

ABSTRACT
The family Flaviviridae consists of four genera, Flavivirus, Pestivirus, Pegivirus, and Hepacivirus, and comprises important pathogens of human and animals. Although the construction of recombinant viruses carrying reporter genes encoding fluorescent and bioluminescent proteins has been reported, the stable insertion of foreign genes into viral genomes retaining infectivity remains difficult. Here, we applied the 11-amino-acid subunit derived from NanoLuc luciferase to the engineering of the Flaviviridae viruses and then examined the biological characteristics of the viruses. We successfully generated recombinant viruses carrying the split-luciferase gene, including dengue virus, Japanese encephalitis virus, hepatitis C virus (HCV), and bovine viral diarrhea virus. The stability of the viruses was confirmed by five rounds of serial passages in the respective susceptible cell lines. The propagation of the recombinant luciferase viruses in each cell line was comparable to that of the parental viruses. By using a purified counterpart luciferase protein, this split-luciferase assay can be applicable in various cell lines, even when it is difficult to transduce the counterpart gene. The efficacy of antiviral reagents against the recombinant viruses could be monitored by the reduction of luciferase expression, which was correlated with that of viral RNA, and the recombinant HCV was also useful to examine viral dynamics in vivo. Taken together, our findings indicate that the recombinant Flaviviridae viruses possessing the split NanoLuc luciferase gene generated here provide powerful tools to understand viral life cycle and pathogenesis and a robust platform to develop novel antivirals against Flaviviridae viruses.
IMPORTANCE The construction of reporter viruses possessing a stable transgene capable of expressing specific signals is crucial to investigations of viral life cycle and pathogenesis and the development of antivirals. However, it is difficult to maintain the stability of a large foreign gene, such as those for fluorescence and bioluminescence, after insertion into a viral genome. Here, we successfully generated recombinant Flaviviridae viruses carrying the 11-amino-acid subunit derived from NanoLuc luciferase and demonstrated that these viruses are applicable to in vitro and in vivo experiments, suggesting that these recombinant Flaviviridae viruses are powerful tools for increasing our understanding of viral life cycle and pathogenesis and that these recombinant viruses will provide a robust platform to develop antivirals against Flaviviridae viruses.
KEYWORDS: Flaviviridae, reporter virus, antiviral screening, in vivo dynamics
INTRODUCTION
The family Flaviviridae comprises single-stranded positive-sense RNA viruses and consists of four genera: Flavivirus, Hepacivirus, Pegivirus, and Pestivirus. All members encode 2 to 4 structural proteins followed by 7 to 8 nonstructural protein genes flanked by 5′ and 3′ untranslated regions (UTRs), with the genera Hepacivirus, Pegivirus, and Pestivirus sharing a much higher degree of similarity (1, 2). Although they share similar genome components, their host ranges and tissue tropisms differ strikingly. The viruses of genus Flavivirus can infect more than 50 species, with a wide host range from reptiles to mammals; these include dengue virus (DENV), Japanese encephalitis virus (JEV), West Nile virus, tick-borne encephalitis virus, and Zika virus (3). The viruses of genus Pestivirus, including bovine viral diarrhea virus (BVDV), classical swine fever virus, and border disease virus, are causative agents for even-toed ungulate animals (4). Hepatitis C virus (HCV) from the genus Hepacivirus infects only humans and chimpanzees (5).
Since the first discovery of a green fluorescent protein (GFP) from the Aequorea victoria jellyfish in the 1960s (6), reporter proteins have been applied for an array of naturally occurring fluorescent proteins and genetically engineered derivatives for the monitoring and visualization of various intracellular events, including gene expression, protein localization, trafficking, interaction, and signaling pathways (7). In virus research, fluorescent proteins have been used to examine viral life cycles, tropism, and transmission (8–10). In addition to visualized fluorescent proteins, bioluminescence has become a powerful tool to investigate viral pathogenesis, immune responses to infection, and the efficacy of therapies in living animals (11). The bioimaging of viral infection has been achieved by using recombinant viruses possessing a reporter protein, allowing imaging to identify the specific sites of viral replication. Although recombinant viruses possessing a reporter protein have been demonstrated to be effective tools for the detection and quantification of viral replication both in vitro and in vivo, there is a size limitation regarding the accommodation of a foreign gene into a viral genome. In studies of Flaviviridae viruses, several groups have attempted to generate reporter-tagged viruses (12–14), but full-length infectious clones of viruses, especially of flavivirus, often are difficult to work with because many of the cDNA clones are deleterious for bacteria (15), and recombinant viruses carrying a large reporter gene are genetically unstable. In addition, the insertion of foreign genes into an irrelevant locus results in the disruption of the structural RNA elements required for viral replication.
To overcome these issues, we employed NanoLuc binary technology (NanoBiT) in this study (16). NanoBiT is a split reporter consisting of two subunits, high-affinity NanoBiT (HiBiT) (17) and large NanoBiT (LgBiT). The individual subunits do not possess enzymatic activity, but when HiBiT and LgBiT associate in cells or in vitro, the complex regains its NanoLuc enzymatic activity. We chose to insert smaller HiBiT subunits into selected viruses from the family Flaviviridae (DENV, JEV, HCV, and BVDV) and investigated optimal sites for the insertion to generate stable recombinant viruses. Our findings revealed that in susceptible cell lines, the propagation of the recombinant viruses possessing the split luciferase gene was comparable to that of parental viruses and significantly higher than that of recombinant viruses that had a full-length luciferase gene. The efficacy of antiviral reagents against the recombinant viruses could be monitored by the reduction of luciferase expression, which was correlated with that of viral RNA in vitro and in vivo. These results suggest that the recombinant Flaviviridae viruses generated in this study are powerful tools that can be used to increase our understanding of the viral life cycle and pathogenesis of Flaviviridae viruses. The recombinant viruses also will provide a robust platform for the development of therapeutic measures against infection with Flaviviridae viruses.
RESULTS
Determination of a suitable locus for insertion of the split-luciferase (HiBiT) gene into HCV genome and characterization of recombinant HCV.
To determine an ideal locus for the insertion of HiBiT into the HCV genome, we constructed 10 recombinant cDNA clones of HCV carrying the HiBiT gene (VSGWRLFKKIS) and a linker sequence (GS) in the N terminus of each viral protein (Fig. 1A). Infectious titers in the culture supernatants and intracellular luciferase activities in Huh7.5.1 cells lentivirally transduced with the other piece of the NanoLuc protein (LgBiT; Fig. 1B) then were determined at 72 h posttransfection with the recombinant HCV clones. Among the 10 HCV clones we examined, three viruses carrying HiBiT in the N terminus of E1, E2, or NS2 succeeded in the recovery of infectious viruses and luciferase expression (Fig. 1C). The highest viral titer and luciferase activity were obtained by the transfection of the HCV clone carrying HiBiT in the N terminus of NS2. In addition, we confirmed that the HiBiT tag was fused to viral NS2 (Fig. 1D). We therefore selected the recombinant virus for further characterization.
FIG 1.

Determination of a suitable locus for insertion of HiBiT into the HCV genome and characterization of recombinant HCV. (A) A schematic representation of HCV and sequence of the HiBiT luciferase and the adjacent viral gene. Arrows indicate the insertion sites of the HiBiT luciferase gene. (B) Expression of the LgBiT protein was determined by immunoblotting at 48 h postransduction of lentiviruses into Huh7.5.1 cells. (C) Infectious titers and luciferase activity were determined upon infection with the recombinant HCV carrying the HiBiT luciferase at the respective N terminus of the viral protein gene. (D) Huh7.5.1 cells were infected with the parental and the recombinant HCV carrying HiBiT at the N terminus of NS2 at an MOI of 1. On day 3 postinfection, cells were lysed and subjected to immunoblotting. (E) Huh7.5.1 cells were inoculated with 100 μl of culture supernatants obtained from the transfected cells. The supernatants were collected at 24, 48, and 72 h postinfection. Virus titers were determined in duplicate in Huh7.5.1 cells. Asterisks indicate significant differences (*, P < 0.05) versus the results of the HiBiT recombinant virus. (F) The recombinant HCV carrying the tag in the N terminus of NS2 was passaged on Huh7.5.1 cells for five rounds, and 100 μl of culture supernatants was used to infect naive Huh7.5.1 cells. At 72 h postinfection, the culture supernatants were collected, virus titers were determined, and samples were subjected to passage. (G) Sequence analyses of the recombinant viruses before (P0) and after five rounds of passage (P5). (H) The luciferase activities in cells infected with the recombinant viruses before and after passage. (I) The intracellular RNA copies and luciferase activity were determined upon infection with the parental and recombinant HCV carrying the HiBiT luciferase at an MOI of 1. Asterisks indicate significant differences (*, P < 0.05) versus the results of the parental virus.
Earlier studies reported that the construction of the HCV recombinants incorporated a gene cassette encoding a full-length luciferase and 2A peptide of picornavirus in frame with the N terminus of the NS2 gene (18, 19). To examine the effects of HiBiT insertion on virus propagation, growth kinetics of the parental HCV JFH-1 strain (WT) and the recombinants carrying HiBiT (HBiT Luc), or a full-length NanoLuc luciferase gene (Nano Luc), was determined in Huh7.5.1 cells (Fig. 1E). The propagation of the HiBiT recombinant was slow but reached levels comparable to that of the parental virus at 72 h postinfection. Importantly, the propagation was significantly higher than that of the recombinant possessing a full-length luciferase, suggesting that the length of the insertion is critical for virus propagation.
To examine the stability of the reporter gene in the HiBiT recombinant, we serially passaged the virus in Huh7.5.1 cells for five rounds. The infectious titers were slightly elevated by the passages and almost reached plateau levels at around 106.6 focus-forming units (FFU)/ml (Fig. 1F). Although two amino acid substitutions were observed (a threonine-to-isoleucine substitution at position 1496 in NS3 and a cysteine-to-serine substitution at position 2460 in NS5A) after the serial passage, the HiBiT luciferase gene was maintained (Fig. 1G). In addition, luciferase activities in cells infected with the recombinant viruses were similar even after five rounds of passage (Fig. 1H). These data suggest that the HiBiT gene in the N terminus of NS2 was stable in the HCV genome.
