: 10.1007/s11064-016-1975-0. [DOI] [PubMed] [Google Scholar]
- 8.Wan C, Han R, Liu L, Zhang F, Li F, Xiang M, Ding W. Role of miR-155 in fluorooctane sulfonate-induced oxidative hepatic damage via the Nrf2-dependent pathway. Toxicol Appl Pharmacol. 2016;295:85–93. doi: 10.1016/j.taap.2016.01.023. [DOI] [PubMed] [Google Scholar]
- 9.Chen J, Zhao X, Li X, Wu Y. Calycosin induces apoptosis by the regulation of ERbeta/miR-17 signaling pathway in human colorectal cancer cells. Food Funct. 2015;6:3091–3097. doi: 10.1039/c5fo00374a. [DOI] [PubMed] [Google Scholar]
- 10.Chen J, Sun L. Formononetin-induced apoptosis by activation of Ras/p38 mitogenactivated protein kinase in estrogen receptorpositive human breast cancer cells. Horm Metab Res. 2012;44:943–948. doi: 10.1055/s-0032-1321818. [DOI] [PubMed] [Google Scholar]
- 11.Zhang X, Bi L, Ye Y, Chen J. Formononetin induces apoptosis in PC-3 prostate cancer cells through enhancing the Bax/Bcl-2 ratios and regulating the p38/Akt pathway. Nutr Cancer. 2014;66:656–661. doi: 10.1080/01635581.2014.894098. [DOI] [PubMed] [Google Scholar]
- 12.Li Z, Dong X, Zhang J, Zeng G, Zhao H, Liu Y, Qiu R, Mo L, Ye Y. Formononetin protects TBI rats against neurological lesions and the underlying mechanism. J Neurol Sci. 2014;338:112–117. doi: 10.1016/j.jns.2013.12.027. [DOI] [PubMed] [Google Scholar]
- 13.Chen J, Sanberg PR, Li Y, Wang L, Lu M, Willing AE, Sanchez-Ramos J, Chopp M. Intravenous administration of human umbilical cord blood reduces behavioral deficits after stroke in rats. Stroke. 2001;32:2682–2688. doi: 10.1161/hs1101.098367. [DOI] [PubMed] [Google Scholar]
- 14.Xiong Y, Mahmood A, Chopp M. Animal models of traumatic brain injury. Nat Rev Neurosci. 2013;14:128–142. doi: 10.1038/nrn3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berry C, Ley EJ, Tillou A, Cryer G, Margulies DR, Salim A. The effect of gender on patients with moderate to severe head injuries. J Trauma. 2009;67:950–953. doi: 10.1097/TA.0b013e3181ba3354. [DOI] [PubMed] [Google Scholar]
- 16.Soltani Z, Khaksari M, Jafari E, Iranpour M, Shahrokhi N. Is genistein neuroprotective in traumatic brain injury? Physiol Behav. 2015;152:26–31. doi: 10.1016/j.physbeh.2015.08.037. [DOI] [PubMed] [Google Scholar]
- 17.Engler-Chiurazzi EB, Brown CM, Povroznik JM, Simpkins JW. Estrogens as neuroprotectants: estrogenic actions in the context of cognitive aging and brain injury. Prog Neurobiol. 2017;157:188–211. doi: 10.1016/j.pneurobio.2015.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Z, Wang H, Shi X, Li L, Zhou M, Ding H, Yang Y, Li X, Ding K. DL-3-n-Butylphthalide (NBP) provides neuroprotection in the mice models after traumatic brain injury via Nrf2-ARE signaling pathway. Neurochem Res. 2017;42:1375–1386. doi: 10.1007/s11064-017-2186-z. [DOI] [PubMed] [Google Scholar]
- 19.Cheng T, Wang W, Li Q, Han X, Xing J, Qi C, Lan X, Wan J, Potts A, Guan F, Wang J. Cerebroprotection of flavanol (-)-epicatechin after traumatic brain injury via Nrf2-dependent and -independent pathways. Free Radic Biol Med. 2016;92:15–28. doi: 10.1016/j.freeradbiomed.2015.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem. 2009;284:13291–13295. doi: 10.1074/jbc.R900010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shu L, Wang C, Wang J, Zhang Y, Zhang X, Yang Y, Zhuo J, Liu J. The neuroprotection of hypoxic preconditioning on rat brain against traumatic brain injury by up-regulated transcription factor Nrf2 and HO-1 expression. Neurosci Lett. 2016;611:74–80. doi: 10.1016/j.neulet.2015.11.012. [DOI] [PubMed] [Google Scholar]
- 22.Lou J, Cao G, Li R, Liu J, Dong Z, Xu L. Betacaryophyllene attenuates focal cerebral ischemia-reperfusion injury by Nrf2/HO-1 pathway in rats. Neurochem Res. 2016;41:1291–1304. doi: 10.1007/s11064-016-1826-z. [DOI] [PubMed] [Google Scholar]
- 23.Zhou Y, Wu H, Zhao M, Chang C, Lu Q. The bach family of transcription factors: a comprehensive review. Clin Rev Allergy Immunol. 2016;50:345–356. doi: 10.1007/s12016-016-8538-7. [DOI] [PubMed] [Google Scholar]
- 24.Reichard JF, Motz GT, Puga A. Heme oxygenase-1 induction by NRF2 requires inactivation of the transcriptional repressor BACH1. Nucleic Acids Res. 2007;35:7074–7086. doi: 10.1093/nar/gkm638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pulkkinen KH, Yla-Herttuala S, Levonen AL. Heme oxygenase 1 is induced by miR-155 via reduced BACH1 translation in endothelial cells. Free Radic Biol Med. 2011;51:2124–2131. doi: 10.1016/j.freeradbiomed.2011.09.014. [DOI] [PubMed] [Google Scholar]
- 26.Sabirzhanov B, Zhao Z, Stoica BA, Loane DJ, Wu J, Borroto C, Dorsey SG, Faden AI. Downregulation of miR-23a and miR-27a following experimental traumatic brain injury induces neuronal cell death through activation of proapoptotic Bcl-2 proteins. J Neurosci. 2014;34:10055–10071. doi: 10.1523/JNEUROSCI.1260-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu L, Sun T, Liu Z, Chen X, Zhao L, Qu G, Li Q. Traumatic brain injury dysregulates microRNAs to modulate cell signaling in rat hippocampus. PLoS One. 2014;9:e103948. doi: 10.1371/journal.pone.0103948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu Z, Yu D, Almeida-Suhett C, Tu K, Marini AM, Eiden L, Braga MF, Zhu J, Li Z. Expression of miRNAs and their cooperative regulation of the pathophysiology in traumatic brain injury. PLoS One. 2012;7:e39357. doi: 10.1371/journal.pone.0039357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang T, Song J, Bu X, Wang C, Wu J, Cai J, Wan S, Fan C, Zhang C, Wang J. Elevated serum miR-93, miR-191, and miR-499 are noninvasive biomarkers for the presence and progression of traumatic brain injury. J Neurochem. 2016;137:122–129. doi: 10.1111/jnc.13534. [DOI] [PubMed] [Google Scholar]