

ABSTRACT
Porcine reproductive and respiratory syndrome virus (PRRSV) infects alveolar macrophages (AMϕ), causing dysregulated alpha interferon (IFN-α) and tumor necrosis factor alpha (TNF-α) production through a mechanism(s) yet to be resolved. Here, we show that AMϕ infected with PRRSV secreted a reduced quantity of IFN-α following exposure of the cell to synthetic double-stranded RNA (dsRNA). This reduction did not correlate with reduced IFNA1 gene transcription. Rather, it coincided with two events that occurred late during infection and that were indicative of translational attenuation, specifically, the activation of eukaryotic translation initiation factor 2α (eIF2α) and the appearance of stress granules. Notably, the typical rapid production of TNF-α by AMϕ exposed to lipopolysaccharide (LPS) was suppressed or enhanced by PRRSV, depending on when the LPS exposure occurred after virus infection. If exposure was delayed until 6 h postinfection (hpi) so that the development of the cytokine response coincided with the time in which phosphorylation of eIF2α by the stress sensor PERK (protein kinase RNA [PKR]-like ER kinase) occurred, inhibition of TNF-α production was observed. However, if LPS exposure occurred at 2 hpi, prior to a detectable onset of eIF2α phosphorylation, a synergistic response was observed due to the earlier NF-κB activation via the stress sensor IRE1α (inositol-requiring kinase 1α). These results suggest that the asynchronous actions of two branches of the unfolded protein response (UPR), namely, IRE1α, and PERK, activated by ER stress resulting from the virus infection, are associated with enhancement or suppression of TNF-α production, respectively.
IMPORTANCE The activation of AMϕ is controlled by the microenvironment to deter excessive proinflammatory cytokine responses to microbes that could impair lung function. However, viral pneumonias frequently become complicated by secondary bacterial infections, triggering severe inflammation, lung dysfunction, and death. Although dysregulated cytokine production is considered an integral component of the exacerbated inflammatory response in viral-bacterial coinfections, the mechanism responsible for this event is unknown. Here, we show that PRRSV replication in porcine AMϕ triggers activation of the IRE1α branch of the UPR, which causes a synergistic TNF-α response to LPS exposure. Thus, the severe pneumonias typically observed in pigs afflicted with PRRSV-bacterial coinfections could result from dysregulated, overly robust TNF-α production in response to opportunistic pathogens that is not commensurate with the typical restrained reaction by uninfected AMϕ. This notion could help in the design of therapies to mitigate the severity of viral and bacterial coinfections.
KEYWORDS: UPR, cytokines, immunomodulation, inflammation, interferons, macrophages, porcine reproductive and respiratory syndrome virus, stress kinases, stress response, tumor necrosis factor
INTRODUCTION
Porcine reproductive and respiratory syndrome virus (PRRSV), a positive-sense single-stranded, enveloped RNA virus that is a member of the family Arteriviridae (1), causes the most economically significant infectious malady afflicting pigs in commercial swine farms worldwide (2). Exposure of the respiratory mucosa of a pig to PRRSV results in virus replication in regional macrophages (Mϕ) and the development of viremia within 12 h after infection, leading to systemic distribution of the virus to other macrophage populations in the body (3, 4). In the lung, PRRSV exploits alveolar macrophages (AMϕ) for its replication, triggering a massive infiltration of the alveolar septa by macrophages, resulting in interstitial pneumonia (5). In the absence of secondary bacterial infections, pneumonias caused by PRRSV are rarely lethal and begin to resolve within 2 weeks (6, 7). While interleukin 1 (IL-1) and IL-6 are amply detected in bronchoalveolar lavage (BAL) fluids obtained from such pneumonic lungs, the presence of alpha interferon (IFN-α) and tumor necrosis factor alpha (TNF-α) is negligible (8–12). In contrast, pneumonias caused by PRRSV that are accompanied by a secondary bacterial infection result in a severe respiratory syndrome characterized by abundant presence of TNF-α in the lung, enhanced lung tissue damage, high morbidity, hypoxia, and a high rate of mortality (6, 7, 13, 14). The mechanism responsible for the apparent pathogenic synergy between PRRSV and bacterial pathogens is not understood (15).
Compared to the profile of innate cytokines elicited by other viruses that cause pneumonia in pigs, such as swine influenza virus and porcine respiratory coronavirus, which trigger the abundant presence of IFN-α and TNF-α in lung tissue (5), the nominal presence of these two cytokines in the lungs of pigs afflicted by PRRSV is intriguing; however, the mechanism responsible for this condition is unclear (16). Given the critical roles that IFN-α and TNF-α play in host immunity, the apparent ability of PRRSV to modulate the production of the two cytokines has been extensively examined. Several studies have relied on measuring transcription factor (TF) activation using reporter gene assays and overexpression of single viral genes. These studies indicate that some PRRSV nonstructural proteins have the ability to modulate cytokine production stimulated by strong agonists, like synthetic double-stranded RNA (dsRNA) or lipopolysaccharide (LPS), by inhibiting the activation of IRF3 or NF-κB (17–20). In the context of virus infection, the modulatory properties ascribed to PRRSV have been found to be disparate. For example, in the case of IFN-α, virus infection has been reported to inhibit the production of the cytokine in response to stimulation with potent type I IFN agonists, such as porcine coronavirus (8) and synthetic dsRNA (21). On the other hand, the production of TNF-α in response to stimulation with LPS has been reported to range from enhancement to inhibition (22). To clarify these disparate modulatory outcomes, we systemically examined the effect of infecting porcine AMϕ (PAMϕ) with PRRSV on their ability to produce IFN-α and TNF-α in response to stimulation with two agonists of the cytokines, namely, synthetic dsRNA [poly(I·C)] and LPS, respectively. Our results indicated that the infection of AMϕ with PRRSV does not impair the activation of the major TFs necessary for type I IFN or TNF-α gene transcription. Rather, we provide evidence that the modulation of cytokine production in PRRSV-infected AMϕ involves the actions of two endoplasmic reticulum (ER) stress sensor proteins, namely, protein kinase RNA-like ER kinase (PERK) and inositol-requiring enzyme 1α (IRE1α).
Viral replication places a major burden on the ER to produce viral proteins, causing ER stress (23, 24). To cope with ER stress and maintain protein homeostasis, cells initiate the unfolded protein response (UPR), which comprises the activation of PERK, IRE1α, and a third stress sensor, namely, activating transcription factor 6 (ATF6). The UPR is aimed at promoting cell survival by reducing misfolded protein levels (25), but it can also promote apoptotic cell death if the ER stress is not alleviated. Initially, activation of IRE1α produces cytoprotective and prosurvival responses that, despite the persistent ER stress, can become attenuated within a few hours. To reduce the burden on the ER with misfolded proteins, PERK promotes translational repression via the phosphorylation of eukaryotic translation initiation factor 2α (eIF2α). However, if the ER stress is not resolved, sustained PERK-mediated phosphorylation of eIF2α leads to C/EBP homologous protein (CHOP)-induced apoptotic cell death through various signaling pathways (26, 27). The infection of AMϕ with PRRSV has been shown to trigger ER stress, including the activation of IRE1α and PERK (28). Our studies revealed that in AMϕ infected with PRRSV, NF-κB is activated by IRE1α early in infection, while eIF2α is activated by PERK late in infection. The phosphorylation of eIF2α occurring late in infection was associated with inhibited production of IFN-α and TNF-α, which could be partially reversed by an inhibitor of PERK. In contrast, early in the infection, IRE1α promoted the activation of NF-κB and enhanced TNF-α production stimulated by LPS and was abolished by an inhibitor of the kinase activity of IREα. The potential role of the UPR in promoting the production of proinflammatory cytokines is discussed as a plausible mechanism to explain the exacerbated and often lethal pneumonia that occurs during PRRSV-bacterial coinfections.
RESULTS
Permissiveness and growth kinetics of PRRSV in the porcine AMϕ line ZMAC.
Due to the high variability in the permissiveness of primary PAMϕ to PRRSV, which can range from 15% to 60% (29, 30), we primarily used the porcine AMϕ line ZMAC, which is readily infected by PRRSV (31). The permissiveness of the ZMAC cells to PRRSV was demonstrated by infecting them with PRRSV strain P129-GFP, which was engineered to transcribe green fluorescent protein (GFP) as an additional subgenomic mRNA with its own transcription regulation sequence, resulting in the efficient expression of GFP in cells productively infected with the virus (32). The percentage of GFP-positive (GFP+) ZMAC cells infected with P129-GFP virus, at a multiplicity of infection (MOI) of 5, was scored directly in unprocessed cell cultures using an inverted fluorescence microscope. At 4 h postinfection (hpi), <1% of the infected ZMAC cells exhibited green fluorescence (Fig. 1A). In contrast, at 8 hpi, clear evidence of GFP expression was observed in 73% of the cells. Approximately 10% of them exhibited evidence of cytopathic effect (CPE), which was characterized by cell rounding, and additionally, a few cells exhibited the presence of membrane blebs (Fig. 1A). By 12 hpi, 80% of the cells were GFP+, and the majority also exhibited CPE, expressed as cell rounding accompanied by the formation of cytoplasmic vacuoles. At this time, approximately 15% of the cells also exhibited the presence of membrane blebs, suggesting that such cells were in the early stages of apoptosis, a process known to occur in AMϕ infected with PRRSV (33), although, as determined by vital-dye exclusion, 95% of the cells were viable (Fig. 1B). After an additional 8 h (20 hpi), the percentage of GFP+ cells had decreased to 50% (Fig. 1A). However, most GFP- cells lacked structural integrity and exhibited extensive disintegration. The nature of the CPE observed at 20 hpi was consistent with an advanced stage of apoptosis termed secondary necrosis, which is also known to occur in AMϕ during the late stages of PRRSV infection (33). The occurrence of PRRSV infection-mediated cell death due to apoptosis at 20 hpi was confirmed by the presence of terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL)-positive cells among virus-infected cells (Fig. 1C).
FIG 1.

Permissiveness of the porcine AMϕ cell line ZMAC to PRRSV. (A) ZMAC cells were infected with the GFP-expressing PRRSV strain P129-GFP (MOI = 5). GFP+ cells in each sample were scored visually at the indicated times postinfection by fluorescence microscopy. The mean percentages of GFP+ cells ± standard deviations (SD) from three independent experiments are indicated. The arrows identify cells exhibiting membrane blebs. (B) ZMAC cells were either mock infected or infected with PRRSV strain FL12 (MOI = 5). At 12 hpi, cell viability was determined by trypan blue (TB) exclusion. The percentages of viable cells were calculated as the TB-negative cells divided by the total counted cells. The results shown are representative of three independent experiments. (C) Monolayers of ZMAC cells were either mock infected or infected with PRRSV strain FL12 (MOI = 5). At 20 hpi, cell monolayers were processed for DeadEnd colorimetric TUNEL assay. In this assay, apoptotic nuclei are stained dark brown. The images are representative of three independent experiments. (D) Single-step growth curve of PRRSV in ZMAC cells. ZMAC cells at 2 × 105/ml were infected with PRRSV strain FL12 (MOI = 5) for 1 h, washed twice, and suspended to the original volume (2 ml). At the indicated times postinfection, a sample of the overlying medium was removed and the titer of infectious virus (TCID50 per milliliter) was determined. The data represent the means ± standard deviations of duplicate measurements of a representative of three independent experiments.
The rate of PRRSV replication in ZMAC cells was determined using a single-step growth curve by infecting ZMAC cells with PRRSV strain FL12 (MOI = 5). After a slight gain in virus titer at 4 hpi, there was a 104-fold increase by 8 hpi and a subsequent 20-fold increase 4 to 6 h later (Fig. 1D). This indicated that the replication cycle of PRRSV in ZMAC cells was completed within a 12- to 16-h period, similar to the reported 12-h period required for the same process to occur in primary PAMϕ (33).
Kinetics of IFN-α and TNF-α responses of porcine AMϕ.
The kinetics of IFN-α and TNF-α responses by ZMAC cells was examined by stimulating the cells with either synthetic dsRNA or LPS, respectively. In response to stimulation with poly(I·C), IFN-α production could be detected by 4 h after stimulation and increased by 5-fold 4 h later, reaching a level of >8 ng/ml (Fig. 2A). As calculated by linear regression, a half-maximal response was reached at approximately 6 h after stimulation. The production of IFN-α was preceded by the phosphorylation of IRF3 (p-IRF3), which became evident at 1 h after poly(I·C) stimulation (Fig. 2B). In contrast, the production of TNF-α in response to LPS stimulation developed with faster kinetics, as indicated by the nearly plateaued production of the cytokine by 6 h after exposure to the agonist. As calculated by linear regression, the half-maximal TNF-α response of ZMAC cells to LPS occurred approximately 3 h after stimulation (Fig. 2C). The response to LPS stimulation was preceded by the phosphorylation of NF-κB, which was detected at 30 min poststimulation and appeared to have dissipated by 1.5 h later (Fig. 2D). Therefore, similar to primary PAMϕ (21, 34), ZMAC cells readily produce IFN-α and TNF-α in response to their stimulation by poly(I·C) and LPS, respectively.
FIG 2.