To evaluate the specificity of the luciferase activity of the recombinant HCV, the parental and recombinant viruses were inoculated into Huh7.5.1 cells, and intracellular HCV RNA and luciferase activity were determined (Fig. 1I). Although the increase of the intracellular HCV RNA levels of the recombinant HCV was comparable to that of the parental virus, the luciferase activity in the cells infected with the recombinant virus, but not with the parental virus, increased in accord with the increase of intracellular viral RNA.
Although only particular human hepatic cell lines are susceptible to the propagation of HCV, nonhepatic 293T cells that exogenously express Claudin1 (CLDN1), microRNA-122 (miR-122), and apolipoprotein E (ApoE) permit the complete propagation of HCV (20, 21). Expression of the LgBiT luciferase and these host factors was examined by immunoblotting in 293T cells upon transduction with lentiviral vectors (Fig. 2A). Although increases of intracellular luciferase activity and viral RNA upon infection with the recombinant HCV were observed in 293T cells expressing the LgBiT, CLDN1, and miR-122, an increase of those in the culture supernatants was only achieved when ApoE was additionally provided (Fig. 2B), suggesting that the HiBiT HCV recombinant can be used to monitor the replication of HCV.
FIG 2.
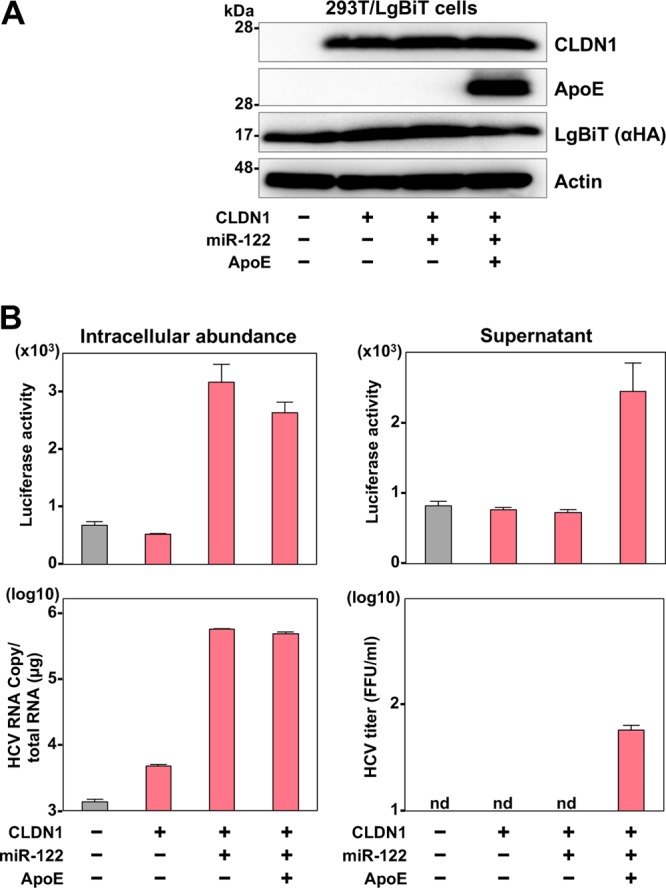
Evaluation of HiBiT recombinant HCV infection in nonhepatic cells. (A) The expression levels of CLDN1, ApoE, and LgBiT luciferase in 293T cells were determined by immunoblot analysis. (B) The cells were infected with the recombinant HCV at an MOI of 10, and the intracellular HCV RNA and luciferase activities at 72 h postinfection were determined by qRT-PCR and luciferase assay, respectively. The culture supernatants were harvested and infectious titers were determined by a focus-forming assay. The supernatants were also inoculated into the naive cells and the luciferase activity was determined by a luciferase assay.
Construction and characterization of the recombinant flaviviruses carrying the HiBiT gene.
To construct the luciferase-tagged recombinant flaviviruses, we used the cDNA clones of JEV strain AT31 (22) from Culex mosquito-borne virus and the DENV-4 strain H241 from Aedes mosquito-borne virus as templates. The nonstructural protein NS1 of flavivirus is a tolerant protein for insertion of foreign peptide (23, 24), and secretory NS1 is utilized as a marker for diagnosis of infection (25). Therefore, we inserted the HiBiT luciferase gene in frame with the N terminus of the NS1 gene of JEV and DENV (Fig. 3A) and examined the biological characteristics of the recombinant viruses. The recombinant JEV was inoculated into LgBiT-expressing and nonexpressing Huh7 cells (Fig. 3B), and infectious titers and luciferase activities in the culture supernatants of the cells were determined (Fig. 3C). The infectious titers in the culture supernatants were slightly lower in the cells infected with the recombinant than in cells infected with the parental JEV (Fig. 3C, left). Luciferase activity was detected not only in the LgBiT-expressing Huh7 cells but also in nonexpressing cells upon infection with the recombinant JEV by addition of the recombinant LgBiT protein to infected cell lysates. This result suggests that expression of LgBiT in infected cells is not required for the utility of this reporter assay and extends the suitability of the assay for use in primary cell lines and/or invertebrate cell lines that may be difficult to transduce.
FIG 3.

Construction and characterization of the recombinant flavivirus carrying the HiBiT gene. (A) A schematic representation of flavivirus and sequence of the insertion site of HiBiT luciferase gene. The arrow indicates an insertion site of the luciferase gene. (B) The expression of the LgBiT protein was determined by immunoblotting at 48 h postransduction of lentiviruses into Huh7 cells. (C) The infectious titers and luciferase activity were evaluated in parental and LgBiT-expressing Huh7 cells upon infection with either wild-type or recombinant JEV. In the parental Huh7 cells, luciferase activity was determined by addition of the recombinant LgBiT protein. (D) The recombinant JEV was passaged on Huh7 cells for five rounds, and 100 μl of culture supernatant was used to infect naive Huh7 cells. At 72 h postinfection, the culture supernatants were collected, virus titers were determined, and viruses were subjected to passage. (E) Sequence analyses of the recombinant viruses before (P0) and after five rounds of passage (P5). (F) The luciferase activities in the supernatants of cells infected with the recombinant viruses before and after passage. (G) The luciferase activity and infectious titers were determined in Huh7 cells and mosquito-derived C6/36 cells upon infection with the recombinant DENV at 120 h postinfection.
To evaluate the genetic stability of the recombinant JEV, we next passaged the virus in Huh7 cells five times, and infectious titers, viral gene sequence, and luciferase activities were determined (Fig. 3D, E, and F). Similar levels of infectious titers and luciferase activities were maintained, and no mutation was detected in the viral sequence of the recombinant, including the inserted gene, after five passages (Fig. 3D, E, and F), suggesting that recombinant JEV carrying the HiBiT gene is genetically stable. In addition, significant luciferase activities were detected in both Huh7 and C6/36 cells upon infection with recombinant DENV-4 possessing the HiBiT gene in the N-terminal region of NS1 (Fig. 3G), suggesting that the HiBiT luciferase-tagged system is applicable to flaviviruses.
Parental and recombinant JEV next were inoculated into Huh7 cells expressing the LgBiT and mosquito-derived C6/36 cells at a multiplicity of infection (MOI) of 0.1, and infectious titers in the culture supernatants, luciferase activities, and intracellular viral RNA levels were determined (Fig. 4A). In the C6/36 cells, the recombinant virus exhibited slow replication in the early phase but reached a level comparable to that of the parental virus in the late phase of infection. In contrast, the replication of the recombinant JEV in the LgBiT-expressing Huh7 cells was lower than that of wild-type JEV. The luciferase activity in both cells infected with recombinant JEV, but not with parental JEV, increased in accord with the intracellular RNA.
FIG 4.
