

ABSTRACT
Infectious bursal disease virus (IBDV) is a bisegmented double-strand RNA (dsRNA) virus of the Birnaviridae family. While IBDV genomic dsRNA lacks a 5′ cap, the means by which the uncapped IBDV genomic RNA is translated effectively is unknown. In this study, we describe a cap-independent pathway of translation initiation of IBDV uncapped RNA that relies on VP1 and VP3. We show that neither purified IBDV genomic dsRNA nor the uncapped viral plus-sense RNA transcripts were directly translated and rescued into infectious viruses in host cells. This defect in translation of the uncapped IBDV genomic dsRNA was rescued by trans-supplementation of the viral proteins VP1 and VP3 which was dependent on both the intact polymerase activity of VP1 and the dsRNA binding activity of VP3. Deletion analysis showed that both 5′ and 3′ untranslated regions (UTRs) of IBDV dsRNA were essential for VP1/VP3-dependent translation initiation. Significantly, VP1 and VP3 could also mediate the recovery of infectious IBDV from the authentic minus-sense strand of IBDV dsRNA. Moreover, downregulation or inhibition of the cap-binding protein eIF4E did not decrease but, rather, enhanced the VP1/VP3-mediated translation of the uncapped IBDV RNA. Collectively, our findings for the first time reveal that VP1 and VP3 compensate for the deficiency of the 5′ cap and replace eIF4E to confer upon the uncapped IBDV RNA the ability to be translated and rescued into infectious viruses.
IMPORTANCE A key point of control for virus replication is viral translation initiation. The current study shows that the uncapped IBDV RNA cannot be translated into viral proteins directly by host translation machinery and is thus noninfectious. Our results constitute the first direct experimental evidence that VP1 and VP3 are required and sufficient to initiate translation of uncapped IBDV genomic RNA by acting as a substitute for cap and replacing the cap-binding protein eIF4E. Significantly, VP1/VP3 mediate the recovery of infectious IBDV not only from the plus-sense strand but also from the minus-sense strand of the IBDV dsRNA. These findings provide not only new insights into the molecular mechanisms of the life cycle of IBDV but also a new tool for an alternative strategy for the recovery of IBDV from both the plus- and the minus-sense strands of the viral genomic dsRNA.
KEYWORDS: IBDV, double-strand RNA, infectious RNA, reverse genetics
INTRODUCTION
Infectious bursal disease (IBD) is an acute and highly contagious disease of young chickens, causing major economic losses to the worldwide poultry industry. IBD manifests mainly by inflammation and subsequent atrophy of the bursa of Fabricius and immunosuppression (1, 2). The causative agent of the disease is the infectious bursal disease virus (IBDV), a nonenveloped virus belonging to the Birnaviridae family. Two distinct serotypes of IBDV, designated serotypes I and II, have been identified (3). Whereas serotype II viruses have caused disease in turkey and are avirulent for chicken, serotype I viruses are pathogenic to chicken (4).
The genome of IBDV is a bisegmented double-strand RNA (dsRNA), which consists of two segments: segment A and segment B. Segment A is 3.2 kb nucleotides in length and carries two partially overlapping open reading frames (ORFs). While the smaller ORF encodes the viral nonstructural protein VP5 which is involved in viral egress and dissemination (5–7), the larger ORF encodes a precursor polyprotein (pVP2-VP4-VP3) (8, 9). VP4 is a serine protease that cleaves pVP2-VP4-VP3 to form separate VP2, VP4, and VP3 proteins (10). VP2 is the major viral structural protein and assembles into 260 trimmers to form a T=13 icosahedral IBDV capsid (11). Segment B is 2.8 kb and contains only one ORF that encodes an RNA-dependent RNA polymerase (RdRp), VP1 (12). VP1 is present in virions both as a free polypeptide (VP1) and as a genome-linked protein (VPg) that is covalently linked to the 5′ ends of the genomic RNA segments (13, 14). VP1 was considered to be involved in the generation of the cap structure in the genome 5′ ends (15), while Dobos reported that VP1 lacks the enzymatic activities for generating a cap structure (16). VP3 interacts with both VP1 and the viral genomic dsRNA to assemble into ribonucleoprotein (RNP) complexes (17, 18) in which VP3 significantly stimulates the RdRp activity of VP1 (19). Further, VP3 is an antiapoptotic protein that prevents the activation of the cellular dsRNA-dependent protein kinase (PKR) (20) and a competitor of MDA5 to bind IBDV genomic dsRNA to prevent the induction of beta interferon (IFN-β) (21).
A key point of control for virus replication is viral translation initiation. Viral protein synthesis from a viral mRNA and/or a positive single-strand RNA (ssRNA) can be initiated by two different known mechanisms: cap-dependent translation initiation, which involves recognition of the cap structure at the 5′ end of the mRNA by the cap-binding protein eIF4E (22), and cap-independent translation initiation. One of the predominant forms of cap-independent translation is driven by RNA sequences called internal ribosome entry sites (IRESs) that functionally replace the RNA cap structure (23). The coding ORFs of IBDV dsRNAs in both segments are flanked by 5′ and 3′ untranslated regions (UTRs) (24). However, on both segments, the 5′-terminal ends of the UTR do not have a cap structure, and the 3′-terminal ends of the UTR lack a poly(A) tail (16). The 5′ UTRs of both segments do not contain IRESs (24). These observations suggest that there may be an atypical mechanism of IBDV dsRNA translation initiation. However, thus far, only very limited data on the factors involved in the translation initiation of the uncapped IBDV RNA have been reported. In particular, the questions regarding the viral and/or host factors involved in the translation initiation of the uncapped IBDV RNA remain to be defined. Here, we show that neither the naked IBDV dsRNA nor the viral plus-sense RNA transcript can be directly translated and rescued into infectious viruses in host cells. This defect in translation of the uncapped IBDV RNA and virus rescue was rescued by trans-supplementation of the viral proteins VP1 and VP3. The VP1/VP3-mediated translation initiation of uncapped IBDV dsRNA requires the intact polymerase activity of VP1, the dsRNA binding activity of VP3, and both the 5′ and 3′ UTRs of IBDV dsRNA but is independent of eIF4E. Significantly, VP1 and VP3 could also mediate the recovery of infectious IBDV from the authentic minus-sense strand of IBDV dsRNA.