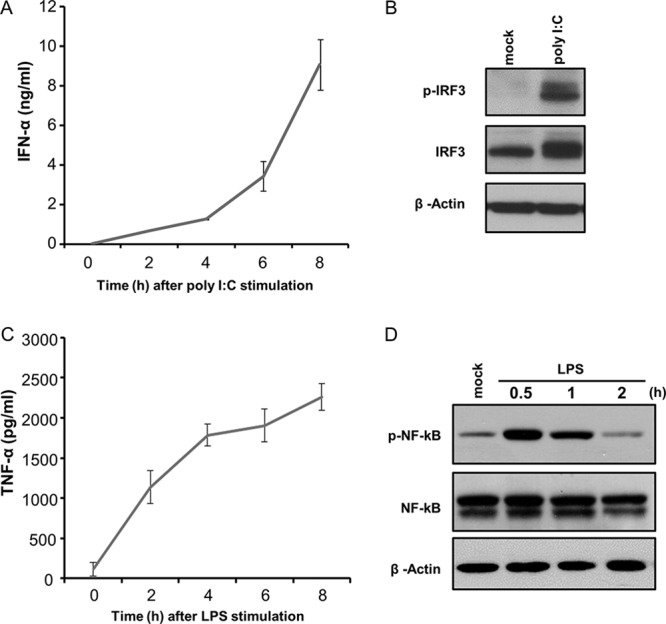
Kinetics of IFN-α and TNF-α responses of porcine AMϕ. (A and C) ZMAC cells were stimulated with poly(I·C) (25 μg/ml) (A) or LPS (100 ng/ml) (C), and the amount of IFN-α (A) or TNF-α (C) present in cell-free culture supernatant at the indicated time after stimulation was determined by ELISA. The results represent the means ± standard deviations of three independent experiments for each agonist. (B) ZMAC cells were either mock treated or exposed to poly(I·C) (25 μg/ml) for 1 h, and their whole-cell lysates were analyzed by Western blotting to detect p-IRF3, total IRF3, and β-actin. (D) ZMAC cells were either mock treated or exposed to LPS (100 ng/ml), and their whole-cell lysates were harvested at the time points indicated and analyzed by Western blotting to detect p-NF-κB, total NF-κB, and β-actin. The results shown are representative of two independent experiments.
Infection of AMϕ with PRRSV inhibits their ability to produce IFN-α in response to stimulation with synthetic dsRNA.
Infection of AMϕ with PRRSV does not bring about significant IFN-α production (9). Rather, infection with PRRSV has been reported to inhibit the IFN-α response of AMϕ to agonists, such as synthetic dsRNA (8, 21). To confirm these observations, we compared the IFN-α responses of ZMAC cells resulting from their exposure to PRRSV or to transmissible gastroenteritis virus (TGEV). The latter is a porcine coronavirus that is a potent stimulator of IFN-α. While the exposure of AMϕ to TGEV triggered a noticeable IFN-α response, their exposure to PRRSV triggered a meager response (Fig. 3A). Regarding the inhibitory effect of PRRSV on IFN-α production, exposure of ZMAC cells to poly(I·C) resulted in strong production of IFN-α, while exposure of the cells to PRRSV strain NADC20 failed to elicit production of the cytokine (Fig. 3B). In contrast, infection of AMϕ with PRRSV 2 h before stimulation with poly(I·C) reduced the production of IFN-α by approximately 50%. Notably, the presence of infectious virus was required for this event to occur, as evidenced by the fact that exposure of the cells to UV-inactivated virus resulted in the release of a quantity of the cytokine similar to that released by cells treated with poly(I·C). Hence, the ability of PRRSV to inhibit the production of IFN-α requires the presence of live virus.
FIG 3.

Infection of AMϕ with PRRSV fails to stimulate IFN-α and inhibits their ability to produce IFN-α in response to stimulation with poly(I·C). (A) ZMAC cells were infected with PRRSV strain FL12 or TGEV strain Purdue, and the frequency of IFN-α-secreting cells after 16 h of culture was determined by ELISPOT assay. (B) Duplicate cultures of ZMAC cells were either mock treated or incubated in the presence of UV light-inactivated or viable PRRSV strain NADC20 for 2 h. Afterward, one member of each pair was mock stimulated or stimulated with poly(I·C) (25 μg/ml) for 8 h, and the amount of IFN-α present in the supernatant was determined by ELISA. The results represent the means ± standard deviations of three independent experiments. The asterisks indicate statistically significant differences (**, P < 0.01) between the cytokine present in the supernatants of infected versus mock-infected cultures stimulated with poly(I·C). NS indicates lack of statistical significance.
Infection of AMϕ with PRRSV inhibits neither the activation IRF3 or STAT1 nor the transcription of IFNA1, IFNB1, and IRF7 genes induced by stimulation with synthetic dsRNA.
The ability of PRRSV to inhibit IFN-α production has been attributed to the capacity of some viral nonstructural proteins to block the transcription of type I IFN genes by interfering with the activation of key transcription factors (20). Consequently, we examined the effect of PRRSV infection on the poly(I·C)-induced activation of TFs known to play key roles in the early phase (IRF3) and positive-feedback regulation (STAT1) of type I IFN gene expression (35, 36). Phosphorylated IRF3 (Fig. 4A) or STAT1 (Fig. 4B) was undetectable in lysates of unstimulated and mock-infected cells. In contrast, similar quantities of phosphorylated IRF3 and STAT1 were readily found in lysates prepared from the poly(I·C)-treated cells regardless of whether they were left uninfected or infected with one of two different PRRSV strains for 1, 3, or 5 h prior to agonist addition (Fig. 4A and B). Hence, the infection of AMϕ with PRRSV does not appear to negatively influence the activation of IRF3 or STAT1 in response to stimulation with synthetic dsRNA.
FIG 4.

Infection of AMϕ with PRRSV inhibits neither the activation of IRF3 or STAT1 nor the transcription of IFNB1, IRF7, and IFNA1 genes induced by stimulation with poly(I·C). (A and B) ZMAC cells were infected with either PRRSV strain FL12 or NADC20 (MOI = 5) and stimulated with poly(I·C) (25 μg/ml) at the indicated times postinfection. At 1 h after stimulation, whole-cell lysates were obtained and analyzed by Western blotting to sequentially detect p-IRF3, IRF3, and β-actin (A) or p-STAT1, STAT1, and β-actin (B). As a control, replicate cell sets were mock infected and cultured for 5 h before an additional 1-h incubation in the presence or absence of poly(I·C) and then harvested. (C, D, and E) ZMAC cells were mock infected or infected with PRRSV strain FL12 or NADC20 and stimulated with poly(I·C) (25 μg/ml) at 2 hpi. After 1, 4, or 7 h of stimulation, total RNA was obtained from each sample and subjected to real-time PCR analysis to detect IFNB1, IRF7, and IFNA1 gene transcripts. As a control, a replicate cell set was mock treated and cultured for 3 h before harvest. The fold changes in the amounts of these RNAs present in the virus-infected and poly(I·C)-stimulated AMϕ, as well as the mock-infected cells exposed to poly(I·C) for 4 or 7 h, relative to that in the mock-infected cells incubated with poly(I·C) for 1 h were determined by using the formula 2−ΔΔCt. The RPL32 gene was used as the reference housekeeping gene. RNA fold increases for IFNB1 and IFNA1 gene transcripts in mock-treated control cell samples cultured for 3 h were undetectable relative to those observed in cells exposed to poly(I·C) for 1 h, while the IRF7 gene transcript levels were approximately 2-fold greater. The asterisk indicates a statistically significant difference (*, P < 0.05).
The PRRSV-mediated inhibition of IFN-α production observed in virus-infected ZMAC cells stimulated with poly(I·C) appeared to occur subsequent to the activation of IRF3 and STAT1. Thus, a possible effect on the transcription of genes whose products are involved in type I IFN induction was assessed. Initially, an early phase representative of this process, IFNB1, was examined. In this case, virus infection of the ZMAC cells was allowed to proceed for 2 h before the addition of poly(I·C), followed by 1, 4, or 7 h of culture. Compared to its undetected activity in mock-infected and untreated cells cultured for 3 h, expression of the IFNB1 gene was observed at 1 h post-agonist exposure and was found to be at a similar rate of expression regardless of whether the cells were uninfected or infected with one of two different PRRSV strains (Fig. 4C). This response appeared to decrease by approximately 80% 3 h later and then may have increased approximately 2-fold after an additional 3 h. Since the early phase of type I interferon induction was not negatively affected by PRRSV (Fig. 4C), we monitored the presence of mRNA of a gene whose expression is known to be a key part of the positive-feedback regulation of type I IFN production, namely, the IRF7 gene (36). In this case, constitutive expression of the IRF7 gene was detected in uninfected, untreated cells at approximately twice the level detected in cells stimulated for 2 h with poly(I·C). However, the relative quantities of IRF7 mRNA had tripled 3 h later and were sustained or slightly increased for the next 3 h. There was no apparent negative effect of PRRSV on the poly(I·C)-induced IRF7 gene expression at either 4 h or 7 h (Fig. 4D). Considering that the expression of IRF7 in poly(I·C)-stimulated cells should promote the expression of IFN-α genes, we monitored the transcription level of one representative, namely, IFNA1. Expression of the IFNA1 gene in uninfected, untreated cells was not detected. Cells that were exposed to poly(I·C) for 1 or 4 h in the presence or absence of the virus exhibited similar small amounts of IFNA1 mRNA. By 7 h poststimulation, the amounts of IFNA1 mRNA had increased 4- to 5-fold regardless of the presence or absence of virus infection (Fig. 4E). It is notable that the expression of the porcine IFNA1 gene in poly(I·C)-stimulated cells became detectable only after expression of the IRF7 gene had occurred. This sequence of events suggests that the regulation of this particular porcine IFN-α gene might be similar to that of other members of the murine IFN-α gene family, which require the presence of IRF7 in order to become transcriptionally active (35). This event is known to be dependent on the synthesis of small amounts of IFN-β/α during the early phase of the type I IFN gene induction pathway. The limited amount of IFN-β/α produced in the early phase engages the type I IFN receptor and initiates a positive-feedback loop of the type I IFN response via the JAK/STAT signaling pathway (36). Evidence that the positive-feedback loop was operational in PRRSV-infected cells was provided by the presence of phosphorylated STAT1, regardless of the length of time after virus infection when the cells were stimulated with poly(I·C) (Fig. 4B). In addition, the observation that the transcription of the IFNA1 gene occurred at 7 h after stimulation with the agonist and corresponded to 9 h after the initiation of the virus infection indicates that the reduced synthesis of IFN-α observed in virus-infected AMϕ occurs after the positive-feedback loop of type I IFN is operational.
The synthetic analog of dsRNA, poly(I·C), is recognized by at least two types of pattern recognition receptors (PRRs): the cytoplasmic RIG-I-like receptors (RLRs) (RIG-I and MDA5) and Toll-like receptor 3 (TLR3) localized in endosomes. While free poly(I·C) is readily sensed by TLR3 as a result of endocytosis (37), its detection by cytosolic sensors is apparently best achieved by the forced delivery of the molecule by way of transfection (38–40). In both cases, IRF3 is the downstream transcription factor that becomes activated following the engagement of these sensors and promotes type I IFN gene expression (36). Since the experiments shown in Fig. 4 were done using free poly(I·C), we sought to determine if the signaling pathways activated by the recognition of dsRNA by cytosolic sensors would also become inhibited during the infection of AMϕ with PRRSV. Accordingly, ZMAC cells that had been infected with PRRSV 2 h earlier were transfected with poly(I·C) and then examined for the presence of phosphorylated IRF3, as well as the production of IFN-α. The infection of AMϕ with PRRSV did not appear to influence the ability of poly(I·C) delivered by either transfection or endocytosis to trigger the phosphorylation of IRF3 (Fig. 5A). The amount of IFN-α produced by AMϕ infected with either strain FL12 or NADC20 of PRRSV in response to stimulation with poly-I·C via transfection was >65% lower than the amount produced by uninfected and identically poly(I·C)-transfected cells (Fig. 5B). Hence, the infection of AMϕ with PRRSV inhibits the production of IFN-α in response to the stimulation of cytosolic or endosomal sensors by dsRNA, but it appears to do so without inhibiting the activation of IRF3. These results indicate that the mechanism by which the infection of AMϕ with PRRSV mediates the inhibition of IFN-α production in response to stimulation with synthetic dsRNA occurs after the positive-feedback loop of type I IFN production has been initiated and is manifested at a late stage (>6 hpi) of virus infection.
FIG 5.