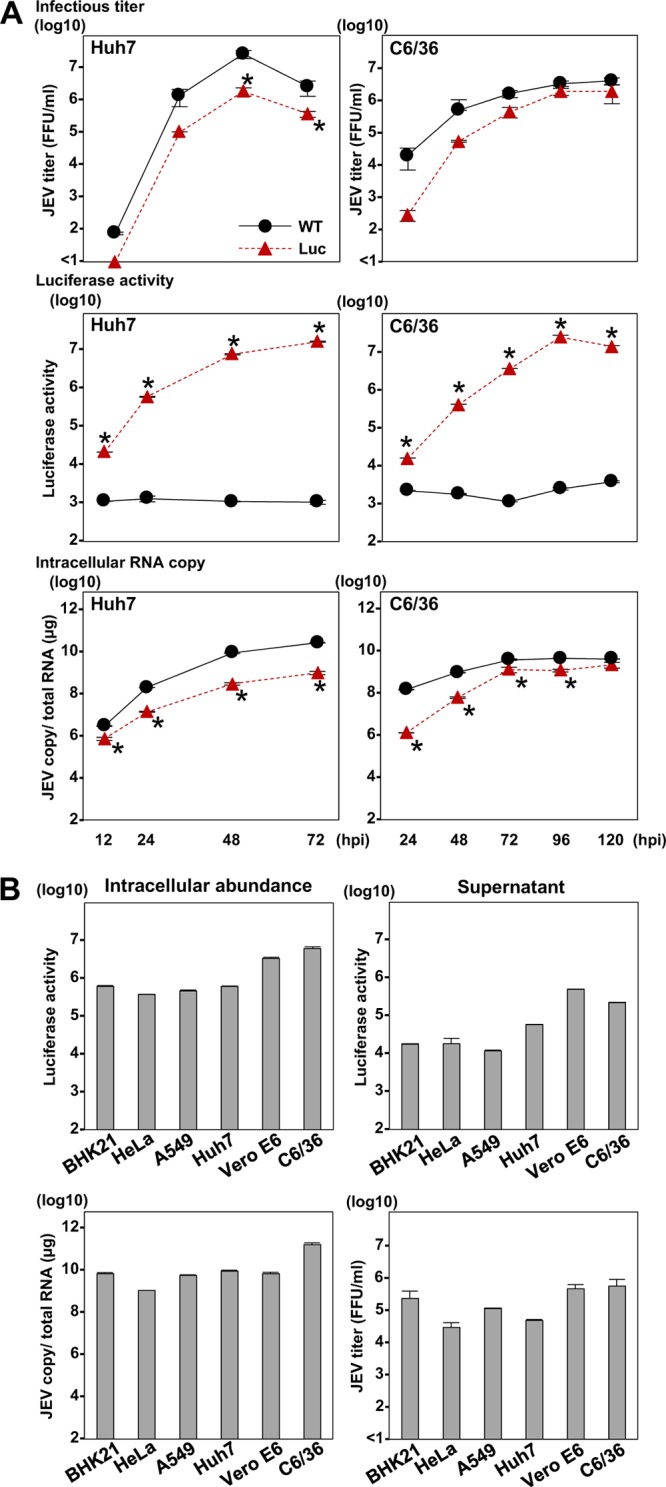
In vitro growth kinetics of the HiBiT recombinant flavivirus and infectivity in susceptible cell lines. (A) Huh7 and C6/36 cells were infected with the parental and recombinant JEV. The intracellular JEV RNA level, virus titers, and luciferase activity were determined at the indicated time points by qRT-PCR, a focus-forming assay, and a luciferase assay, respectively. Asterisks indicate significant differences (*, P < 0.05) versus the results of the parental virus. (B) The indicated species of cells were inoculated with the recombinant JEV. At 72 h postinfection, the intracellular viral RNA levels, virus titers in supernatants, and luciferase signals were determined by qRT-PCR, a focus-forming assay, and a luciferase assay, respectively.
Because flaviviruses, especially JEV, can infect various cell lines derived from invertebrate and vertebrate animals (1), we first examined the infectivity of the recombinant JEV to other cell lines derived from various mammals, including BHK-21 (hamster), HeLa (human), A549 (human), and Vero E6 (monkey) cells, in addition to the Huh7 and C6/36 cell lines (Fig. 4B). The recombinant virus exhibited a high infectivity to all of the cell lines examined, and the intracellular luciferase activity was correlated with the intracellular viral RNA. In addition, because the flavivirus NS1 is not only a component of viral replicase but also a secretory protein used as a marker for the diagnosis of flavivirus infection (25), we examined the luciferase activity in the culture supernatants of cells infected with recombinant virus. Luciferase activities in the supernatants were correlated with the intracellular viral RNA and extracellular infectious titers. These results indicate that recombinant JEV is infectious to various cell lines and that its infection can be monitored by the expression of both intracellular and extracellular luciferase.
Construction and characterization of the recombinant pestivirus carrying the HiBiT gene.
The construction of a recombinant BVDV carrying a FLAG tag in the N terminus of the envelope proteins has been reported to exhibit characteristics similar to those of parental virus (26). Thus, we have generated a recombinant BVDV possessing HiBiT luciferase in the N terminus of the envelope protein E2 (Fig. 5A) and have determined its virological properties. Bovine MDBK cells were infected with parental and recombinant BVDV at an MOI of 0.1, and the infectious titers in the culture supernatants and the luciferase activities and viral RNA in the cells were determined. Because of the difficulty of achieving the exogenous expression of LgBiT protein in MDBK cells, luciferase activity was determined by the addition of recombinant LgBiT protein into cell lysates. The recombinant BVDV showed propagation that was comparable to that of the wild-type virus and produced increasing luciferase activity in cells upon infection (Fig. 5B).
FIG 5.

Construction and characterization of the recombinant pestivirus carrying the HiBiT gene. (A) Schematic representation of pestivirus and sequence of the insertion site of HiBiT luciferase gene. (B) MDBK cells were inoculated at an MOI of 0.1 of the parental and recombinant BVDV. The intracellular viral RNA, virus titers, and luciferase activity were determined at the indicated time points by qRT-PCR, TCID50 determination, and luciferase assay, respectively. Asterisks indicate significant differences (*, P < 0.05) versus the results of the parental virus. (C) The culture supernatants of cells upon infection with the parental and recombinant BVDVs at an MOI of 1 were subjected to density gradient fractionation, and the infectious titers (upper) and viral RNA copies (lower) for each fraction were determined. (D) The recombinant BVDV was passaged on MDBK cells for five rounds, and 100 μl of culture supernatant was used to infect naive cells. At 72 h postinfection, the culture supernatants were collected, virus titers were determined, and viruses were subjected to passaging. (E) The luciferase activities in cells infected with the recombinant viruses before (P0) and after five rounds of passage (P5). (F) Sequence analyses of the recombinant viruses before and after passage.
Because HiBiT was inserted into the E2 envelope protein of BVDV, we examined the effect of insertion on physical properties of the viral particles. The parental and recombinant BVDV particles in the supernatants were analyzed by buoyant density ultracentrifugation, and infectious titers and viral RNA in each fraction were determined (Fig. 5C). The highest infectious titers and viral RNA of wild-type and recombinant BVDV were detected at densities of 1.07 and 1.08 g/ml, suggesting that the insertion of HiBiT in the N-terminal E2 region has no effect on particle formation of BVDV.
To evaluate the genetic stability of the recombinant BVDV, we next passaged the virus in MDBK cells five times, and infectious titers, luciferase activities, and viral genome sequences were determined. Similar levels of infectious titers at around 107 50% tissue culture infective doses (TCID50)/ml and luciferase activities were maintained, and no mutation was detected in the viral sequence of the recombinant, including the inserted gene after five passages (Fig. 5D, E, and F), suggesting that recombinant BVDV carrying the HiBiT gene is genetically stable.
Application of HiBiT Flaviviridae viruses for drug screening and investigation of in vivo viral dynamics.
Direct-acting antiviral (DAA) agents have been applied in a clinical setting for chronic hepatitis C patients (27). In the present study, to determine the sensitivity of the HiBiT recombinant HCV to treatment with antiviral drugs, Huh7.5.1 cells were treated with various concentrations of DAAs (sofosbuvir [SOF], daclatasvir [DCV], and telaprevir [TVR]) or type I interferon (IFN-α) at 2 h postinfection with recombinant HCV, and intracellular HCV RNA and luciferase activities were determined at 48 h postinfection (Fig. 6A). The intracellular HCV RNA was reduced in a dose-dependent manner for each reagent and was correlated with luciferase activity, suggesting that the sensitivity of luciferase expression upon infection with recombinant HCV is comparable to that of the viral RNA determined by quantitative reverse transcription-PCR (qRT-PCR).
FIG 6.

Application of the HiBiT Flaviviridae viruses for drug screening. (A) Huh7.5.1 cells infected with the recombinant HCV at an MOI of 1 were treated with SOF, DCV, TVR, and IFN-α at 2 h postinfection. The intracellular HCV RNA and luciferase activity levels were determined by qRT-PCR and a luciferase assay at 48 h postinfection. (B) Huh7 cells infected with the recombinant JEV at an MOI of 0.1 were treated with MPA and IFN-α at 1 h postinfection. The intracellular viral RNA and luciferase signals were determined at 48 h postinfection. (C) MDBK cells infected with the recombinant BVDV at an MOI of 0.1 were treated with MPA at 1 h postinfection. The recombinant BVDV was treated with various concentrations of polyclonal antibodies at 1 h and inoculated with the complex into naive MDBK cells. The intracellular viral RNA and luciferase activity were determined by qRT-PCR and luciferase assay at 48 h postinfection. Recombinant HCV (D) and JEV (E) were subjected to a chemical library of 69 drugs to assess sensitivity. The relative luciferase activity compared with the activity of nontreated cells (M) is shown as bar graphs. (F) The sensitivity of IFN-α, MPA, and compound 65 to recombinant DENV-4 was determined as described above. (G) Huh7 cells and Huh7.5.1 cells cotransfected with the expression plasmids encoding HiBiT or LgBiT were treated with the compounds. The relative luciferase activity was determined as described above.
To evaluate the sensitivity of recombinant JEV to treatment with antiviral reagents, Huh7 cells next were treated with mycophenolic acid (MPA) and IFN-α at 1 h postinfection with recombinant JEV, and intracellular viral RNA and luciferase activities were determined at 48 h postinfection (Fig. 6B). Intracellular viral RNA and luciferase activities were reduced in a dose-dependent manner for each reagent, as seen in the infection with recombinant HCV. MPA also exhibits antiviral activity against pestivirus (28); therefore, MDBK cells were treated with various concentrations of MPA at 1 h postinfection. Intracellular BVDV RNA and luciferase activities were determined at 48 h postinfection (Fig. 6C). In addition, recombinant BVDV was neutralized by various concentrations of polyclonal antibody against BVDV and inoculated into MDBK cells. Intracellular viral RNA and luciferase activities were reduced in a dose-dependent manner for each reagent. Collectively, these data indicate that the HiBiT recombinant Flaviviridae viruses generated in this study are useful for the screening of antiviral therapeutics.
To confirm the usefulness of the recombinant viruses for the screening of antiviral compounds, we next examined the commercially available protease inhibitor library (Table 1) by using recombinant HCV and JEV (Fig. 6D and E). Among the compounds we examined, three compounds (number 12, fluorouracil; number 20, pterostilbene; and number 39, DCV) exhibited more than 10-fold suppression of the luciferase expression in cells infected with recombinant HCV, comparable to suppression by IFN-α and the DAAs, including DCV and SOF. DCV (compound 39) is a well-known HCV inhibitor (29, 30). We observed here that compound 65 (DBeQ) was the only compound that could suppress luciferase expression by more than 10-fold in cells infected with recombinant JEV, i.e., comparably to IFN-α. DBeQ is a reversible inhibitor of p97/valosin-containing protein, a member of the ATPases associated with diverse cellular activities (31). Thus, we assessed the antiviral activity of DBeQ in cells infected with recombinant DENV-4 (Fig. 6F). DBeQ also showed suppression of luciferase expression comparable to that by IFN-α and MPA, suggesting that HiBiT recombinant flaviviruses are useful tools for the screening of antiviral reagents. We also confirmed that the compounds that inhibited viral replication exhibit no inhibitory effect on NanoBiT luciferase (Fig. 6G).
TABLE 1.
Compound library of protease inhibitors used in this study
| No. | Identity | Chemical name | CAS no. | Molecular formula | Molecular wt (g/mol) |
|---|---|---|---|---|---|
| 1 | T2316 | MK3102 | 1226781-44-7 | C17H20F2N4O3S | 398.43 |
| 2 | T1772 | Apoptosis activator 2 | 79183-19-0 | C15H9Cl2NO2 | 306.14 |
| 3 | T1581 | Picolamine | 3731-52-0 | C6H8N2 | 108.14 |
| 4 | T2893 | Muscone | 541-91-3 | C16H30O | 238.41 |
| 5 | T0429 | Glucosamine | 3416-24-8 | C6H13NO5 | 179.17 |
| 6 | T0372 | Gabexate mesylate | 56974-61-9 | C17H27N3O7 | 417.48 |
| 7 | T0087L | Sulfacetamide sodium | 127-56-0 | C8H9N2NaO3S | 236.22 |
| 8 | T0127 | Glimepiride | 93479-97-1 | C24H34N4O5S | 490.62 |
| 9 | T0178 | Saxagliptin hydrate | 945667-22-1 | C18H27N3 | 333.43 |
| 10 | T0191 | Linagliptin | 668270-12-0 | C25H28N8O2 | 472.54 |
| 11 | T0242 | Sitagliptin | 486460-32-6 | C16H15F6N5O | 407.32 |
| 12 | T0984 | Fluorouracil (5-FU) | 51-21-8 | C4H3FN2O2 | 130.08 |
| 13 | T1140 | Doxycycline HCl | 10592-13-9 | C22H25ClN2O8 | 480.896 |
| 14 | T1149 | Fenofibrate | 49562-28-9 | C20H21ClO4 | 360.83 |
| 15 | T1366 | 3-Pyridylacetic acid hydrochloride | 6419-36-9 | C7H8ClNO2 | 173.6 |
| 16 | T2731 | Usnic acid | 125-46-2 | C18H16O7 | 344.32402 |
| 17 | T2728 | Limonin | 1180-71-8 | C26H30O8 | 470.5242 |
| 18 | T2830 | Betulinic acid | 472-15-1 | C29H46O3 | 442.68817 |
| 19 | T2754 | Oxymatrine | 16837-52-8 | C15H24N2O2 | 264.37073 |
| 20 | T2888 | Pterostilbene | 537-42-8 | C16H16O3 | 256.30412 |
| 21 | T0789 | Phenylmethylsulfonyl fluoride | 329-98-6 | C7H7FO2S | 174.19 |
| 22 | T0951 | Hydroxychloroquine sulfate | 747-36-4 | C18H28ClN3O5S | 433.95 |
| 23 | T1402 | Fenofibric acid | 42017-89-0 | C17H15ClO4 | 318.75 |
| 24 | T1462 | Captopril | 62571-86-2 | C9H15NO3S | 217.29 |
| 25 | T1525 | Ritonavir | 155213-67-5 | C37H48N6O5S2 | 720.96 |
| 26 | T1564 | Cisplatin | 15663-27-1 | H6Cl2N2Pt | 300.05 |
| 27 | T2843 | Aloe-emodin | 481-72-1 | C15H10O5 | 270.24 |
| 28 | T2401 | Alogliptin benzoate | 850649-62-6 | C25H27N5O4 | 461.51 |
| 29 | T2399 | Bortezomib (PS-341) | 179324-69-7 | C19H25BN4O4 | 384.24 |
| 30 | T1592 | Acetohydroxamic acid | 546-88-3 | C2H5NO2 | 75.07 |
| 31 | T1623 | Lopinavir | 192725-17-0 | C37H48N4O5 | 628.8 |
| 32 | T2296 | SYR472 | 1029877-94-8 | C22H26FN5O6 | 475.47 |
| 33 | T2262 | GHF-5074 | 749269-83-8 | C16H11Cl2FO2 | 325.162 |
| 34 | T2016 | MLN9708 | 1201902-80-8 | C20H23BCl2N2O9 | 517.12 |
| 35 | T2122 | MLN2238 (Ixazomib) | 1072833-77-2 | C14H19BCl2N2O4 | 361.03 |
| 36 | T2239 | Raltegravir potassium | 871038-72-1 | C20H20FKN6O5 | 482.511 |
| 37 | T2117 | PSI6206 | 863329-66-2 | C10H13FN2O5 | 260.22 |
| 38 | T2392 | Nafamostat mesylate | 82956-11-4 | C21H25N5O8S2 | 539.58 |
| 39 | T1786 | Daclatasvir, BMS790052 | 1009119-65-6 | C40H52Cl2N8O6 | 811.8 |
| 40 | T2324 (T3335) | Darunavir ethanolate | 635728-49-3 | C29H43N3O8S | 593.73 |
| 41 | T2743 | Ilomastat (GM6001, Galardin) | 142880-36-2 | C20H28N4O4 | 388.46 |
| 42 | T2332 | Elvitegravir (GS-9137, JTK-303) | 697761-98-1 | C23H23ClFNO5 | 447.88 |
| 43 | T2329 | Dolutegravir (GSK1349572) | 1051375-19-9 | C20H18F2N3NaO5 | 441.36 |
| 44 | T2834 | Nobiletin | 478-01-3 | C21H22O8 | 402.40469 |
| 45 | T3028 | Celastrol | 34157-83-0 | C29H38O4 | 450.62381 |
| 46 | T2792 | Glucosamine sulfate | 29031-19-4 | C6H15NO9S | 277.25 |
| 47 | T0100 | Atazanavir sulfate | 229975-97-7 | C38H54N6O11S | 802.93 |
| 48 | T1853 | NMS 873 | 1418013-75-8 | C27H28N4O3S2 | 520.67 |
| 49 | T1822 | Clemizole | 442-52-4 | C19H20ClN3 | 325.84 |
| 50 | T1795 | Carfilzomib (PR-171) | 868540-17-4 | C40H57N5O7 | 719.91 |
| 51 | T0100L | Atazanavir | 198904-31-3 | C38H52N6O7 | 704.87 |
| 52 | T1502 | Vildagliptin (LAF-237) | 274901-16-5 | C17H25N3O2 | 303.4 |
| 53 | T2030 | Tiplaxtinin(PAI-039) | 393105-53-8 | C24H16F3NO4 | 439.38 |
| 54 | T2009 | SB-3CT | 292605-14-2 | C15H14O3S2 | 306.4 |
| 55 | T1757 | ML323 | 1572414-83-5 | C23H24N6 | 384.48 |
| 56 | T2424 | P22077 | 1247819-59-5 | C12H7F2NO3S2 | 315.32 |
| 57 | T2493 | PD 151746 | 179461-52-0 | C11H8FNO2S | 237.25 |
| 58 | T2503 | PAC1 | 315183-21-2 | C23H28N4O2 | 392.49 |
| 59 | T2393 | Efavirenz | 154598-52-4 | C14H9ClF3NO2 | 315.68 |
| 60 | T1883 | Des(benzylpyridyl) atazanavi | 1192224-24-0 | C26H43N5O7 | 537.65 |
| 61 | T1862 | PR-619 | 2645-32-1 | C7H5N5S2 | 223.328 |
| 62 | T2625 | MK0752 | 471905-41-6 | C21H21ClF2O4S | 442.9 |
| 63 | T2639 | LY2811376 | 1194044-20-6 | C15H14F2N4S | 320.36 |
| 64 | T3075 | FLI-06 | 313967-18-9 | C25H30N2O5 | 438.52 |
| 65 | T1969 | DBeQ | 177355-84-9 | C22H20N4 | 340.42 |
| 66 | T1932 | B-AP15 | 1009817-63-3 | C22H17N3O6 | 419.39 |
| 67 | T1924 | LDN-57444 | 668467-91-2 | C17H11Cl3N2O3 | 397.64 |
| 68 | T1891 | NSC 405020 | 7497-07-6 | C12H15Cl2NO | 260.16 |
| 69 | T2154 | MG-132 | 133407-82-6 | C26H41N3O5 | 475.62 |
Moreover, to obtain the HiBiT recombinant virus without preparing cDNA clones, we used the circular polymerase extension reaction (CPER) method (32–35) for obtaining the recombinant flavivirus. The DENV-2 16681 strain in our laboratory was used to generate the HiBiT recombinant virus (Fig. 7). Following our published protocol (34, 35), we successfully obtained the infectious recombinant DENV-2, suggesting that this reporter system can be extensively applied to Flaviviridae viruses, including clinical isolates, without the construction of cDNA plasmids.
FIG 7.
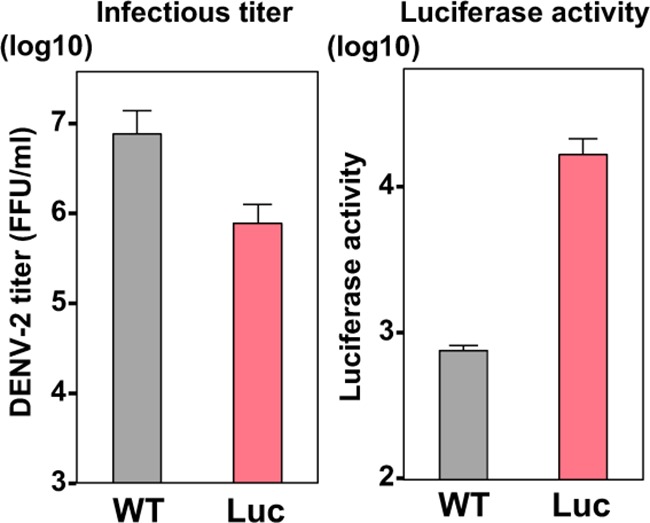
Bacterium-free generation of an HiBiT recombinant flavivirus. Wild-type and HiBiT recombinant DENV-2 were generated by the CPER method. Luciferase activity and virus titer are shown as bar graphs.