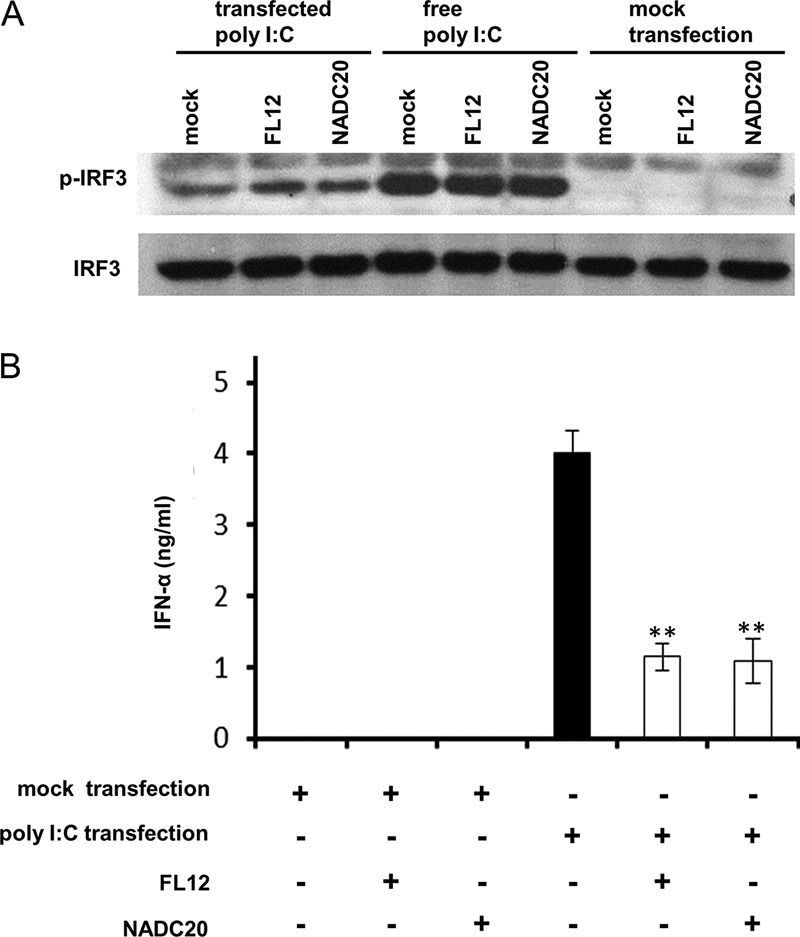
Infection of AMϕ with PRRSV does not inhibit the activation of IRF3 induced by transfection with poly(I·C) despite the inhibition of IFN-α production. (A) ZMAC cells were mock infected or infected with PRRSV strain NADC20 or FL12 (MOI = 5). At 2 hpi, the cells were exposed to poly(I·C) either by transfection (400 ng/ml) or in free form (25 μg/ml). At 2 h after poly(I·C) treatment, whole-cell lysates were analyzed by Western blotting for the presence of p-IRF3 and total IRF3. Identical cultures were treated with the transfection reagent without poly(I·C) (mock transfection). (B) Duplicate cultures of ZMAC cells were mock infected or infected with PRRSV strain FL12 or NADC20 (MOI = 5). At 2 hpi, one member of each pair was exposed to either poly(I·C) (400 ng/ml) complexed with transfection reagent or mock transfected [exposed to transfection reagent without poly(I·C)]. After 8 h of culture, the amounts of IFN-α present in cell-free supernatants were determined by ELISA. The data represent the means ± standard deviations of two independent experiments. Statistical comparisons were made between the amounts of the cytokine present in the supernatants of infected versus mock-infected cultures stimulated with poly(I·C). The asterisks indicate statistical significance (**, P < 0.01).
Infection of AMϕ with PRRSV triggers the unfolded protein response.
Considering the evidence indicating that PRRSV infection-mediated inhibition of IFN-α production by AMϕ was due neither to a negative influence on the activation of the transcription factor IRF3 or STAT1 nor to the inhibition of transcription of the IFNA1, IFNB1, and IRF7 genes, we sought to uncover an alternative explanation which would involve an event occurring at a late stage of virus infection. The replication of RNA viruses typically places an inordinate stress on the protein-folding machinery of the host cell, causing ER stress (23, 24). ER stress activates a series of adaptive mechanisms known as the UPR. The adaptive response occurring in the UPR aims to rebalance protein-folding homeostasis by activating three ER stress sensor proteins: IRE1α, PERK, and ATF6. Activation of these sensors induces signal transduction events that alleviate the accumulation of misfolded proteins in the ER by increasing expression of ER chaperones, inhibiting protein entry into the ER by arresting mRNA translation, and stimulating retrograde transport of misfolded proteins from the ER into the cytosol for ubiquitination and destruction by a process named ERAD (ER-assisted degradation) (25). Accordingly, we focused on examining the activation status of the ER stress sensors IRE1α and PERK in PRRSV-infected cells. Mock-infected AMϕ exhibited the presence of a relatively low-level expression of phosphorylated IRE1α (p-IRE1α), which was slightly increased by their exposure to the ER stress-inducing agent dithiothreitol (DTT) (Fig. 6A). Exposure of AMϕ to the same chemical in the presence of the drug IRE1α kinase-inhibiting RNase attenuator 6 (KIRA6), which blocks IRE1α trans-autophosphorylation during ER stress (41), reduced p-IRE1α below the level exhibited by untreated or DTT-treated cells. This result indicates that the drug KIRA6 can block the kinase domain of the ER transmembrane stress sensor IRE1α in porcine AMϕ. Examination of the presence of p-IRE1α in PRRSV-infected AMϕ revealed a marked increase in the level of p-IRE1α at 2 hpi, which gradually decreased over the ensuing 4 h (Fig. 6A). PERK is a transmembrane protein kinase that under ER stress conditions dimerizes and autophosphorylates, favoring the phosphorylation of eIF2α. To examine the activation of this stress sensor, lysates of PRRSV-infected AMϕ were collected at several times after virus infection and probed by Western blotting for the presence of phosphorylated PERK (p-PERK). We found that p-PERK could be detected in the PRRSV-infected AMϕ as early as 3 hpi and increased gradually over the next 6 h, becoming maximal by 9 hpi (Fig. 6B). Activation of PERK causes the phosphorylation of eIF2α, which in turn causes global translation attenuation and favors selective translation (42). Temporal screening for the presence of phosphorylated eIF2α (p-eIF2α) in PRRSV-infected ZMAC cells revealed that, whereas p-eIF2α was either undetected or present in small amounts in mock-infected cells, it became detectable by 6 h after virus infection and gradually increased for the next 6 h (Fig. 6C). The UPR has a primary function in stress adaption and cell survival; however, under irremediable ER stress, a switch from prosurvival to proapoptotic signaling events can occur, resulting in apoptotic death of damaged cells. One major event responsible for this switch is the expression of CHOP, in which PERK plays an essential role. CHOP expression is low under nonstressed conditions, but its expression markedly increases in response to ER stress through IRE1α-, PERK-, and ATF6-dependent transcriptional induction (43). Analysis of the expression of CHOP in virus-infected cells revealed that its expression became evident by 6 hpi (Fig. 6D). Combined, the results presented above indicate that the infection of AMϕ with PRRSV triggers the UPR response.
FIG 6.

Infection of AMϕ with PRRSV induces the UPR and the formation of SGs. (A) ZMAC cells were infected with PRRSV strain FL12 (MOI = 5) and harvested at the indicated times postinfection. Replicate cell sets were mock infected and cultured for 6 h before an additional 1-h incubation without treatment (mock), treated with DTT (DTT), or treated with both DTT and the IRE1α kinase inhibitor KIRA6 (DTT+KIRA). Whole-cell lysates were prepared and analyzed by Western blotting to detect p-IRE1α. (B) ZMAC cells were infected with PRRSV strain FL12 (MOI = 5) and harvested at the indicated times postinfection. Whole-cell lysates were analyzed by Western blotting for p-PERK and β-actin. Negative- and positive-control cell lysates were prepared from mock-infected cells cultured for 8 h or cultured for 2 h in the presence of DTT, respectively. (C) ZMAC cells were infected with PRRSV strain FL12 (MOI = 5) and harvested at the indicated times postinfection. Whole-cell lysates were analyzed by Western blotting for p-eIF2α and total eIF2α. Negative- and positive-control cell lysates were prepared from mock-infected cells cultured for 12 h or cultured for 1 h in the presence of the ER stress-inducing chemical DTT, respectively. (E) (Top row) Monolayers of ZMAC cells were either mock infected for 8 h, treated with DTT for 1 h, or infected with PRRSV strain FL12 (MOI = 5) for 4 or 8 h. Afterward, the cell monolayers were fixed and processed for immunofluorescence staining using anti-TIAR (red fluorescence) or anti-dsRNA (green fluorescence) antibodies. (Bottom row) Higher magnifications (×3) of the original boxed areas of the ×40 images. The percentages of dsRNA+ (red) and SG+ (green) cells were calculated from the examination of 100 cells per treatment. The averages ± SD of three independent experiments are indicated below. (F) ZMAC cells were infected with PRRSV strain FL12 (MOI = 5) and harvested at the indicated times postinfection. Whole-cell lysates were analyzed by Western blotting for phosphorylated PKR and β-actin. Negative- and positive-control cell lysates were prepared from mock-treated cells cultured for 8 h or cells treated with poly(I·C) (25 μg/ml) and cultured for 2 or 6 h, respectively.
Infection of AMϕ with PRRSV induces stress granule formation.
The phosphorylation eIF2α should cause the accumulation of stalled translation initiation complexes that would associate with RNA binding proteins to create stress granules (SGs) (44). Accordingly, we examined PRRSV-infected ZMAC cells for the presence of SGs by using indirect immunofluorescence with an antibody (Ab) specific for the TIA-1 related protein (TIAR), which is a primary component of these structures (45). At 4 hpi, the percentage of TIAR+ cells (3.8%) was comparable to that observed in the uninfected, untreated cells (3.5%). However, by 4 h later (8 hpi), the percentage of TIAR+ cells had increased >11-fold to 44%, and this value was similar to the 51% measured in cells treated with the ER stress-inducing chemical DTT (Fig. 6E). The timing of the appearance of TIAR+ SGs in PRRSV-infected cells at 8 hpi coincided with the appearance of p-eIF2α. To examine the relationship between the presence of TIAR+ SGs and virus infection, ZMAC cells were dually stained with anti-TIA-1 and anti-dsRNA antibodies to detect viral dsRNA produced during PRRSV replication. While not identified in the mock-infected cells or in cells exposed to DTT, dsRNA was observed in 13% and 72% of the virus-infected cells at 4 and 8 hpi, respectively, and all of the TIAR+ cells were also dsRNA+ (Fig. 6E). These results suggest that in AMϕ, PRRSV infection results in the formation of stress granules and, by implication, translational attenuation. Notably, despite the presence of dsRNA in the virus-infected cells, there was no evidence that the dsRNA-dependent protein kinase R (PKR) became phosphorylated at either 2, 5, or 8 hpi (Fig. 6F). These results indicate that the observed phosphorylation of eIF2α in PRRSV-infected AMϕ does not appear to be mediated via the action of PKR.
The repressed ability of PRRSV-infected AMϕ to produce IFN-α in response to synthetic dsRNA coincides with the timing of eIF2α phosphorylation induced by virus infection.
To ascertain the involvement of translation attenuation in the PRRSV-mediated inhibition of IFN-α production, the kinetics of eIF2α phosphorylation and inhibition of IFN-α production were simultaneously determined in PRRSV-infected AMϕ. Compared to mock-infected cells stimulated with poly(I·C), a reduction in the amount of IFN-α released by the infected ZMAC cells became apparent by 8 hpi. This difference increased through the ensuing hours, reaching a 69% level of inhibition at 10 hpi (Fig. 7A). The presence of p-eIF2α became evident by 5 hpi and increased gradually in intensity over the ensuing 3 h (Fig. 7B). At all times examined, the uninfected cells exhibited negligible presence of p-eIF2α (Fig. 7C). The same experiment was performed using primary PAMϕ, in which case strong inhibition of IFN-α production was evident at 12 hpi (Fig. 7D), at the time when p-eIF2α was patently present (Fig. 7E). To assess the effect of p-eIF2α on viral mRNA translation, lysates of ZMAC cells (Fig. 8A) or primary PAMϕ (Fig. 8B) infected with P129-GFP virus were probed for the presence of GFP, eIF2α, and p-eIF2α. The presence of p-eIF2α increased over time. Notably, the expression of GFP became evident at 10 hpi despite the pronounced presence of p-eIF2α. These results indicate that PRRSV mRNAs, to a measurable extent, are resistant to translational attenuation mediated by p-eIF2α.
FIG 7.

The repressed ability of AMϕ infected with PRRSV to produce IFN-α in response to poly(I·C) coincides with the timing of eIF2α phosphorylation induced by virus infection. ZMAC cells (A to C) or primary PAMϕ (D to F) were either mock infected or infected with the indicated strain of PRRSV (MOI = 5) for 2 h prior to their stimulation with poly(I·C) (25 μg/ml). (A and D) The amounts of IFN-α present in the culture supernatants at the indicated times after stimulation were determined by ELISA. The data represent the means ± SD of triplicate values obtained in a representative of three (A) or two (D) independent experiments. For virus-infected cultures, the times postinfection at which the cell culture supernatants and cells were harvested for analysis are indicated. The bottom rows indicate the percent inhibition of IFN-α production in virus-infected cultures relative to that detected in identically stimulated mock-infected cultures incubated for a corresponding length of time. This value was calculated using the following formula: 100 − [(IFN-α in virus-infected culture supernatant/IFN-α in mock-infected culture supernatant) × 100]. At each of the time points analyzed, whole-cell lysates were also prepared from the virus-infected (B and E) and mock-infected (C and F) cell cultures and proved by Western blotting for p-eIF2α and total eIF2α. The statistical comparisons in panels A and C were made between the amounts of cytokine present in the supernatants from mock-infected versus virus-infected cultures stimulated with poly(I·C). The asterisks indicate statistical significance (**, P < 0.01; *, P < 0.05).