Finally, to evaluate the in vivo utility of recombinant HCV for monitoring viral dynamics and the efficacy of antiviral compounds, the HiBiT recombinant HCV was inoculated into human liver-transplanted chimeric mice (n = 15 mice total), and viral RNA and luciferase activity were determined after treatment with antivirals. Upon the inoculation of recombinant HCV into chimeric mice, the HiBiT luciferase gene was maintained in the virus and all mice became viremic (Fig. 8A), indicating that recombinant HCV can establish chronic infection in vivo. After treatment with a DAA (ombitasvir, or OBV) and pegylated IFN-α (PEG-IFN-α), HCV RNA was decreased in correlation with the luciferase activity (Fig. 8B), indicating that the in vivo viral dynamics and efficacy of antiviral reagents can be determined by using the recombinant HCV.
FIG 8.
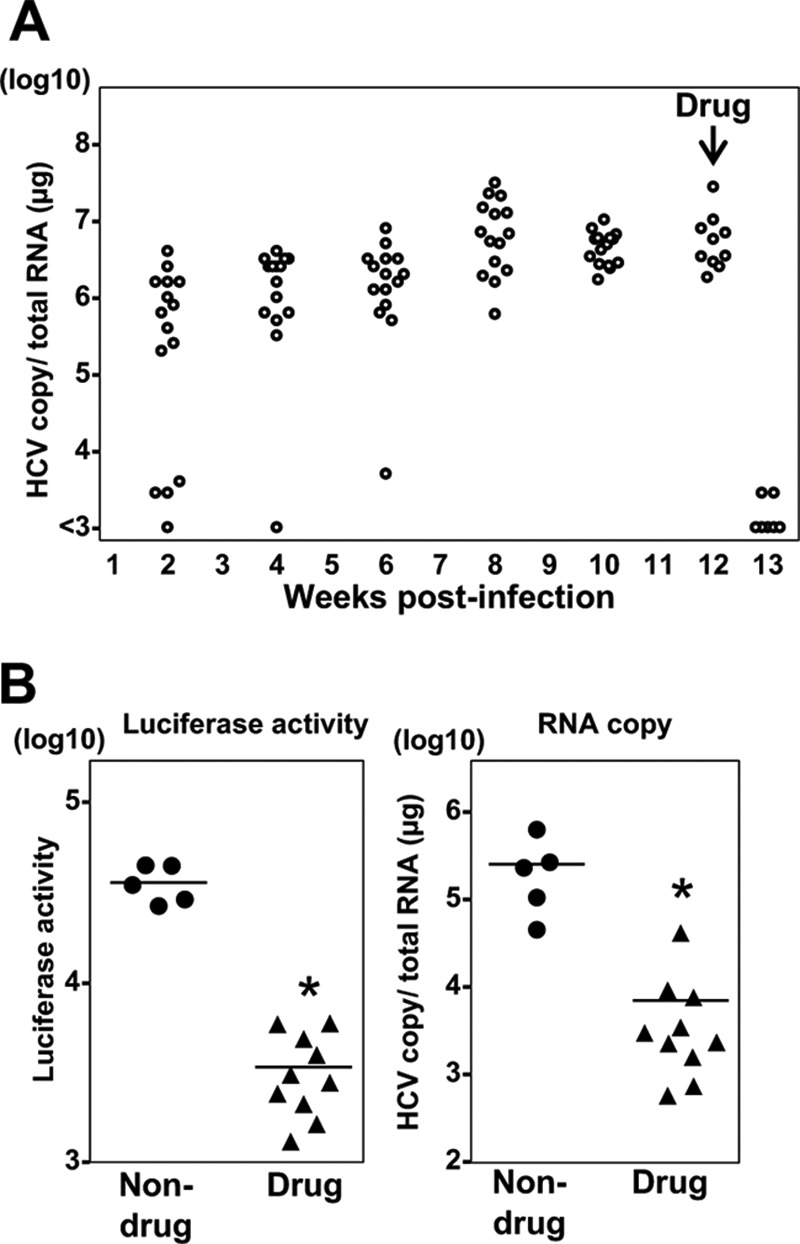
Investigation of HiBiT Flaviviridae in vivo viral dynamics. Fifteen human liver-transplanted chimeric mice were injected intravenously with 105.1 FFU (1 ml) of the reporter HCV. After the HCV RNA in blood reached a plateau, the chimeric mice were treated with the antiviral OBV and PEG-IFN-α, and mice were euthanized before and at 1 week posttreatment. Blood samples were collected for the detection of viral RNA (A), and the liver samples were subjected to determination of HCV RNA and luciferase signals (B).
DISCUSSION
Reporter proteins, including fluorescent and bioluminescent proteins, have been utilized to monitor and visualize intracellular events. Due to the progress in the design of reverse genetic systems of viruses, various reporter proteins have been incorporated into many viral particles in order to elucidate viral dynamics and pathogenesis and to develop antiviral reagents. In this study, we developed the recombinant viruses of the family Flaviviridae possessing the HiBiT luciferase subunit. The most important issue for the construction of recombinant viruses is the identification of a suitable gene locus for the insertion of the foreign gene. To date, however, the protein structures of only a few viruses have been revealed, making the construction of the virus recombinants possible. Thus, we inserted the HiBiT luciferase in the N terminus of each viral protein of HCV without consideration of the structure, and we determined the infectivity of the recombinant viruses. Our findings revealed that the recombinants carrying HiBiT in the N terminus of E1, E2, and NS2, but not of other viral proteins, exhibited significant replication. Previous studies revealed crucial RNA sequence and secondary structure for viral propagation, such as cis-replication elements in HCV NS5B (36) and the conserved complementary cyclization sequence in the capsid and 3′-UTR of flaviviruses (37). It was also shown that insertions in the N terminus of E2 are tolerated by insertion of the HCV chimeric strain J6/JFH1 (38, 39). In the present study, we obtained the recombinant JFH1 strain tagged in the viral envelopes, but recombinants showed a lower replication rate than that of the parental virus, suggesting that subtle structural changes in association with host and/or other viral proteins participate in viral replication (40).
The reporter viruses constructed in previous studies used a full-length fluorescent or bioluminescent protein followed by an autoprotease, such as 2A of foot-and-mouth disease virus (18, 41, 42). The insertion of a large fragment encoding the reporter protein together with the cleavage sequences (reaching approximately 300 amino acids) into a viral genome presents the risk of impairment of viral growth and genetic stability. In this study, we observed that the recombinant HCV carrying the full-length luciferase gene exhibited a significant growth defect compared growth of the parental HCV (Fig. 1E). Recently, Eyre et al. showed that recombinant DENV carrying the full-length NanoLuc luciferase in NS1 impaired virus replication (23). Therefore, we chose to insert a smaller 11-amino-acid HiBiT peptide from the split NanoBiT luciferase system (16, 17) for generation of reporter viruses. The length of these genes is similar to that of commonly used tags, such as hemagglutinin (HA) and FLAG. We demonstrated that the NanoBiT luciferase system is applicable to Flaviviridae viruses, and the HiBiT recombinants were genetically stable and exhibited virological characteristics comparable to those of the parental viruses.
Although a split reporter assay is required in order to express a counterpart protein for the examination of the protein-protein interaction (43), it is generally difficult to express foreign proteins in primary and nonmammalian cells. In the present study, we showed that the NanoBiT luciferase assay can be used for cells that do not express LgBiT by the addition of the purified recombinant LgBiT protein to infected cell lysates, indicating that the NanoBiT luciferase assay also is applicable to primary and insect cells. In addition, the expression of LgBiT protein in cells showed no effect on virus replication in vitro. Although we have generated the recombinant Flaviviridae viruses carrying HiBiT in the N terminus of viral proteins, further studies are needed to determine the other suitable sites for the insertion of a foreign protein into viral protein genes. Indeed, Eyre et al. successfully generated the recombinant DENVs carrying the FLAG tag or split GFP tag within the middle site of NS1, showing high similarity to the parental DENV (23).
Reverse genetics techniques are necessary for virus research seeking to elucidate viral life cycle and pathogenesis and for the development of antivirals. To date, however, only a limited number of virus strains is available for reverse genetics. Moreover, it is generally difficult to construct cDNA clones of Flaviviridae viruses, especially flavivirus, and thereby to obtain the infectious viruses (15). In the present study, we initially generated the recombinant viruses using infectious full-length cDNA clones of HCV, JEV, DENV, and BVDV. A bacterium-free CPER method for the generation of infectious flaviviruses was developed recently (33–35). We therefore used the CPER method for the generation of an HiBiT recombinant DENV-2 16681 strain, and we successfully obtained a replication-competent DENV-2 virus possessing the HiBiT gene, suggesting that the recombinant reporter flaviviruses can be generated by CPER, and it is envisaged that this approach also can be applied to clinical isolates.
The development of effective prophylactics and therapeutics to control infectious diseases such as dengue and Zika is an urgent medical need, because no licensed reagents are clinically available at this time. We screened the commercially available compound library by using the recombinant Flaviviridae viruses described here, and we identified the compounds that significantly suppressed the viral replication and reported their antiviral activities (29, 30, 44), suggesting that the recombinant Flaviviridae viruses generated in the present study are applicable to the high-throughput screening of antiviral compounds against infection with flaviviruses. In addition, the viral dynamics and the sensitivity to the antivirals used in clinical settings were evaluated in human liver-transplanted chimeric mice by using recombinant HCV. Infection of recombinant HCV in mice could be monitored by luciferase activity, which was correlated with viral infectivity and sensitivity to the antivirals. Additional investigations of not only drug sensitivity but also tissue tropism by using animal models for other Flaviviridae viruses are necessary for further evaluation of the usefulness of the recombinant flaviviruses.
In summary, we constructed the recombinant viruses of the family Flaviviridae, possessing a small luciferase subunit in viral proteins, and our analyses indicated their usefulness for the screening of antiviral reagents and for investigations of viral dynamics in vitro and in vivo. Our findings will contribute to further studies, including those of other RNA and DNA virus families, toward the engineering of recombinant viruses. The development of novel biological assays is required to improve our understanding of the molecular mechanisms of virus replication and pathogenesis and to continue advances in the discovery of antiviral reagents.
MATERIALS AND METHODS
Ethical statement.
The animal experiments described here were approved by the Committee for Animal Experiment of Hiroshima University (A14-195) and were conducted in compliance with national and international animal experimentation guidelines.
Plasmids.
The cDNA clones of CLDN1, miR-122, ApoE, LgBiT luciferase, and AcGFP were inserted between the XhoI and XbaI sites of the lentiviral vector pCSII-EF-RfA by using the infusion technique, and the resulting plasmids were designated pCSII-EF-CLDN1, pCSII-EF-miR-122, pCSII-EF-ApoE, pCSII-EF-LgBiT, and pCSII-EF-GFP, respectively. The plasmid pHH-JFH1 encodes the full-length cDNA of the JFH1 strain (GenBank accession number AB047639) (45). pHH-JFH1-E2p7NS2mt contains three adaptive mutations in pHH-JFH1 (46). pJFH1-E2p7NS2mt-Nlucsec possesses full-length NanoLuc luciferase generated previously (19). BVDV was derived from full-length cDNA of BVDV strain NCP7 (GenBank accession number AF220247) (47). The plasmids pMW119-DV4 and pMWJEAT (22) encode a full-length infectious clone of the DENV serotype 4 H241 strain (GenBank accession number AY947539) and JEV AT31 strain, respectively. The cDNA clones encoding the viral sequence for transfection were flanked by a modified T7 promoter sequence at the 5′ end and a NotI or KpnI restriction site at the 3′ end. The cDNA clones encoding the HiBiT luciferase gene were constructed by using a KOD-plus-mutagenesis kit (Toyobo) and the respective oligonucleotide primers. The plasmids used in this study were confirmed by sequencing with an ABI 3130 genetic analyzer (Thermo Fisher Scientific).
Cell lines.
All of the mammalian cell lines were cultured at 37°C under the conditions of a humidified atmosphere and 5% CO2. The human hepatocellular carcinoma-derived Huh7 cells, human embryonic kidney-derived 293T cells, human alveolar adenocarcinoma-derived A549 cells, human cervical cancer-derived HeLa cells, baby hamster kidney fibroblast-derived BHK-21 cells, and African green monkey kidney-derived Vero E6 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (Nakarai Tesque) supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, and 10% fetal bovine serum (FBS). The Huh7-derived cell line Huh7.5.1 was kindly provided by Frank Chisari. The bovine kidney-derived MDBK cells were propagated in DMEM supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, 5% BVDV antibody-free FBS (Japan Bio Serum), and 5% horse serum (Thermo Fisher Scientific). The Aedes albopictus mosquito-derived cell line C6/36 was grown in Leibovitz's L-15 medium (Thermo Fisher Scientific) with 10% tryptose phosphate broth and FBS at 28°C.
Antibodies and reagents.
Mouse monoclonal antibodies to β-actin, double-stranded RNA (dsRNA), ApoE, and pestiviral NS3 were purchased from Sigma-Aldrich, English & Scientific Consulting Kft, Santa Cruz Biotechnology, and TropBio, respectively. Rabbit anti-CLDN1 and Alexa Fluor (AF) 488-conjugated anti-rabbit IgG antibodies were purchased from Thermo Fisher Scientific. Rabbit polyclonal antibody against HCV NS2 was obtained from Gene Tex. Rat anti-HA antibody was purchased from Roche Diagnostics. Rabbit anti-HCV NS5A antibody and anti-BVDV polyclonal antibody were generated previously (48, 49). The compounds DCV and SOF were purchased from Shanghai Haoyuan Chemexpress. OBV, PEG-IFN-α, TVR, IFN-α, and MPA were obtained from Chemscene, Roche, ChemStep, PBL Biomedical Laboratories, and Sigma-Aldrich, respectively. A chemical inhibitor library of 69 drugs (L1100; Protease Inhibitor Library) was purchased from TargetMol.
Immunoblotting.
Cells lysed on ice in lysis buffer (20 mM Tris-HCl [pH 7.4], 135 mM NaCl, 1% Triton X-100, 10% glycerol) supplemented with a protease inhibitor cocktail, cOmplete Mini (Roche), were boiled in loading buffer and subjected to 5 to 20% gradient SDS-PAGE. The proteins were transferred to polyvinylidene difluoride membranes (Millipore) and incubated with the appropriate antibodies. The immune complexes were visualized with SuperSignal West Femto substrate (Thermo Fisher Scientific) and detected by use of an LAS-4000 image analyzer system (Fujifilm).
Preparation of viruses.
All of the cDNA-derived JEV and BVDV were rescued as described previously (22, 50). The supernatants were collected from the electroplated cells, and the infectious titers were determined and expressed as focus-forming units (FFU) or expressed as 50% tissue culture infective doses (TCID50) per milliliter. pHH-JFH1-E2p7NS2mt and mutants thereof were introduced into Huh7.5.1 cells. HCV in the supernatant was collected, and infectious titers were determined by a focus-forming assay and are expressed in FFU. The supernatants were collected and subjected to virus titration. The infectious DENV-4 H241 clone linearized with NotI was transcribed by using an mMESSAGE mMACHINE T7 Ultra kit (Thermo Fisher Scientific), and the in vitro-transcribed RNA (5 μg) was electroporated into cells at 5 × 106 cells/0.5 ml under conditions of 190 V and 950 μF using a Gene Pulser Xcell electroporation system (Bio-Rad) and then plated on DMEM containing 10% FBS. The supernatants were collected from the electroplated cells, and the infectious titers were determined by a focus-forming assay. JEV and DENV were propagated in C6/36 cells in order to reap sufficient virus yields.
Luciferase assay.
Luciferase activity was measured by using a Bright-Glo luciferase assay system (Promega) and Nano-Glo HiBiT lytic detection system (Promega) according to the protocol provided by the manufacturer.
Virus replication kinetics.
In vitro growth kinetics of the parental and recombinant viruses was evaluated in the susceptible cell lines. In the case of HCV, 100 μl of the culture supernatants obtained from the transfected cells was inoculated with the naive Huh7.5.1 cells, and the cell culture supernatants were collected at 12, 24, 48, and 72 h postinoculation. Huh7 and C6/36 cells were inoculated with JEV at an MOI of 0.1. The supernatants of Huh7 cells were collected at 12, 24, 48, and 72 h postinfection, and the supernatants of C6/36 cells were collected at 24, 48, 72, 96, and 120 h postinfection. The replication kinetics of BVDV were determined in MDBK cells by inoculation at an MOI of 0.1 with the collection of cell culture supernatants at 12, 24, 48, and 72 h postinoculation. The virus titers were determined in duplicate using the respective cell lines.
Quantitative RT-PCR.
For the quantification of viral RNA copies, total RNA was extracted from cells by using a PureLink RNA minikit (Thermo Fisher Scientific), and then first-strand cDNA synthesis and quantitative RT-PCR were performed by using a TaqMan RNA-to-Ct one-step kit and ViiA7 real-time PCR system (Thermo Fisher Scientific), respectively, according to the manufacturer's protocols. For quantification of viral RNA, the primer sets for the detection of the noncoding region reported in previous studies (51–53) were used. Fluorescent signals were determined by the ViiA7 system.
Neutralization assay.
The polyclonal antibody against BVDV was 4-fold diluted and incubated with virus (8,000 TCID50/ml) for 1 h and then inoculated into MDBK cells. Intracellular BVDV RNA levels and luciferase activity at 48 h postinfection were determined by qRT-PCR and a luminometer, respectively.
Buoyant density fractionation.
The culture supernatants of cells infected with the parental and recombinant BVDV were concentrated with the use of Spin-X UF centrifugal concentrators (Corning), applied to the top of a linear gradient formed from 10% to 40% OptiPrep (Axis-Shield) in phosphate-buffered saline (PBS), and spun at 32,000 rpm for 16 h at 4°C by using an SW41 rotor (Beckman Coulter). Aliquots of 12 consecutive fractions were collected from top to bottom, and the density, infectious titer, and viral RNA level were determined for each fraction.
Generation of HiBiT recombinant flavivirus by CPER.
The HiBiT recombinant flavivirus was generated by CPER as described previously (35), with some modifications. The viral RNA was obtained from the culture supernatants of the infected cells of the DENV-2 Thailand/16681/84 strain (GenBank accession number U87411) with the use of a PureLink RNA minikit. The viral RNA was reverse transcribed with a PrimeScript RT reagent kit (Perfect Real Time) (TaKaRa Bio) for cDNA. Seven fragments covering the entire gene were amplified by the respective primers and PrimeSTAR GXL DNA polymerase (TaKaRa Bio). The seven PCR products and UTR linker were cloned into pCR-Blunt II-TOPO vectors (Thermo Fisher Scientific). The plasmids were completely sequenced as described above. Eight PCR fragments next were generated with PrimeSTAR GXL DNA polymerase and primer pairs that have complementary ends with a 24- to 30-nucleotide overlap. The resulting eight DNA fragments then were mixed in equimolar amounts (0.1 pmol each) and subjected to CPER with PrimeSTAR GXL DNA polymerase (an initial 2 min of denaturation at 98°C; 20 cycles of 10 s at 98°C, 15 s at 55°C, and 12 min at 68°C; and a final extension for 12 min at 68°C) to generate circular DNA. The CPER products were transfected into Huh7 cells with Trans IT LT-1 transfection reagent (Mirus). The culture supernatants were harvested 15 days posttransfection.