FIG 8.
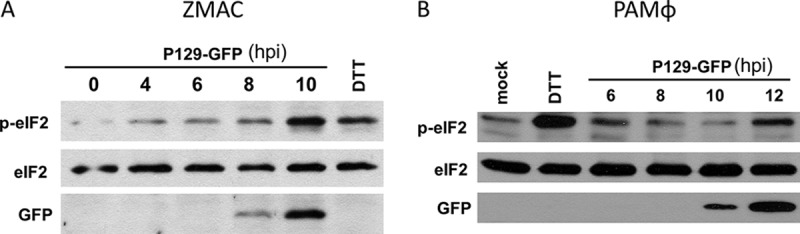
Time course of viral mRNA translation and phosphorylation of eIF2α. ZMAC cells (A) or primary PAMϕ (B) were infected with PRRSV strain P129-GFP (MOI = 5) and harvested at the indicated times postinfection. Whole-cell lysates were analyzed by Western blotting for the detection of p-eIF2α, total eIF2α, and virus-encoded GFP. Positive-control cell lysates were prepared from cells cultured in the presence of the ER stress-inducing chemical DTT for 1 h. Negative-control cell lysates were prepared from cells exposed to mock-infected medium for 12 h. The images are representative of three (A) or two (B) independent experiments.
Infection of AMϕ with PRRSV initially enhances but later suppresses the TNF-α response to LPS.
Due to the relatively slow kinetics of the IFN-α response of AMϕ to poly(I·C) (Fig. 2A), the inhibitory effect mediated by the infection of AMϕ with PRRSV on the production of the cytokine became manifest by >6 h after virus infection. To further examine the temporal association between the phosphorylation of eIF2α and the inhibition of cytokine production, we decided to test the TNF-α response of virus-infected cells stimulated with LPS. This decision was based on the fact that the response to this agonist has faster kinetics, resulting in half-maximal TNF-α production within 3 h after stimulation (Fig. 2C). Furthermore, the infection of AMϕ with PRRSV triggers negligible production of the cytokine. Two time points after virus infection, 2 and 6 h, were selected to stimulate the cells with LPS. In this setup, at 2 hpi and for the ensuing 4 h, the virus infection-induced phosphorylation of eIF2α should be minimal, while at 6 hpi, and for the ensuing 4 h, it would be substantial. Compared to the amount of TNF-α released by mock-infected AMϕ cultures stimulated with LPS at 2 hpi, a 1.7-fold increase of the cytokine was measured 2 and 4 h later, corresponding to 4 and 6 hpi, respectively (Fig. 9A). In contrast, when the TLR4 agonist was provided at 6 hpi, a >50% reduction in the measured amount of TNF-α was detected 2 and 4 h later, corresponding to 8 and 10 hpi, respectively (Fig. 9A).
FIG 9.

Infection of AMϕ with PRRSV initially enhances but later suppresses the TNF-α response to LPS. (A) ZMAC cells were either mock infected or infected with PRRSV strain FL12 (MOI = 5) for 2 or 6 h prior to their stimulation with LPS (100 ng/ml). After 2 or 4 h of additional culture, the presence of TNF-α in the supernatants was determined by ELISA. The data shown are the means ± SD of a representative of three independent experiments. Statistically significant differences between the amounts of cytokine present in supernatants from infected versus mock-infected cultures stimulated with LPS are indicated with asterisks (**, P < 0.01). (B and C) Status of NF-κB in AMϕ that were infected with PRRSV with no further treatment (B) or infected and stimulated with LPS (100 ng/ml) for 1 h before harvest (C). Whole-cell lysates obtained at the indicated times postinfection were analyzed by Western blotting for p-NF-κB, total NF-κB, and β-actin. Lysates of mock-infected AMϕ cultured for 5 h before an additional 1-h incubation in the presence or absence of LPS were also examined. The blots shown are representative of three independent experiments.
Examination of the status of NF-κB-p65 in virus-infected cells revealed that the phosphorylated version of this molecule, which is a subunit of the NF-κB transcription complex that regulates the expression of TNF-α, was detectable in PRRSV-infected AMϕ as early as 2 h after infection and remained at the same or slightly increased levels of activation for the ensuing 4 h (Fig. 9B). Furthermore, assessment of the presence of phosphorylated NF-κB-p65 in cell cultures infected with PRRSV for 1, 3, or 5 h before stimulation with LPS and cultured for 1 h before preparation of cell lysates revealed no apparent differences between the levels of phosphorylated NF-κB-p65 present in those lysates and in lysates obtained from mock-infected cells stimulated with LPS (Fig. 9C). Hence, unlike the delayed negative influence of PRRSV on the IFN-α response by AMϕ stimulated with poly(I·C), its effect on TNF-α production by virus-infected cells in response to LPS ranged from intensification to reduction, which was dependent on the temporal extent of virus infection prior to their stimulation with the TLR4 agonist. Furthermore, the inhibitory effect of PRRSV infection on the production of TNF-α late in infection occurred even though the activation of NF-κB-p65 by LPS was not impaired.
The PERK-eIF2α signaling pathway is involved in the PRRSV infection-induced inhibition of the TNF-α and IFN-α responses of AMϕ.
Our results revealed that the repressed production of TNF-α exhibited by PRRSV-infected AMϕ stimulated with LPS occurred only when the cells were exposed to the TLR4 agonist at 6 hpi (Fig. 9A), and this event occurred without impairing the activation of NF-κB (Fig. 9C). These observations, combined with the lack of an inhibitory effect on cytokine gene expression induced by the stimulation of virus-infected cells with poly(I·C) (Fig. 4E), as well as the abundant presence of p-eIF2α at the late stage (>6 hpi) of virus infection (Fig. 7B), prompted us to explore the possibility that the inhibitory effect on cytokine production could be due to translational attenuation occurring at a late phase of virus infection, as suggested by the appearance of SGs (Fig. 6E). To test this notion, we simultaneously examined the production of TNF-α and the phosphorylation status of eIF2α in AMϕ stimulated with LPS at either 2 or 6 h after the initiation of the virus infection. A marked inhibition (>60%) of TNF-α production was observed in AMϕ infected with either of two different strains of PRRSV (FL12 or NADC20) for 6 h before being exposed to LPS and then cultured for 2 h. In contrast, the inhibitory effect was absent when the cells were exposed to LPS at 2 hpi and cultured for 2 h (Fig. 10A). Examination of the phosphorylation status of eIF2α in the cell lysates obtained from cells stimulated with LPS at 6 hpi and cultured for 2 h revealed the abundant presence of p-eIF2α at levels comparable to those observed in cells exposed for 1 h to DTT (Fig. 10B). In contrast, only scarcely detectable levels of p-eIF2α were present in the lysates of cells that had been infected with either strain of PRRSV for only 2 h before being stimulated with LPS and harvested 2 h later, at 4 hpi (Fig. 10B). These results indicate that the ability of PRRSV to inhibit TNF-α production by AMϕ stimulated with LPS coincides with the time after infection when p-eIF2α is markedly present.
FIG 10.
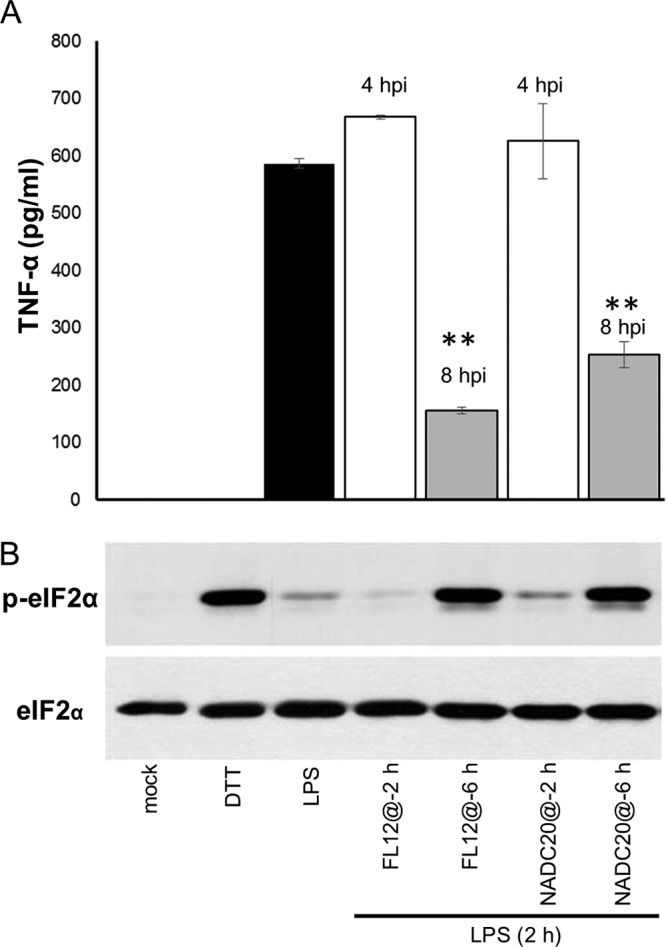
The ability of PRRSV to inhibit TNF-α production by AMϕ stimulated with LPS coincides with the time after infection when p-eIF2α is markedly present. (A and B) ZMAC cells were either mock infected or infected with PRRSV strain FL12 or NADC20 (MOI = 5) for 2 (@-2) or 6 (@-6) h and then exposed to LPS (100 ng/ml). Cell-free supernatants and cells from the same cultures were harvested 2 h later. (A) The amounts of TNF-α present in the supernatants were determined by ELISA. (B) The presence of p-eIF2α and total eIF2α in whole-cell lysates was assessed by Western blotting. Additional replicate sets were mock infected and cultured for 2 h before an additional 2- or 1-h incubation in the presence of LPS or DTT, respectively. Shown are the means ± standard deviations (A) and corresponding immunoblots representative of three independent experiments (B). Statistical comparisons in panel A were made between the amounts of cytokine present in the supernatants of virus-infected versus mock-infected cultures stimulated with LPS. The asterisks indicate statistical significance (**, P < 0.01).
Considering that the infection of AMϕ with PRRSV did not trigger the activation of PKR (Fig. 6F), we examined the involvement of PERK in the generation of p-eIF2α by testing the effect of the PERK inhibitor (PERKi) GSK2606414 (46) on the ability of the PRRSV infection to induce the phosphorylation of eIF2α and its inhibitory effect on TNF-α production. AMϕ were infected with PRRSV for either 4 or 6 h prior to their exposure to PERKi, followed by an additional 5 or 3 h of culture, respectively, so that all of the samples would be harvested at 9 hpi, at the time when p-eIF2α would be patently present (Fig. 6C). The addition of PERKi at either time post-virus infection resulted in the generation of smaller amounts of p-eIF2α, as indicated by a ≥50% reduction in the p-eIF2α/eIF2α ratios compared to ratios observed in the PRRSV-infected AMϕ not treated with the PERK inhibitor (Fig. 11A). To evaluate the effect of the PERKi on the virus infection-induced suppression of TNF-α production, AMϕ were either mock infected or infected with PRRSV for 2 h prior to a 5-h incubation in the absence or presence of the PERK inhibitor, followed by 2 h of incubation in the presence of LPS. Virus infection alone caused an approximately 38% reduction in the amount of TNF-α produced. In contrast, in the presence of the PERKi, this repressive ability was reduced by more than half (P < 0.01), exhibiting only a 16% inhibition of TNF-α production (Fig. 11B). Similar results were obtained when the same experiment was performed but instead using poly(I·C) as the stimulus and measuring IFN-α production. In this case, the presence of PERKi diminished the PRRSV-mediated inhibition of IFN-α production by almost half, to 28% compared to 51% in its absence (Fig. 11C). The presence of PERKi during the infection did not affect PRRSV replication (Fig. 11D). Together, these results provide evidence that PERK-mediated phosphorylation of eIF2α, which occurs at a late phase of virus infection (>6 hpi), is involved in the virus-mediated inhibition of TNF-α and IFN-α responses of AMϕ to their respective agonists.
FIG 11.