Animal experiments.
For HCV infection, the generation of uPA+/+/SCID+/+ mice and the transplantation of human hepatocytes were performed as described previously (54, 55). All mice were transplanted with frozen human hepatocytes obtained from the same donor. The mice were anesthetized during infection, extraction of serum samples, and euthanasia. The concentration of human serum albumin was measured as described previously (54). Fifteen to 17 weeks after the hepatocyte transplantation, 15 mice were injected intravenously with 105.1 FFU (1 ml) of the recombinant virus. Mouse serum samples were obtained at 1- or 2-week intervals after HCV infection, and the HCV RNA levels were measured. After HCV RNA levels in blood reached a plateau, OBV (5 mg/kg of body weight, per os) and pegylated IFN-α (30 μg/kg, subcutaneously) were administered into the mice, and the liver samples were collected after euthanization for the detection of viral RNA and luciferase signals. The total RNA of the liver samples was purified with a PureLink RNA minikit and was subjected to qRT-PCR as described above. The remaining liver samples (150 mg) were homogenized by using an ultrasonic disintegrator in 1 ml of lysis buffer (100 mM Tris-HCl, 2 mM EDTA, 0.1% Triton X-100, pH 7.8), and the supernatants obtained after centrifugation at 15,000 × g for 30 min at 4°C were used for luciferase assay as described above.
Statistical analysis.
Results are expressed as the means ± standard deviations or standard errors. The significance of differences in the means was determined by Student's t test.
ACKNOWLEDGMENTS
We thank M. Tomiyama and J. Higuchi for their secretarial work and O. Isken, M. Ishibashi, and Y. Sugiyama for their technical assistance.
This work was supported in part by grants-in-aid for Scientific Research on Innovative Areas from the Ministry of Education, Culture, Science, Sports, and Technology (MEXT; http://www.mext.go.jp/) of Japan (16H06429, 16K21723, and 16H06432) and for Scientific Research (B) from MEXT of Japan (15H04736), from the Ministry of Health, Labor and Welfare of Japan and the Japan Agency for Medical Research and Development (AMED; http://www.amed.go.jp/) Research Program on Hepatitis, 17fk0210106h0002 and 16fk0310515h0105, and Research Program on Emerging and Reemerging Infectious Diseases, 17fk0108109h0001, and from the JSPS KAKENHI (https://www.jsps.go.jp/english/e-grants/), grant number 16J02628. T. Tamura is supported by a JSPS Research Fellowships for young scientists (https://www.jsps.go.jp/english/e-grants/).
We have no conflicts of interest to declare.
REFERENCES
- 1.Lindenbach BD MC, Thiel HJ, Rice CM. 2013. Flaviviridae, p 712–794. Fields virology, 6th ed In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed, vol 1 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Simmonds P, Becher P, Bukh J, Gould EA, Meyers G, Monath T, Muerhoff S, Pletnev A, Hesse R, Smith DB, Stapleton JT, ICTV Report Consortium. 2017. ICTV virus taxonomy profile: Flaviviridae. J Gen Virol 98:2–3. doi: 10.1099/jgv.0.000672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kitchen A, Shackelton LA, Holmes EC. 2011. Family level phylogenies reveal modes of macroevolution in RNA viruses. Proc Natl Acad Sci U S A 108:238–243. doi: 10.1073/pnas.1011090108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thiel HJ, Plagemann PGW, Moenning V. 1996. Pestiviruses, p 1059–1073. In Fields BN, Knipe DM, Howley PM (ed), Fields virology, 3rd ed, vol 1 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 5.Bukh J. 2004. A critical role for the chimpanzee model in the study of hepatitis C. Hepatology 39:1469–1475. doi: 10.1002/hep.20268. [DOI] [PubMed] [Google Scholar]
- 6.Shimomura O, Johnson FH, Saiga Y. 1962. Extraction, purification and properties of aequorin, a bioluminescent protein from the luminous hydromedusan, Aequorea. J Cell Comp Physiol 59:223–239. doi: 10.1002/jcp.1030590302. [DOI] [PubMed] [Google Scholar]
- 7.Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC. 1994. Green fluorescent protein as a marker for gene expression. Science 263:802–805. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- 8.Mueller S, Wimmer E. 1998. Expression of foreign proteins by poliovirus polyprotein fusion: analysis of genetic stability reveals rapid deletions and formation of cardioviruslike open reading frames. J Virol 72:20–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elliott G, O'Hare P. 1999. Live-cell analysis of a green fluorescent protein-tagged herpes simplex virus infection. J Virol 73:4110–4119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Manicassamy B, Manicassamy S, Belicha-Villanueva A, Pisanelli G, Pulendran B, Garcia-Sastre A. 2010. Analysis of in vivo dynamics of influenza virus infection in mice using a GFP reporter virus. Proc Natl Acad Sci U S A 107:11531–11536. doi: 10.1073/pnas.0914994107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Doyle TC, Burns SM, Contag CH. 2004. In vivo bioluminescence imaging for integrated studies of infection. Cell Microbiol 6:303–317. doi: 10.1111/j.1462-5822.2004.00378.x. [DOI] [PubMed] [Google Scholar]
- 12.Moser C, Tratschin JD, Hofmann MA. 1998. A recombinant classical swine fever virus stably expresses a marker gene. J Virol 72:5318–5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pierson TC, Diamond MS, Ahmed AA, Valentine LE, Davis CW, Samuel MA, Hanna SL, Puffer BA, Doms RW. 2005. An infectious West Nile virus that expresses a GFP reporter gene. Virology 334:28–40. doi: 10.1016/j.virol.2005.01.021. [DOI] [PubMed] [Google Scholar]
- 14.Koutsoudakis G, Kaul A, Steinmann E, Kallis S, Lohmann V, Pietschmann T, Bartenschlager R. 2006. Characterization of the early steps of hepatitis C virus infection by using luciferase reporter viruses. J Virol 80:5308–5320. doi: 10.1128/JVI.02460-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ruggli N, Rice CM. 1999. Functional cDNA clones of the Flaviviridae: strategies and applications. Adv Virus Res 53:183–207. doi: 10.1016/S0065-3527(08)60348-6. [DOI] [PubMed] [Google Scholar]
- 16.Dixon AS, Schwinn MK, Hall MP, Zimmerman K, Otto P, Lubben TH, Butler BL, Binkowski BF, Machleidt T, Kirkland TA, Wood MG, Eggers CT, Encell LP, Wood KV. 2016. NanoLuc complementation reporter optimized for accurate measurement of protein interactions in cells. ACS Chem Biol 11:400–408. doi: 10.1021/acschembio.5b00753. [DOI] [PubMed] [Google Scholar]
- 17.Schwinn MK, Machleidt T, Zimmerman K, Eggers CT, Dixon AS, Hurst R, Hall MP, Encell LP, Binkowski BF, Wood KV. 21 September 2017. CRISPR-mediated tagging of endogenous proteins with a luminescent peptide. ACS Chem Biol doi: 10.1021/acschembio.7b00549. [DOI] [PubMed] [Google Scholar]
- 18.Jones CT, Murray CL, Eastman DK, Tassello J, Rice CM. 2007. Hepatitis C virus p7 and NS2 proteins are essential for production of infectious virus. J Virol 81:8374–8383. doi: 10.1128/JVI.00690-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Puig-Basagoiti F, Fukuhara T, Tamura T, Ono C, Uemura K, Kawachi Y, Yamamoto S, Mori H, Kurihara T, Okamoto T, Aizaki H, Matsuura Y. 2016. Human cathelicidin compensates for the role of apolipoproteins in hepatitis C virus infectious particle formation. J Virol 90:8464–8477. doi: 10.1128/JVI.00471-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Da Costa D, Turek M, Felmlee DJ, Girardi E, Pfeffer S, Long G, Bartenschlager R, Zeisel MB, Baumert TF. 2012. Reconstitution of the entire hepatitis C virus life cycle in nonhepatic cells. J Virol 86:11919–11925. doi: 10.1128/JVI.01066-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hueging K, Weller R, Doepke M, Vieyres G, Todt D, Wolk B, Vondran FW, Geffers R, Lauber C, Kaderali L, Penin F, Pietschmann T. 2015. Several human liver cell expressed apolipoproteins complement HCV virus production with varying efficacy conferring differential specific infectivity to released viruses. PLoS One 10:e0134529. doi: 10.1371/journal.pone.0134529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao Z, Date T, Li Y, Kato T, Miyamoto M, Yasui K, Wakita T. 2005. Characterization of the E-138 (Glu/Lys) mutation in Japanese encephalitis virus by using a stable, full-length, infectious cDNA clone. J Gen Virol 86:2209–2220. doi: 10.1099/vir.0.80638-0. [DOI] [PubMed] [Google Scholar]
- 23.Eyre NS, Johnson SM, Eltahla AA, Aloi M, Aloia AL, McDevitt CA, Bull RA, Beard MR. 27 September 2017. Genome-wide mutagenesis of dengue virus reveals plasticity of the NS1 protein and enables generation of infectious tagged reporter viruses. J Virol doi: 10.1128/JVI.01455-17. [DOI] [PMC free article] [PubMed]
- 24.Fulton BO, Sachs D, Schwarz MC, Palese P, Evans MJ. 19 July 2017. Transposon mutagenesis of the Zika virus genome highlights regions essential for RNA replication and restricted for immune evasion. J Virol doi: 10.1128/JVI.00698-17. [DOI] [PMC free article] [PubMed]