The PERK-eIF2α signaling pathway is involved in the PRRSV infection-induced inhibition of the TNF-α and IFN-α response of AMϕ. (A) ZMAC cells were either mock infected or infected with PRRSV (FL12) and cultured for 9 h. Replicate cell cultures were infected with PRRSV for 4 h (@-4 h) or 6 h (@-6 h) prior to the addition of 2 μM the PERKi GSK2606414 and then cultured for an additional 5 or 3 h, respectively, before cell harvest. Additional replicate cell sets were cultured for 1 h in the presence or absence of the PERKi before being exposed to DTT and cultured for 1 h before cell harvest. Whole-cell lysates were analyzed by Western blotting for p-eIF2α and eIF2α, and the densities of the resulting bands were used to calculate the relative amounts of eIF2α phosphorylation (p-eIF2α/eIF2α). The results of two experiments are shown. (B) ZMAC cells were either mock infected or infected with PRRSV for 2 h prior to a 5-h incubation in the absence or presence of GSK2606414, followed by 2 h in the presence of LPS (100 ng/ml) before the culture supernatants were harvested. (C) ZMAC cells were either mock infected or infected with PRRSV for 2 h prior to stimulation with poly(I·C) (25 μg/ml) in the absence or presence of GSK2606414, followed by 8 h of incubation before the culture supernatants were harvested. The results shown are the means ± SD of the percent reduction in the quantity of TNF-α (B) or IFN-α (C) released by virus-infected versus mock-infected AMϕ stimulated with LPS (B) or poly(I·C) (C) in the absence or presence of the PERKi. The data shown in panels B and C are representative of three independent experiments each. Statistical comparisons were done between the levels of inhibition obtained in the presence and absence of the PERKi and are indicated by asterisks (*, P < 0.05). (D) Single-step growth curve of PRRSV in the presence of PERKi. ZMAC cells at 2 × 105 cells/ml were infected with PRRSV strain P129-GFP (MOI = 5). After 1 h of incubation, the cells were washed twice and suspended to the original volume (2 ml). At the indicated times postinfection, a sample of the overlying medium was removed and the titer of infectious virus (TCID50 per milliliter) was determined. The data represent the means ± standard deviations of three independent experiments.
The IRE1α-NF-κB signaling pathway is involved in the synergistic TNF-α response of PRRSV-infected AMϕ to LPS.
While the infection of AMϕ with PRRSV stimulated negligible production of TNF-α, stimulation of AMϕ with LPS at 2 hpi resulted in TNF-α production that was 1.5-fold greater than the response generated by identically stimulated mock-infected cells (Fig. 9A), suggesting the presence of a synergistic effect. Previously, synergistic production of TNF-α in response to LPS had been shown to occur in macrophages undergoing pharmacologically induced activation of the stress sensor kinase IRE1α (47). Considering that the infection of AMϕ with PRRSV triggered the activation of IRE1α (Fig. 6A), we investigated the plausible involvement of this kinase in mediating the virus infection-induced enhancement of TNF-α production in response to LPS. The phosphorylation of IRE1α triggers its association with TNF-α receptor-associated factor 2 (TRAF2), an adaptor protein in the TNF-α signaling pathway (48). The IRE1α-TRAF2 complex recruits IκB kinase (IKK), which in turn leads to phosphorylation and degradation of IκB, resulting in NF-κB activation and production of inflammatory cytokines (49). AMϕ infected with PRRSV exhibited the presence of phosphorylated NF-κB-p65 as early as 2 hpi (Fig. 9B), and hence, we examined the involvement of PRRSV infection-induced activation of IRE1α in the phosphorylation of this transcription factor. If involved, the inclusion of the IRE1α kinase inhibitor KIRA6 should negatively impact the ability of PRRSV to promote the phosphorylation of NF-κB, as well as to lessen the enhanced production of TNF-α following the stimulation of PRRSV-infected AMϕ with LPS 2 h after infection. Inclusion of KIRA6 in the virus-infected AMϕ cultures resulted in inhibition of the phosphorylation of NF-κB-p65 and in a >50% decrease in the p-NF-κB-p65/NF-κB-p65 ratio compared to that exhibited by virus-infected cells cultured in the absence of the IRE1α kinase inhibitor (Fig. 12A). In macrophages, the early-phase activation of NF-κB in response to signaling through TLR4 by LPS occurs via an alternate entity, namely, the intracellular adaptor protein MyD88 (50). Accordingly, as a specificity control for the inhibitory effect of KIRA6 on the activation of NF-κB-p65 via the IRE1α pathway, cell cultures were stimulated with LPS in the presence of the drug. As expected, there was no noticeable effect on the relative amounts of phosphorylated NF-κB-p65 when the LPS-treated cells were cultured in the presence of KIRA6. This was demonstrated by the absence of change in the p-NF-κB-p65/NF-κB-p65 ratio exhibited by LPS-treated cells in the presence or absence of the drug (Fig. 12A).
FIG 12.

The IRE1α-NF-κB signaling pathway is involved in the synergistic TNF-α response of PRRSV-infected AMϕ to LPS. (A) ZMAC cells were infected with PRRSV for 3 h in the presence or absence of KIRA6. Replicate cell sets were mock infected for 1 h before an additional 1-h incubation either without further treatment (mock) or with treatment with KIRA6 only, LPS (100 ng/ml) only, or both KIRA6 and LPS. Whole-cell lysates were analyzed by Western blotting for p-NF-κB and total NF-κB, and the densities of the resulting bands were used to calculate the relative amounts of NF-κB phosphorylation (p-NF-κB/NF-κB). (B) ZMAC cells were either mock infected or infected with PRRSV for 2 h before an additional 2 or 4 h of incubation without further treatment, with treatment with LPS (100 ng/ml), or with treatment with both LPS and KIRA6. Afterward, the amount of TNF-α present in the supernatant was determined by ELISA. The means ± SD of a representative of three independent experiment are presented. The presence of a synergistic TNF-α response caused by the interaction between PRRSV and LPS was determined using two-way ANOVA (51) by comparing the amounts of TNF-α present in supernatants of cultures that were either mock infected, PRRSV infected, LPS treated, or LPS treated and infected with PRRSV. The effect of KIRA6 on the synergism was analyzed in the same way, except that the last two treatment groups consisted of cultures performed in the presence or absence of the IRE1α inhibitor. The asterisks indicate statistical significance (*, P < 0.05); NS indicates lack of statistical significance.
The amount of TNF-α produced by AMϕ stimulated with LPS at 2 h after PRRSV infection was again found to be greater (approximately 30%) than that produced by mock-infected cells that were treated only with LPS and cultured for the same length of time (Fig. 12B). The presence of a synergistic effect was assessed by analyzing the data in a two-way analysis of variance (ANOVA) (51). This analysis revealed the presence of a statistically significant (P = 0.03) synergistic effect between the virus infection and the TLR4 agonist in eliciting a TNF-α response. Notably, in the presence of the IRE1α kinase inhibitor KIRA6, the synergistic TNF-α response of virus-infected AMϕ stimulated by LPS was abolished (Fig. 12B). Together, these results indicate that the activation of NF-κB in AMϕ during the early stages of PRRSV infection triggers a synergistic TNF-α response to LPS, which appears to be dependent on the activation of the ER stress sensor IRE1α.
DISCUSSION
Our results showed that the infection of porcine AMϕ with PRRSV inhibits their ability to produce IFN-α in response to their stimulation with dsRNA via either cytosolic or endosomal sensors. The inhibitory effect took place even though the virus infection did not interfere with the poly(I·C)-induced phosphorylation of IRF3 (Fig. 5A). The mechanism by which PRRSV mediates the repression of IFN-α is widely considered to act at the transcriptional level via the action of nonstructural PRRSV proteins, which seemingly have the ability to block type I IFN signaling pathways, including the activation of IRF3 (20). The evidence supporting this notion was not generated in the context of virus infection, but rather by overexpressing single viral genes via transfection and, using reporter gene assays, examining their effects on the activity of transcription factors. This approach, however, might not necessarily reflect the events that occur during the infection of AMϕ with PRRSV. Therefore, the discrepancies between the results presented here and those from previous studies could be ascribed to the fact that we examined the influence of PRRSV infection on the signaling pathway for type I IFN production in the context of the multiphasic induction of type I IFN genes triggered by synthetic dsRNA in AMϕ. The novel contribution of our work is the analysis of the inhibitory effect of PRRSV on IFN-α synthesis in the context of PRRSV infection of its natural host cell. The performance of a stepwise analysis of the type I IFN gene induction pathway, culminating in the actual measurement of IFN-α production, enabled us to determine at what point during the virus infection of a macrophage the inhibition of IFN-α production occurred. Our results revealed that the virus infection impaired neither the activation of IRF3 and STAT1 nor the transcription of the IFNB1, IRF7, or IFNA1 gene in response to simulation of AMϕ with poly(I·C) (Fig. 4). The activation of STAT1 and the transcription of the IRF7 gene are both components of late-phase type I IFN production, in which small amounts of type I IFN, produced early in the response, engage the type I IFN receptor (a heterodimer of IFNAR1 and IFNAR2) in an autocrine and a paracrine fashion. Binding of IFN-β/α to its receptor leads to the activation of ISGF3 (a heterotrimer of STAT1, STAT2, and IRF9), which promotes the transcription of the IRF7 gene, leading to further and robust induction of IFN-α genes and enhanced production of IFN-α (52, 53). Hence, it was intriguing to observe that even though the signaling pathways involved in both the early and late phases of type I IFN-α gene induction were operational, the production of IFN-α was significantly suppressed.
The results described above directed us to search for an alternative mechanism responsible for the inhibited production of IFN-α in virus-infected AMϕ. Such a mechanism should influence the late-phase type I IFN signaling pathway, should be operational >6 h after the initiation of virus infection, and should not involve the inhibition of transcriptional events necessary for TLR agonist-induced cytokine production. Like that of other RNA viruses, such as coronaviruses, the replication of PRRSV is functionally associated with the ER and places an inordinate stress on the protein-folding machinery of the organelle, thus triggering ER stress (24, 54). To survive ER stress, a virus-infected cell mounts the UPR, which includes activation of the stress sensor PERK, which, by phosphorylating the key translation regulator eIF2α, results in translational attenuation (24). The results of our experiments revealed that the virus infection-induced inhibitory effect on IFN-α production in response to poly(I·C) temporally coincided with the kinetics of the appearance of phosphorylated eIF2α (Fig. 7), which became patently manifested at >6 hpi and was accompanied by the appearance of SGs (Fig. 6E). Since these two events are hallmarks of stalled translation (55), it seemed likely that translation attenuation could be involved in the observed inhibitory effect of IFN-α synthesis. To further investigate this notion, we examined the effect of virus infection on the production of TNF-α in response to LPS, which yields a half-maximal response within 3 h. We reasoned that since the virus infection-induced inhibitory effect on cytokine production seemed to become operational >6 hpi, the response should not be inhibited during the first few hours after virus infection. Our results revealed that the inhibitory effect on LPS-induced TNF-α production occurs only when the virus-infected cells are exposed to the TLR4 agonist at 6 hpi. In this scenario, the bulk of the TNF-α synthesis, which occurs between 2 and 4 h after stimulation and corresponds to 8 to 10 hpi, coincides with the time at which there is an ample supply of phosphorylated eIF2α in the virus-infected cells (Fig. 10A and B). On the other hand, if the stimulation of the infected AMϕ with LPS was initiated at 2 hpi, at which time the presence of phosphorylated eIF2α is negligible, there was no inhibition and a synergistic response was even observed (Fig. 9A). Thus, we speculated that such disparate effects could be partially attributed to completion of TNF-α synthesis prior to the onset of p-eIF2α-regulated translational attenuation. Evidence of direct involvement of p-eIF2α in virus infection-induced inhibition of the TNF-α response to LPS was provided by the reduction of eIF2α phosphorylation by PERKi-treated PRRSV-infected AMϕ (Fig. 11B) and by a significant reduction in the inhibition of TNF-α and IFN-α production in similarly treated virus-infected cells (Fig. 11C and D). Together, our results indicate that the phosphorylation of eIF2α, at a late stage of the infection of AMϕ with PRRSV, involves the activation of the ER stress sensor PERK. We propose that the resulting translational attenuation is at least partially responsible for the impaired ability of PRRSV-infected AMϕ to produce IFN-α and TNF-α in response to TLR agonists. Our results are in agreement with the observations by Zhang et al. (56), in which the poly(I·C)-induced synthesis of IFN-α by porcine monocyte-derived dendritic cells infected with PRRSV was found to be curtailed posttranscriptionally. Although the mechanism was not examined, it was proposed that it could occur via translation inhibition (57). Raaben et al. described evidence suggesting that the UPR, triggered by the replication of murine hepatitis virus (MHV) in fibroblasts, caused host translational shutoff, as indicated by the phosphorylation of eIF2α and the formation of SGs (58). Versteeg et al. reported that the inhibition of chemokine synthesis in fibroblasts infected with MHV was due to translational attenuation triggered by ER stress (59). To our knowledge, this is the first report describing the involvement of the ER stress sensor PERK in the inhibition of cytokine production in response to a TLR agonist in virus-infected macrophages.
Our results also revealed that translation of viral mRNA occurs late in infection despite the marked presence of phosphorylated eIF2α (Fig. 8). Arteriviruses, like other nidoviruses, synthesize a 3′-coterminal nested set of segmented mRNAs that contain a common 5′-end “leader sequence” (60) from which the structural proteins are translated, presumably by cap-dependent translation (61). Assuming that arterivirus mRNAs, like coronavirus mRNA, share important structural features with the host mRNA [such as the 5′ cap structure and 3′ poly(A) tail], they should be equally sensitive to inhibition by factors that control translational steps, including the translational attenuation resulting from the presence of p-eIF2α. Efficient translation of coronavirus mRNA in the presence of phosphorylated eIF2α has been shown to occur in cells infected with MHV (62) and severe acute respiratory syndrome (SARS) coronavirus (63). Possible mechanisms that have been suggested to explain the translation of coronavirus mRNA, despite the presence of phosphorylated eIF2α, include the extreme abundance of viral mRNA, which could counterbalance the effects of translational attenuation, and the use of a slightly different repertoire of translation factors (64). It will require further studies to ascertain the mechanism by which PRRSV translates its mRNA in the presence of p-eIF2α.
Our results showed that AMϕ infected with PRRSV exhibit an increased level of phosphorylated IRE1α within 2 h after infection (Fig. 6A), and their stimulation with LPS at such a time would result in a synergistic TNF-α response (Fig. 9A). The phosphorylated cytosolic kinase effector domain of IRE1α interacts with the C terminus of TRAF2 (48), ultimately resulting in NF-κB activation and upregulation of its downstream inflammatory pathways (65–67). Direct evidence of the role of IRE1α in the virus infection-induced activation of NF-κB was provided by the observed reduction in NF-κB activation by KIRA6 (Fig. 12A), an inhibitor of the kinase domain of IRE1α (41). Similarly, direct evidence of the participation of IRE1α in virus infection-induced synergistic TNF-α response to LPS was provided by the abolition of the synergistic TNF-α response (Fig. 12B) mediated by the same IRE1α inhibitor. This observation is consistent with the report that in macrophages undergoing chemically induced ER stress, the activation of IRE1α amplifies TNF-α production in response to LPS (47).
In their role as sentinels against pulmonary infections (68), the inflammatory response of AMϕ to cellular debris or to inhaled innocuous particles is relatively limited compared to a sufficiently strong proinflammatory response to respiratory pathogens that nevertheless must not compromise the vital gas exchange function of the lung (69). However, alterations in the regulatory mechanisms that maintain a delicate balance between pro- and anti-inflammatory functional phenotypes can trigger macrophage-directed immune overreactions resulting in lung immunopathology (70). For instance, overly robust proinflammatory cytokine responses are thought to be involved in exacerbated lung injury in bacterial coinfections with viruses, including human influenza (71). As the most intensely studied proinflammatory cytokine, TNF-α is now considered to be a central factor in acute viral diseases, including influenza, and is prominently mentioned in cytokine storm literature (72). In its respiratory mode, PRRSV targets AMϕ for replication and produces an interstitial pneumonia that eventually resolves (73). Frequently, however, a PRRSV infection becomes complicated with opportunistic bacteria (74, 75) that commonly reside in the upper respiratory tract of pigs without producing overt disease (76). A dual PRRSV-bacterial coinfection manifests as a severe clinical syndrome, which is characterized by an enhanced proinflammatory cytokine response, severe lung tissue damage, high morbidity, hypoxia, and often death (6, 7, 13). Notably, BAL fluids collected from the lungs of animals undergoing a dual PRRSV-bacterial infection have been shown to contain relatively large quantities of TNF-α, which was considered the most likely factor responsible for the severity of the pneumonia (14). An indication that dysregulated cytokine production in the lung could be involved in the severe respiratory syndrome observed during PRRSV-bacterial coinfections is suggested by the observation that PRRSV sensitizes the lung to respond with enhanced pulmonary proinflammatory cytokine production and severe respiratory disease upon exposure to LPS (10). The enhanced TNF-α response of AMϕ to LPS during the early stages of PRRSV infection suggests that the activation of the stress sensor IRE1α by PRRSV infection results in a functional reprogramming of AMϕ activity toward a proinflammatory phenotype. Based on the observations presented here and those of Van Gucht et al. (10), it seems reasonable to propose that the activation of NF-κB via the IRE1α branch of the UPR in PRRSV-infected AMϕ could dysregulate the normally moderate proinflammatory cytokine (TNF-α) response to bacterial pathogen-associated molecular patterns (PAMPs) derived from opportunistic bacteria commonly present in the respiratory tracts of conventionally raised swine (76). In this scenario, we speculate that in the absence of PRRSV infection, AMϕ normally produce a regulated proinflammatory cytokine response to bacterial PAMPs of sufficient intensity to contain the microbes while maintaining homeostasis between the host and the resident and potentially pathogenic microbes. In the event of a respiratory infection with PRRSV, AMϕs could overreact to their exposure to bacterial PAMPs during the early stages of PRRSV infection, resulting in dysregulated production of TNF-α, promoting the development of severe inflammation and lung dysfunction. Future studies will be focused on examining the role of IRE1α in the pathogenic synergy between PRRSV and secondary bacterial pathogens. Our observations provide support to the emerging concept that the UPR directly activates proinflammatory TFs (49) and is involved in microbe sensing by cells of the immune system (77) and are consistent with the notion that the activation of IRE1α can act in synergy with TLR activation of proinflammatory cytokine production in macrophages (47).
MATERIALS AND METHODS
Cells.
The porcine AMϕ cell line ZMAC (created in our laboratory at the University of Illinois at Urbana-Champaign [UIUC]), was derived from the lungs of porcine fetuses (78) and consists of phagocytic cells that express several surface markers characteristic of AMϕ (79), including CD14, CD45, CD163, and CD172 (78). The ZMAC cell line has been shown to efficiently support the growth of PRRSV (31, 80). Primary PAMϕ were obtained from the lavage fluid from lungs harvested from euthanized specific-pathogen-free pigs. The lung lavage fluid was obtained by infusing the trachea under aseptic conditions with 50 ml phosphate-buffered saline (PBS) per lung. The collected lavage fluid was centrifuged at 500 × g and 4°C for 10 min. The cell pellets were washed twice with Hanks' buffered sterile saline (HBSS) and suspended in RPMI medium supplemented with 10% fetal bovine serum (FBS) (Gibco, Invitrogen, Grand Island, NY, USA) and processed for freezing using dimethyl sulfoxide (DMSO). Aliquots of the resulting suspensions were stored in liquid nitrogen until further use. A single batch of primary PAMϕ was used for this study, which was chosen based on the cell population exhibiting >50% permissiveness to PRRSV. Both PAMϕ and ZMAC cells were cultured in RPMI 1640 medium containing l-glutamine (Mediatec, Herndon, VA, USA) and supplemented with 10% FBS (Gibco), 1 mM sodium pyruvate, and 1× nonessential amino acids (Mediatec) and kept at 37°C in a 5% CO2 atmosphere. Maintenance of ZMAC cells also required the inclusion of 10 ng/ml recombinant mouse macrophage colony-stimulating factor (Shenandoah Biotechnology, Inc., Warwick, PA). MARC-145 cells (kindly provided by William Laegreid, University of Wyoming) were grown as previously described (81).
Viruses.
Genotype 2 PRRSV strains NADC20 (82) and FL12 (83) were propagated in MARC-145 cells (81). The Purdue strain of TGEV, kindly provided by Linda Saif (Ohio State University), was grown in swine testicle (ST) cells. Cell-free preparations of PRRSV were obtained from the medium overlying infected cell monolayers showing ≥80% CPE by centrifugation at 4°C and 350 × g for 10 min. Approximately 25 ml of the clarified virus suspension was layered on top of a 3-ml cushion of 15% iodixanol (OptiPrep; Sigma-Aldrich, St. Louis, MO, USA) and subjected to ultracentrifugation at 64,100 × g at 4°C for 3 h. The resulting virus-containing pellets were suspended in 1 ml of TNE buffer (10 mM Tris, pH 7.6, 100 mM NaCl, 1 mM EDTA). The purified virus stocks were titrated in monolayers of ZMAC cells (50% tissue culture infective dose [TCID50]). When required, purified NADC20 virus was inactivated by exposure to short-wave (254-nm) UV light for 3 min. Loss of viability was verified by the inability of the UV light-exposed viruses to produce CPE in monolayers of MARC-145 cells. GFP-expressing PRRSV (P129-GFP virus) was kindly provided by D. Yoo (University of Illinois) (32) and propagated in ZMAC cells. To obtain the single-step virus growth curve, ZMAC cells were infected with PRRSV strain NADC20 at an MOI of 5 to ensure a high degree of synchronous viral infection. After 1 h of infection at 37°C, the cell cultures were washed twice, and thereafter, samples of cell-free supernatants were collected at specified time intervals. The time postinfection was set at zero after the 1-h absorption. The amount of infectious virus present in samples containing PRRSV was determined in ZMAC cells according to the Reed and Muench method and expressed as TCID50 per milliliter.
Infection, treatment of porcine AMϕ, and viability monitoring.
For viral challenge, 2 × 105 ZMAC cells were cultured in sterile 12- by 75-mm round-bottom polypropylene tubes (Corning, New York) and were either mock infected or infected with PRRSV at an MOI of 5 to obtain a synchronized infection. At the time of harvest, the cell viability was determined visually by microscopy using vital-dye exclusion. The occurrence of cellular DNA fragmentation, indicative of late stages of apoptosis, was detected by TUNEL using the DeadEnd colorimetric TUNEL system (Promega, Madison, WI). For cytokine responses, uninfected or PRRSV-infected cells cultured as described above were treated at the indicated times postinfection with either 25 μg/ml of poly(I·C) (Amersham Pharmacia Biotech Inc., Piscataway, NY) or 100 ng/ml of LPS (purified from Escherichia coli 011:B4; Sigma, St. Louis, MO) and further cultured for the indicated length of time. When indicated, the cell cultures were also treated with either 1 μM the IRE1α inhibitor KIRA6 (41) (EMD Millipore, Darmstadt, Germany) or 1 to 2 μM the PERK inhibitor GSK2606414 (46) (EMD Millipore, Darmstadt, Germany). As a positive control for eIF2α activation, the AMϕ cultures were treated with 2 mM DTT (Sigma-Aldrich) for 1 h. For measurement of intracellular protein status by Western blotting, 1 × 106 to 2 × 106 ZMAC cells were cultured in a 6-well tissue culture plate in a 2-ml volume. To deliver poly(I·C) into the cytoplasm, in some experiments, ZMAC cells were transfected with poly(I·C) using jetPEI-Macrophage transfection reagent (Polyplus Transfection, Illkirch, France) following the manufacturer's recommendations. Briefly, 400 μg of poly(I·C) in a 50-μl volume was mixed with 50 μl of a 150 mM NaCl solution containing 1 μl of jetPEI-Macrophage. Following a 30-min incubation at room temperature (RT), this mixture was added to a well of a 24-well plate containing 2.5 × 105 ZMAC cells in a 0.5-ml volume.
Quantitation of IFN-α and TNF-α.
Individual IFN-α-secreting cells (SC) and the presence of IFN-α in cell-free culture supernatants were detected by enzyme-linked immunosorbent spot (ELISPOT) assay and enzyme-linked immunosorbent assay (ELISA), respectively, as previously described (84). For TNF-α detection, the same ELISA procedure was followed except that the wells were coated with 50 μl of 4-μg/ml anti-pig TNF-α monoclonal antibody (MAb) (clone103304; R&D Systems, Minneapolis, MN, USA). The captured cytokine was detected with 50 μl of 2.5-μg/ml biotin-labeled, anti-pig TNF-α MAb (clone 103302; R&D Systems). The optical densities at 450 nm (OD450s) of triplicate wells were averaged, and the amounts of TNF-α were determined based on a curve generated from the values obtained using serial dilutions of a standard (R&D Systems). The lowest levels of detection for the IFN-α and TNF-α assays were 80 pg/ml and 120 pg/ml, respectively.
Western blot analysis.
Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with a protease inhibitor cocktail (Amresco, Solon, OH, USA), and the protein concentrations of the resulting lysates were determined by using a bicinchoninic acid (BCA) protein assay kit (Pierce, Rockford, IL, USA). Equivalent protein amounts of each extract (25 to 60 μg per well) were subjected to separation in an SDS-10% PAGE gel and subsequently transferred onto a 0.2-μm polyvinylidene difluoride (PVDF) membrane (Bio-Rad, Hercules, CA, USA) for Western blot analysis. The membranes were incubated in blocking buffer (2% fish gelatin in TBST solution [50 mM Tris, pH 7.5, 500 mM NaCl, and 0.5% Tween 20]) at RT for 1 h. Afterward, the membranes were incubated at 4°C overnight with one of the following unconjugated primary Abs (a 1:1,000 dilution of the manufacturer's original concentration in TBST with 5% BSA): anti-IRF3 (clone D83B9; Cell Signaling, Danvers, MA, USA), anti-phospho-IRF3 (Ser396) (clone 4D4G; Cell Signaling), anti-NF-κB-p65 (3034; Cell Signaling), anti-NF-κB-p65 (Sc109; Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-phospho NF-κB-p65 (Ser536) (clone 93H1; Cell Signaling), anti-STAT1 (sc346), anti-phospho STAT1 (Tyr701) (SC-7988; Santa Cruz Biotechnology), anti-eIF2 (9722; Cell Signaling), anti-phospho-eIF2 (9721; Cell Signaling), anti-CHOP (clone L63F7; Cell Signaling), or anti-β-actin (4967; Cell Signaling). The membranes were then washed four times in TBST solution and incubated with horseradish peroxidase (HRP)-conjugated anti-rabbit immunoglobulin (IgG) secondary Ab (sc2004; Santa Cruz Biotechnology, Santa Cruz, CA, USA; 1:8,000 in blocking buffer) at RT for 1 h. After being washed again 4 times with TBST, the membranes were incubated with a chemiluminescence reagent (GE Healthcare, Little Chalfont, Buckinghamshire, United Kingdom) to enable detection of bound secondary Ab. Screening for the presence of a specific phosphorylated protein was always performed prior to detection of the corresponding, nonphosphorylated form on membranes that had been incubated in stripping buffer (21059; Thermo Fisher Scientific, Waltham, MA, USA) at RT for 15 min to remove any bound Ab.