- 25.Peeling RW, Artsob H, Pelegrino JL, Buchy P, Cardosa MJ, Devi S, Enria DA, Farrar J, Gubler DJ, Guzman MG, Halstead SB, Hunsperger E, Kliks S, Margolis HS, Nathanson CM, Nguyen VC, Rizzo N, Vazquez S, Yoksan S. 2010. Evaluation of diagnostic tests: dengue. Nat Rev Microbiol 8:S30–S38. doi: 10.1038/nrmicro2459. [DOI] [PubMed] [Google Scholar]
- 26.Wegelt A, Reimann I, Granzow H, Beer M. 2011. Characterization and purification of recombinant bovine viral diarrhea virus particles with epitope-tagged envelope proteins. J Gen Virol 92:1352–1357. doi: 10.1099/vir.0.029330-0. [DOI] [PubMed] [Google Scholar]
- 27.Scheel TK, Rice CM. 2013. Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nat Med 19:837–849. doi: 10.1038/nm.3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stangl JR, Carroll KL, Illichmann M, Striker R. 2004. Effect of antimetabolite immunosuppressants on Flaviviridae, including hepatitis C virus. Transplantation 77:562–567. doi: 10.1097/01.TP.0000114610.40412.C6. [DOI] [PubMed] [Google Scholar]
- 29.Banerjee A, Saito K, Meyer K, Banerjee S, Ait-Goughoulte M, Ray RB, Ray R. 2009. Hepatitis C virus core protein and cellular protein HAX-1 promote 5-fluorouracil-mediated hepatocyte growth inhibition. J Virol 83:9663–9671. doi: 10.1128/JVI.00872-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gastaminza P, Whitten-Bauer C, Chisari FV. 2010. Unbiased probing of the entire hepatitis C virus life cycle identifies clinical compounds that target multiple aspects of the infection. Proc Natl Acad Sci U S A 107:291–296. doi: 10.1073/pnas.0912966107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chou TF, Brown SJ, Minond D, Nordin BE, Li K, Jones AC, Chase P, Porubsky PR, Stoltz BM, Schoenen FJ, Patricelli MP, Hodder P, Rosen H, Deshaies RJ. 2011. Reversible inhibitor of p97, DBeQ, impairs both ubiquitin-dependent and autophagic protein clearance pathways. Proc Natl Acad Sci U S A 108:4834–4839. doi: 10.1073/pnas.1015312108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Quan J, Tian J. 2011. Circular polymerase extension cloning for high-throughput cloning of complex and combinatorial DNA libraries. Nat Protoc 6:242–251. doi: 10.1038/nprot.2010.181. [DOI] [PubMed] [Google Scholar]
- 33.Edmonds J, van Grinsven E, Prow N, Bosco-Lauth A, Brault AC, Bowen RA, Hall RA, Khromykh AA. 2013. A novel bacterium-free method for generation of flavivirus infectious DNA by circular polymerase extension reaction allows accurate recapitulation of viral heterogeneity. J Virol 87:2367–2372. doi: 10.1128/JVI.03162-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Setoh YX, Prow NA, Rawle DJ, Tan CS, Edmonds JH, Hall RA, Khromykh AA. 2015. Systematic analysis of viral genes responsible for differential virulence between American and Australian West Nile virus strains. J Gen Virol 96:1297–1308. doi: 10.1099/vir.0.000069. [DOI] [PubMed] [Google Scholar]
- 35.Setoh YX, Prow NA, Peng N, Hugo LE, Devine G, Hazlewood JE, Suhrbier A, Khromykh AA. 2017. De novo generation and characterization of new Zika virus isolate using sequence data from a microcephaly case. mSphere 2:e00190-17. doi: 10.1128/mSphereDirect.00190-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.You S, Stump DD, Branch AD, Rice CM. 2004. A cis-acting replication element in the sequence encoding the NS5B RNA-dependent RNA polymerase is required for hepatitis C virus RNA replication. J Virol 78:1352–1366. doi: 10.1128/JVI.78.3.1352-1366.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Khromykh AA, Meka H, Guyatt KJ, Westaway EG. 2001. Essential role of cyclization sequences in flavivirus RNA replication. J Virol 75:6719–6728. doi: 10.1128/JVI.75.14.6719-6728.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Merz A, Long G, Hiet MS, Brugger B, Chlanda P, Andre P, Wieland F, Krijnse-Locker J, Bartenschlager R. 2011. Biochemical and morphological properties of hepatitis C virus particles and determination of their lipidome. J Biol Chem 286:3018–3032. doi: 10.1074/jbc.M110.175018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prentoe J, Bukh J. 2011. Hepatitis C virus expressing flag-tagged envelope protein 2 has unaltered infectivity and density, is specifically neutralized by flag antibodies and can be purified by affinity chromatography. Virology 409:148–155. doi: 10.1016/j.virol.2010.10.034. [DOI] [PubMed] [Google Scholar]
- 40.Pietschmann T, Kaul A, Koutsoudakis G, Shavinskaya A, Kallis S, Steinmann E, Abid K, Negro F, Dreux M, Cosset FL, Bartenschlager R. 2006. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc Natl Acad Sci U S A 103:7408–7413. doi: 10.1073/pnas.0504877103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fan ZC, Dennis JC, Bird RC. 2008. Bovine viral diarrhea virus is a suitable viral vector for stable expression of heterologous gene when inserted in between N(pro) and C genes. Virus Res 138:97–104. doi: 10.1016/j.virusres.2008.08.015. [DOI] [PubMed] [Google Scholar]
- 42.Zou G, Xu HY, Qing M, Wang QY, Shi PY. 2011. Development and characterization of a stable luciferase dengue virus for high-throughput screening. Antiviral Res 91:11–19. doi: 10.1016/j.antiviral.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 43.Luker KE, Smith MC, Luker GD, Gammon ST, Piwnica-Worms H, Piwnica-Worms D. 2004. Kinetics of regulated protein-protein interactions revealed with firefly luciferase complementation imaging in cells and living animals. Proc Natl Acad Sci U S A 101:12288–12293. doi: 10.1073/pnas.0404041101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Phongphaew W, Kobayashi S, Sasaki M, Carr M, Hall WW, Orba Y, Sawa H. 2017. Valosin-containing protein (VCP/p97) plays a role in the replication of West Nile virus. Virus Res 228:114–123. doi: 10.1016/j.virusres.2016.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Krausslich HG, Mizokami M, Bartenschlager R, Liang TJ. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med 11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Russell RS, Meunier JC, Takikawa S, Faulk K, Engle RE, Bukh J, Purcell RH, Emerson SU. 2008. Advantages of a single-cycle production assay to study cell culture-adaptive mutations of hepatitis C virus. Proc Natl Acad Sci U S A 105:4370–4375. doi: 10.1073/pnas.0800422105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meyers G, Tautz N, Becher P, Thiel HJ, Kummerer BM. 1996. Recovery of cytopathogenic and noncytopathogenic bovine viral diarrhea viruses from cDNA constructs. J Virol 70:8606–8613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moriishi K, Shoji I, Mori Y, Suzuki R, Suzuki T, Kataoka C, Matsuura Y. 2010. Involvement of PA28gamma in the propagation of hepatitis C virus. Hepatology 52:411–420. doi: 10.1002/hep.23680. [DOI] [PubMed] [Google Scholar]
- 49.Abe Y, Tamura T, Torii S, Wakamori S, Nagai M, Mitsuhashi K, Mine J, Fujimoto Y, Nagashima N, Yoshino F, Sugita Y, Nomura T, Okamatsu M, Kida H, Sakoda Y. 2016. Genetic and antigenic characterization of bovine viral diarrhea viruses isolated from cattle in Hokkaido, Japan. J Vet Med Sci 78:61–70. doi: 10.1292/jvms.15-0186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Isken O, Langerwisch U, Schonherr R, Lamp B, Schroder K, Duden R, Rumenapf TH, Tautz N. 2014. Functional characterization of bovine viral diarrhea virus nonstructural protein 5A by reverse genetic analysis and live cell imaging. J Virol 88:82–98. doi: 10.1128/JVI.01957-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Takeuchi T, Katsume A, Tanaka T, Abe A, Inoue K, Tsukiyama-Kohara K, Kawaguchi R, Tanaka S, Kohara M. 1999. Real-time detection system for quantification of hepatitis C virus genome. Gastroenterology 116:636–642. doi: 10.1016/S0016-5085(99)70185-X. [DOI] [PubMed] [Google Scholar]
- 52.Shirato K, Miyoshi H, Kariwa H, Takashima I. 2005. Detection of West Nile virus and Japanese encephalitis virus using real-time PCR with a probe common to both viruses. J Virol Methods 126:119–125. doi: 10.1016/j.jviromet.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 53.La Rocca SA, Sandvik T. 2009. A short target real-time RT-PCR assay for detection of pestiviruses infecting cattle. J Virol Methods 161:122–127. doi: 10.1016/j.jviromet.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 54.Tateno C, Yoshizane Y, Saito N, Kataoka M, Utoh R, Yamasaki C, Tachibana A, Soeno Y, Asahina K, Hino H, Asahara T, Yokoi T, Furukawa T, Yoshizato K. 2004. Near completely humanized liver in mice shows human-type metabolic responses to drugs. Am J Pathol 165:901–912. doi: 10.1016/S0002-9440(10)63352-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tateno C, Kawase Y, Tobita Y, Hamamura S, Ohshita H, Yokomichi H, Sanada H, Kakuni M, Shiota A, Kojima Y, Ishida Y, Shitara H, Wada NA, Tateishi H, Sudoh M, Nagatsuka S, Jishage K, Kohara M. 2015. Generation of novel chimeric mice with humanized livers by using hemizygous cDNA-uPA/SCID mice. PLoS One 10:e0142145. doi: 10.1371/journal.pone.0142145. [DOI] [PMC free article] [PubMed] [Google Scholar]