RNA preparation and real-time reverse transcription-PCR.
Samples of 105 uninfected or PRRSV-infected ZMAC cells were cultured in the presence or absence of poly(I·C) for the indicated length of time. Afterward, each sample was lysed in buffer RLT, and the total RNAs were purified, DNase treated, converted into cDNA, and subjected to real-time PCR, as previously described (80). Primers and probes for the amplification/detection of porcine IFNA1 and IFNB1 gene transcripts have been described previously (80), whereas those associated with the amplification/detection of IRF7 and ribosomal protein L32 (RPL32) gene transcripts were designed and provided by H. Dawson (U.S. Department of Agriculture [USDA], Beltsville, MD) and are described in the DGIL Porcine Translational Research Database (http://www.ars.usda.gov/Services/docs.htm?docid=6065). Changes in the extent of expression of the IFNA1, IFNB1, and IRF7 genes were determined by using the comparative threshold cycle (CT) method and the formula 2−ΔΔCt (85), where the RPL32 gene was used as the reference housekeeping gene.
Detection of stress granules and dsRNA in virus-infected AMϕ.
A total of 2 × 105 ZMAC cells were grown in individual wells of a Nunc LabTekII 8-well chamber slide and were either mock infected for 8 h, treated with DTT for 1 h, or infected with PRRSV strain FL12 (MOI = 5) for 4 or 8 h. Afterward, the cell monolayers were fixed in PBS containing 4% paraformaldehyde for 20 min at RT, washed with PBS, incubated with blocking buffer (PBS containing 0.3% Triton X-100 and 3% normal goat serum) for 1 h at RT, and incubated in MAb dilution buffer (PBS containing 0.3% Triton X-100 and 1% BSA) containing rabbit anti-TIAR MAb (1:200 dilution of the manufacturer's original concentration; cloneD32D3; Cell Signaling) overnight at 4°C. Afterward, the cells were washed 3 times in PBS and incubated with MAb dilution buffer containing 5 μg/ml goat anti-rabbit IgG conjugated to DyLight594 (35560; Thermo Scientific) at RT for 1 h. After being washed 3 times with PBS, the monolayers were incubated at RT in blocking buffer for 1 h and in MAb dilution buffer containing mouse anti-dsRNA monoclonal antibody (1:200 dilution of the manufacturer's original concentration; cloneJ2; Scicons, Szirák, Hungary) for 2 h, washed 3 times with PBS, and incubated with Ab dilution buffer containing 2.5 μg/ml goat anti-mouse IgG conjugated to fluorescein isothiocyanate (FITC) (62-6511; Zymed, Life Technologies) at RT for 1 h. Afterward, the chamber was removed and the glass slide was wet mounted in antifading medium (Invitrogen, Life Technologies). Fluorescent signals were observed with an immunofluorescence microscope (DMII 4000B; Leica, Wetzler, Germany).
Statistical analysis.
An unpaired Student's t test was used to determine if significant differences existed in the cytokine gene expression or synthesis exhibited by AMϕ between treatment groups. To determine the presence of statistically significant synergism, the interaction effects between PRRSV, LPS, and the IRE1α inhibitor were tested using two-way ANOVA (51). A P value of <0.05 was considered statistically significant.
Ethics statement.
All experimental procedures requiring the use of animals were performed under protocols 06082 and 13064 approved by the Institutional Animal Care and Use Committee (IACUC) of UIUC. The animal care and use protocols were those followed by the Agricultural Animal Care and Use Program (AACUP), which adheres to the Federation of Animal Science Societies (FASS) Guide to the Care and Use of Agricultural Animals in Research and Teaching. The AACUP at UIUC is accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care International (AAALAC).
ACKNOWLEDGMENTS
This work was supported by USDA Hatch grants ILLU-888377 and ILLU-888331 and by USDA NIFA award no. 2017-67015-26629.
We are very grateful to Levon Abrahamyan, Université de Montréal, and Daniel L. Rock and Joanna L. Shisler, University of Illinois, for their advice and helpful suggestions. We also thank Dongwan Yoo, University of Illinois, for providing the GFP-P129 virus; Linda Saif, The Ohio State University, for providing TGEV; and Christine Zuckermann for editorial assistance.
REFERENCES
- 1.King AM, Adams MJ, Lefkowitz EJ (ed). 2011. Virus taxonomy. Classification and nomenclature of viruses. Ninth Report of the International Committee on Taxonomy of Viruses, vol 9 Elsevier Academic Press, San Diego, CA. [Google Scholar]
- 2.Holtkamp DJ, Kliebenstein JB, Neumann EJ, Zimmerman JJ, Rotto HF, Yoder TK, Wang C, Yeske PE, Mowrer CL, Haley CA. 2013. Assessment of the economic impact of porcine reproductive and respiratory syndrome virus on United States pork producers. J Swine Health Prod 21:72–84. [Google Scholar]
- 3.Rossow KD. 1998. Porcine reproductive and respiratory syndrome. Vet Pathol 35:1–20. doi: 10.1177/030098589803500101. [DOI] [PubMed] [Google Scholar]
- 4.Rossow KD, Collins JE, Goyal SM, Nelson EA, Christopher-Hennings J, Benfield DA. 1995. Pathogenesis of porcine reproductive and respiratory syndrome virus infection in gnotobiotic pigs. Vet Pathol 32:361–373. doi: 10.1177/030098589503200404. [DOI] [PubMed] [Google Scholar]
- 5.van Reeth K, Nauwynck H. 2000. Proinflammatory cytokines and viral respiratory disease in pigs. Vet Res 31:187–213. doi: 10.1051/vetres:2000113. [DOI] [PubMed] [Google Scholar]
- 6.Thanawongnuwech R, Brown G, Halbur P, Roth J, Royer R, Thacker B. 2000. Pathogenesis of porcine reproductive and respiratory syndrome virus-induced increase in susceptibility to Streptococcus suis infection. Vet Pathol 37:143–152. doi: 10.1354/vp.37-2-143. [DOI] [PubMed] [Google Scholar]
- 7.Xu M, Wang S, Li L, Lei L, Liu Y, Shi W, Wu J, Li L, Rong F, Xu M. 2010. Secondary infection with Streptococcus suis serotype 7 increases the virulence of highly pathogenic porcine reproductive and respiratory syndrome virus in pigs. Virol J 7:184. doi: 10.1186/1743-422X-7-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Albina E, Carrat C, Charley B. 1998. Interferon-alpha response to swine arterivirus (PoAV), the porcine reproductive and respiratory syndrome virus. J Interferon Cytokine Res 18:485–490. doi: 10.1089/jir.1998.18.485. [DOI] [PubMed] [Google Scholar]
- 9.Van Gucht S, Labarque G, Van Reeth K. 2004. The combination of PRRS virus and bacterial endotoxin as a model for multifactorial respiratory disease in pigs. Vet Immunol Immunopathol 102:165–178. doi: 10.1016/j.vetimm.2004.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Van Gucht S, Van Reeth K, Pensaert M. 2003. Interaction between porcine reproductive-respiratory syndrome virus and bacterial endotoxin in the lungs of pigs: potentiation of cytokine production and respiratory disease. J Clin Microbiol 41:960–966. doi: 10.1128/JCM.41.3.960-966.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van Reeth K, Labarque G, Nauwynck H, Pensaert M. 1999. Differential production of proinflammatory cytokines in the pig lung during different respiratory virus infections: correlations with pathogenicity. Res Vet Sci 67:47–52. doi: 10.1053/rvsc.1998.0277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van Reeth K, Van Gucht S, Pensaert M. 2002. Correlations between lung proinflammatory cytokine levels, virus replication, and disease after swine influenza virus challenge of vaccination-immune pigs. Viral Immunol 15:583–594. doi: 10.1089/088282402320914520. [DOI] [PubMed] [Google Scholar]
- 13.Guo B, Lager KM, Henningson JN, Miller LC, Schlink SN, Kappes MA, Kehrli ME, Brockmeier SL, Nicholson TL, Yang H-C. 2013. Experimental infection of United States swine with a Chinese highly pathogenic strain of porcine reproductive and respiratory syndrome virus. Virology 435:372–384. doi: 10.1016/j.virol.2012.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Han D, Hu Y, Li L, Tian H, Chen Z, Wang L, Ma H, Yang H, Teng K. 2014. Highly pathogenic porcine reproductive and respiratory syndrome virus infection results in acute lung injury of the infected pigs. Vet Microbiol 169:135–146. doi: 10.1016/j.vetmic.2013.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brockmeier SL, Halbur PG, Thacker EL. 2002. Porcine respiratory disease complex, p 231–258, polymicrobial diseases. American Society for Microbiology, Washington, DC. [Google Scholar]
- 16.Loving CL, Osorio FA, Murtaugh MP, Zuckermann FA. 2015. Innate and adaptive immunity against porcine reproductive and respiratory syndrome virus. Vet Immunol Immunopathol 167:1–14. doi: 10.1016/j.vetimm.2015.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beura LK, Sarkar SN, Kwon B, Subramaniam S, Jones C, Pattnaik AK, Osorio FA. 2010. Porcine reproductive and respiratory syndrome virus nonstructural protein 1β modulates host innate immune response by antagonizing IRF3 activation. J Virol 84:1574–1584. doi: 10.1128/JVI.01326-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang C, Zhang Q, Guo X-K, Yu Z-B, Xu A-T, Tang J, Feng W-H. 2014. Porcine reproductive and respiratory syndrome virus nonstructural protein 4 antagonizes beta interferon expression by targeting the NF-κB essential modulator. J Virol 88:10934–10945. doi: 10.1128/JVI.01396-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun Y, Han M, Kim C, Calvert JG, Yoo D. 2012. Interplay between interferon-mediated innate immunity and porcine reproductive and respiratory syndrome virus. Viruses 4:424–446. doi: 10.3390/v4040424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang R, Zhang Y-J. 2014. Antagonizing interferon-mediated immune response by porcine reproductive and respiratory syndrome virus. BioMed Res Int 2014:315470. doi: 10.1155/2014/315470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miller LC, Lager KM, Kehrli ME. 2009. Role of Toll-like receptors in activation of porcine alveolar macrophages by porcine reproductive and respiratory syndrome virus. Clin Vaccine Immunol 16:360–365. doi: 10.1128/CVI.00269-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gómez-Laguna J, Salguero FJ, Pallarés FJ, Carrasco L. 2013. Immunopathogenesis of porcine reproductive and respiratory syndrome in the respiratory tract of pigs. Vet J 195:148–155. doi: 10.1016/j.tvjl.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.He B. 2006. Viruses, endoplasmic reticulum stress, and interferon responses. Cell Death Differ 13:393–403. doi: 10.1038/sj.cdd.4401833. [DOI] [PubMed] [Google Scholar]
- 24.Jheng JR, Ho JY, Horng JT. 2014. ER stress, autophagy, and RNA viruses. Front Microbiol 5:388. doi: 10.3389/fmicb.2014.00388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walter P, Ron D. 2011. The unfolded protein response: from stress pathway to homeostatic regulation. Science 334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 26.Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, Shokat KM, Lavail MM, Walter P. 2007. IRE1 signaling affects cell fate during the unfolded protein response. Science 318:944–949. doi: 10.1126/science.1146361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin JH, Li H, Zhang Y, Ron D, Walter P. 2009. Divergent effects of PERK and IRE1 signaling on cell viability. PLoS One 4:e4170. doi: 10.1371/journal.pone.0004170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huo Y, Fan L, Yin S, Dong Y, Guo X, Yang H, Hu H. 2013. Involvement of unfolded protein response, p53 and Akt in modulation of porcine reproductive and respiratory syndrome virus-mediated JNK activation. Virology 444:233–240. doi: 10.1016/j.virol.2013.06.015. [DOI] [PubMed] [Google Scholar]
- 29.Duan X, Nauwynck HJ, Pensaert MB. 1997. Effects of origin and state of differentiation and activation of monocytes/macrophages on their susceptibility to porcine reproductive and respiratory syndrome virus (PRRSV). Arch Virol 142:2483–2497. doi: 10.1007/s007050050256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gaudreault N, Rowland R, Wyatt C. 2009. Factors affecting the permissiveness of porcine alveolar macrophages for porcine reproductive and respiratory syndrome virus. Arch Virol 154:133–136. doi: 10.1007/s00705-008-0271-y. [DOI] [PubMed] [Google Scholar]
- 31.Du Y, Yoo D, Paradis M, Scherba G. 2011. Antiviral activity of tilmicosin for type 1 and type 2 porcine reproductive and respiratory syndrome virus in cultured porcine alveolar macrophages. J Antivir Antiretrovir 3:28–33. doi: 10.4172/jaa.1000031. [DOI] [Google Scholar]
- 32.Pei Y, Hodgins DC, Wu J, Welch S-KW, Calvert JG, Li G, Du Y, Song C, Yoo D. 2009. Porcine reproductive and respiratory syndrome virus as a vector: Immunogenicity of green fluorescent protein and porcine circovirus type 2 capsid expressed from dedicated subgenomic RNAs. Virology 389:91–99. doi: 10.1016/j.virol.2009.03.036. [DOI] [PubMed] [Google Scholar]
- 33.Costers S, Lefebvre DJ, Delputte PL, Nauwynck HJ. 2008. Porcine reproductive and respiratory syndrome virus modulates apoptosis during replication in alveolar macrophages. Arch Virol 153:1453–1465. doi: 10.1007/s00705-008-0135-5. [DOI] [PubMed] [Google Scholar]
- 34.Qiao S, Feng L, Bao D, Guo J, Wan B, Xiao Z, Yang S, Zhang G. 2011. Porcine reproductive and respiratory syndrome virus and bacterial endotoxin act in synergy to amplify the inflammatory response of infected macrophages. Vet Microbiol 149:213–220. doi: 10.1016/j.vetmic.2010.11.006. [DOI] [PubMed] [Google Scholar]
- 35.Marie I, Durbin JE, Levy DE. 1998. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. EMBO J 17:6660–6669. doi: 10.1093/emboj/17.22.6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Honda K, Takaoka A, Taniguchi T. 2006. Type I interferon gene induction by the interferon regulatory factor family of transcription factors. Immunity 25:349–360. doi: 10.1016/j.immuni.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 37.Matsumoto M, Funami K, Tatematsu M, Azuma M, Seya T. 2014. Assessment of the Toll-like receptor 3 pathway in endosomal signaling. Methods Enzymol 535:149–165. doi: 10.1016/B978-0-12-397925-4.00010-9. [DOI] [PubMed] [Google Scholar]
- 38.Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ. 2006. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 39.Watanabe A, Tatematsu M, Saeki K, Shibata S, Shime H, Yoshimura A, Obuse C, Seya T, Matsumoto M. 2011. Raftlin is involved in the nucleocapture complex to induce poly(I:C)-mediated TLR3 activation. J Biol Chem 286:10702–10711. doi: 10.1074/jbc.M110.185793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zou J, Kawai T, Tsuchida T, Kozaki T, Tanaka H, Shin K-S, Kumar H, Akira S. 2013. Poly IC triggers a cathepsin D-and IPS-1-dependent pathway to enhance cytokine production and mediate dendritic cell necroptosis. Immunity 38:717–728. doi: 10.1016/j.immuni.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 41.Ghosh R, Wang L, Wang ES, Perera BG, Igbaria A, Morita S, Prado K, Thamsen M, Caswell D, Macias H, Weiberth KF, Gliedt MJ, Alavi MV, Hari SB, Mitra AK, Bhhatarai B, Schurer SC, Snapp EL, Gould DB, German MS, Backes BJ, Maly DJ, Oakes SA, Papa FR. 2014. Allosteric inhibition of the IRE1alpha RNase preserves cell viability and function during endoplasmic reticulum stress. Cell 158:534–548. doi: 10.1016/j.cell.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hetz C. 2012. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol 13:89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- 43.Oyadomari S, Mori M. 2004. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ 11:381–389. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- 44.Kedersha NL, Gupta M, Li W, Miller I, Anderson P. 1999. RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2α to the assembly of mammalian stress granules. J Cell Biol 147:1431–1442. doi: 10.1083/jcb.147.7.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Anderson P, Kedersha N. 2008. Stress granules: the Tao of RNA triage. Trends Biochem Sci 33:141–150. doi: 10.1016/j.tibs.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 46.Axten JM, Medina JR, Feng Y, Shu A, Romeril SP, Grant SW, Li WH, Heerding DA, Minthorn E, Mencken T, Atkins C, Liu Q, Rabindran S, Kumar R, Hong X, Goetz A, Stanley T, Taylor JD, Sigethy SD, Tomberlin GH, Hassell AM, Kahler KM, Shewchuk LM, Gampe RT. 2012. Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1H-indol-5-yl)-7H-p yrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J Med Chem 55:7193–7207. doi: 10.1021/jm300713s. [DOI] [PubMed] [Google Scholar]
- 47.Martinon F, Chen X, Lee A-H, Glimcher LH. 2010. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat Immunol 11:411–418. doi: 10.1038/ni.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D. 2000. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 49.Zhang K, Kaufman RJ. 2008. From endoplasmic-reticulum stress to the inflammatory response. Nature 454:455–462. doi: 10.1038/nature07203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Akira S, Takeda K. 2004. Toll-like receptor signalling. Nat Rev Immunol 4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 51.Slinker BK. 1998. The statistics of synergism. J Mol Cell Cardiol 30:723–731. doi: 10.1006/jmcc.1998.0655. [DOI] [PubMed] [Google Scholar]
- 52.Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, Taniguchi T. 2005. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 434:772–777. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- 53.Levy DE, Marie I, Smith E, Prakash A. 2002. Enhancement and diversification of IFN induction by IRF-7-mediated positive feedback. J Interferon Cytokine Res 22:87–93. doi: 10.1089/107999002753452692. [DOI] [PubMed] [Google Scholar]
- 54.Fung TS, Liu DX. 2014. Coronavirus infection, ER stress, apoptosis and innate immunity. Front Microbiol 5:296. doi: 10.3389/fmicb.2014.00296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kimball SR, Horetsky RL, Ron D, Jefferson LS, Harding HP. 2003. Mammalian stress granules represent sites of accumulation of stalled translation initiation complexes. Am J Physiol Cell Physiol 284:C273–C284. doi: 10.1152/ajpcell.00314.2002. [DOI] [PubMed] [Google Scholar]
- 56.Zhang H, Guo X, Nelson E, Christopher-Hennings J, Wang X. 2012. Porcine reproductive and respiratory syndrome virus activates the transcription of interferon alpha/beta (IFN-alpha/beta) in monocyte-derived dendritic cells (Mo-DC). Vet Microbiol 159:494–498. doi: 10.1016/j.vetmic.2012.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang X, Christopher-Hennings J. 2012. Post-transcriptional control of type i interferon induction by porcine reproductive and respiratory syndrome virus in its natural host cells. Viruses 4:725–733. doi: 10.3390/v4050725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Raaben M, Groot Koerkamp MJA, Rottier PJM, De Haan CAM. 2007. Mouse hepatitis coronavirus replication induces host translational shutoff and mRNA decay, with concomitant formation of stress granules and processing bodies. Cell Microbiol 9:2218–2229. doi: 10.1111/j.1462-5822.2007.00951.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Versteeg GA, van de Nes PS, Bredenbeek PJ, Spaan WJ. 2007. The coronavirus spike protein induces endoplasmic reticulum stress and upregulation of intracellular chemokine mRNA concentrations. J Virol 81:10981–10990. doi: 10.1128/JVI.01033-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Snijder EJ, Kikkert M, Fang Y. 2013. Arterivirus molecular biology and pathogenesis. J Gen Virol 94:2141–2163. doi: 10.1099/vir.0.056341-0. [DOI] [PubMed] [Google Scholar]
- 61.van den Born E, Posthuma CC, Gultyaev AP, Snijder EJ. 2005. Discontinuous subgenomic RNA synthesis in arteriviruses is guided by an RNA hairpin structure located in the genomic leader region. J Virol 79:6312–6324. doi: 10.1128/JVI.79.10.6312-6324.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bechill J, Chen Z, Brewer JW, Baker SC. 2008. Coronavirus infection modulates the unfolded protein response and mediates sustained translational repression. J Virol 82:4492–4501. doi: 10.1128/JVI.00017-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Krahling V, Stein DA, Spiegel M, Weber F, Muhlberger E. 2009. Severe acute respiratory syndrome coronavirus triggers apoptosis via protein kinase R but is resistant to its antiviral activity. J Virol 83:2298–2309. doi: 10.1128/JVI.01245-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nakagawa K, Lokugamage KG, Makino S. 2016. Viral and cellular mRNA translation in coronavirus-infected cells. Adv Virus Res 96:165–192. doi: 10.1016/bs.aivir.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hu P, Han Z, Couvillon AD, Kaufman RJ, Exton JH. 2006. Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1α-mediated NF-κB activation and down-regulation of TRAF2 expression. Mol Cell Biol 26:3071–3084. doi: 10.1128/MCB.26.8.3071-3084.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kaneko M, Niinuma Y, Nomura Y. 2003. Activation signal of nuclear factor-κB in response to endoplasmic reticulum stress is transduced via IRE1 and tumor necrosis factor receptor-associated factor 2. Biol Pharmaceut Bull 26:931–935. doi: 10.1248/bpb.26.931. [DOI] [PubMed] [Google Scholar]
- 67.Tam AB, Mercado EL, Hoffmann A, Niwa M. 2012. ER stress activates NF-κB by integrating functions of basal IKK activity, IRE1 and PERK. PLoS One 7:e45078. doi: 10.1371/journal.pone.0045078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bowden DH. 1984. The alveolar macrophage. Environ Health Perspect 55:327–341. doi: 10.1289/ehp.8455327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hussell T, Bell TJ. 2014. Alveolar macrophages: plasticity in a tissue-specific context. Nat Rev Immunol 14:81–93. doi: 10.1038/nri3600. [DOI] [PubMed] [Google Scholar]
- 70.Gwyer Findlay E, Hussell T. 2012. Macrophage-mediated inflammation and disease: a focus on the lung. Mediators Inflamm 2012:140937. doi: 10.1155/2012/140937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McCullers JA. 2014. The co-pathogenesis of influenza viruses with bacteria in the lung. Nat Rev Microbiol 12:252–262. doi: 10.1038/nrmicro3231. [DOI] [PubMed] [Google Scholar]
- 72.Tisoncik JR, Korth MJ, Simmons CP, Farrar J, Martin TR, Katze MG. 2012. Into the eye of the cytokine storm. Microbiol Mol Biol Rev 76:16–32. doi: 10.1128/MMBR.05015-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Halbur PG, Paul PS, Meng X-J, Lum MA, Andrews JJ, Rathje JA. 1996. Comparative pathogenicity of nine US porcine reproductive and respiratory syndrome virus (PRRSV) isolates in a five-week-old cesarean-derived, colostrum-deprived pig model. J Vet Diagn Invest 8:11–20. doi: 10.1177/104063879600800103. [DOI] [PubMed] [Google Scholar]
- 74.Opriessnig T, Gimenez-Lirola L, Halbur P. 2011. Polymicrobial respiratory disease in pigs. Anim Health Res Rev 12:133–148. doi: 10.1017/S1466252311000120. [DOI] [PubMed] [Google Scholar]
- 75.Zeman DH. 1996. Concurrent respiratory-infections in 221 cases of Prrs virus pneumonia: 1992-1994. Swine Health Prod 4:143–145. [Google Scholar]
- 76.Kernaghan S. 2014. Characterization of the bacterial communities of the tonsil of the soft palate of swine. M.S. thesis University of Guelph, Guelph, Ontario, Canada. [Google Scholar]
- 77.Smith JA. 2014. A new paradigm: innate immune sensing of viruses via the unfolded protein response. Front Microbiol 5:222. doi: 10.3389/fmicb.2014.00222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zuckermann F. 19 June 2012. Non-simian cells for growth of porcine reproductive and respiratory syndrome (PRRS) virus. US patents 8,202,717; 9,169,465. [Google Scholar]
- 79.Ezquerra A, Revilla C, Alvarez B, Perez C, Alonso F, Dominguez J. 2009. Porcine myelomonocytic markers and cell populations. Dev Comp Immunol 33:284–298. doi: 10.1016/j.dci.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 80.Calzada-Nova G, Schnitzlein WM, Husmann RJ, Zuckermann FA. 2011. North American porcine reproductive and respiratory syndrome viruses inhibit type I interferon production by plasmacytoid dendritic cells. J Virol 85:2703–2713. doi: 10.1128/JVI.01616-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Meier WA, Galeota J, Osorio FA, Husmann RJ, Schnitzlein WM, Zuckermann FA. 2003. Gradual development of the interferon-γ response of swine to porcine reproductive and respiratory syndrome virus infection or vaccination. Virology 309:18–31. doi: 10.1016/S0042-6822(03)00009-6. [DOI] [PubMed] [Google Scholar]
- 82.Harms PA, Sorden SD, Halbur PG, Bolin SR, Lager KM, Morozov I, Paul PS. 2001. Experimental reproduction of severe disease in CD/CD pigs concurrently infected with type 2 porcine circovirus and porcine reproductive and respiratory syndrome virus. Vet Pathol 38:528–539. doi: 10.1354/vp.38-5-528. [DOI] [PubMed] [Google Scholar]
- 83.Truong HM, Lu Z, Kutish GF, Galeota J, Osorio FA, Pattnaik AK. 2004. A highly pathogenic porcine reproductive and respiratory syndrome virus generated from an infectious cDNA clone retains the in vivo virulence and transmissibility properties of the parental virus. Virology 325:308–319. doi: 10.1016/j.virol.2004.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Calzada-Nova G, Schnitzlein W, Husmann R, Zuckermann FA. 2010. Characterization of the cytokine and maturation responses of pure populations of porcine plasmacytoid dendritic cells to porcine viruses and Toll-like receptor agonists. Vet Immunol Immunopathol 135:20–33. doi: 10.1016/j.vetimm.2009.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]