RESULTS
Neither the purified IBDV genomic dsRNA nor the viral authentic plus-sense transcript can be directly translated into viral proteins in host cells.
While the plus-sense viral RNA of ssRNA viruses is commonly infectious because it can serve as mRNA and can be directly translated into viral proteins in the host cells by host translation machinery, it has not been clear whether the IBDV genomic dsRNA is infectious by itself. To address this, the naked IBDV genomic dsRNAs were purified from the concentrated viruses by proteinase K digestion and phenol-chloroform extraction, and the integrity of purified IBDV genomic dsRNA was confirmed by agarose gel electrophoresis analysis, showing two obvious segments with a size of around 3.0 kb (Fig. 1A, lane 1). DF-1 cells were transfected with the purified IBDV genomic dsRNA. The supernatant of transfected cells was harvested at 0, 24, 36, and 48 h posttransfection and passaged. Cell extracts from DF-1 cells inoculated with the passaged supernatant or IBDV (IBDV wild type [wt])-infected cells or cells cotransfected with pVP1 and pVP3 were subjected to Western blot analysis. The viral proteins VP1 and VP3 were detected in the lysate from cells infected with wild-type IBDV or cotransfected with the plasmids of pVP1 and pVP3 (Fig. 1B, lanes 5 and 6) but not detected in the lysate of DF-1 cells inoculated with the passaged supernatant derived from cells transfected with IBDV dsRNA for a time course up to 72 h (Fig. 1B, lanes 2, 3, and 4). These data demonstrate that the purified naked IBDV genomic dsRNA could not be directly translated into viral proteins in the host cells.
FIG 1.
The purified IBDV genomic dsRNA or the authentic plus-sense IBDV RNA transcript cannot be translated by itself in host cells. (A) Agarose gel electrophoresis analysis of the purified IBDV genomic dsRNA. (B) The supernatants of DF-1 cells transfected with the purified IBDV genomic dsRNA were collected at various time points (0, 24, 48, and 72 h) posttransfection and used for three passages on DF-1 cells. Cell extracts from the DF-1 cells inoculated with the passaged supernatant or IBDV wild-type (IBDV/wt)-infected cells or cells cotransfected with pVP1 and pVP3 were subjected to Western blot analysis with antibodies against VP1, VP3, and GAPDH. (C) Schematic representation of the constructs containing the cDNA of segment A (p1-mA) or segment B (p1-mB), flanked immediately by Pol I and Ter I. (D) Western blot analysis of the cell lysate from HEK 293T cells cotransfected with p1-mA and p1-mB or infected with wild-type IBDV, using antibodies against VP1 and VP3. Alternatively, total RNA was extracted and subjected to RT-PCR amplification of vp1, vp3, and gapdh. The amplified vp1 fragment was subjected to EcoRI digestion. (E) The total RNA extracted from DF-1 cells either mock-infected or infected with IBDV (multiplicity of infection of 1) for 12 h was subjected to immunoprecipitation with the anti-cap MAb (H20) or with a mouse IgG control. RNA obtained from the precipitate or the total RNA (1 μg, input control) was subjected to RT-PCR amplification of vp1, vp3, or gapdh. (F) Western blot analysis was performed to detect the expression of GFP and RFP in cell lysate of HEK 293T cells transfected with the indicated plasmids.
Next, we examined whether the plus-sense IBDV RNA transcripts can be translated in host cells. First, we generated constructs where the cDNA of IBDV segment A or segment B was precisely under the control of the RNA polymerase I (Pol I) promoter and terminator I (Ter I) to generate the authentic uncapped plus-sense viral RNAs (25, 26) (Fig. 1C). A synonymous mutation was introduced by site-directed mutagenesis to create an EcoRI site at nucleotide (nt) 1657 of segment B as a molecular marker so that discrimination between IBDV wild-type virus and the rescued virus could be easily achieved by EcoRI digestion. The resulting plasmids were confirmed by DNA sequencing and were named p1-mA (segment A) and p1-mB (segment B). While the viral proteins of both VP1 and VP3, encoded by segment B and Segment A, respectively, were detected in the cell lysates of the IBDV-infected cells, no VP1 and VP3 was detected in the lysates of cells cotransfected with p1-mA and p1-mB (Fig. 1D, lane 2 versus lane 3 in the Western blot). To exclude the possibility that the defect in the expression of viral proteins was due to unsuccessful transcription from the transfected plasmids, total RNA was extracted from the transfected cells and subjected to reverse transcription PCR (RT-PCR) amplification of vp1 and vp3. The fragments of both vp1 and vp3 could be specifically amplified from the transfected cells and from the IBDV-infected cells. Further, the vp1 amplified from the RNA extracts of the cells cotransfected with p1-mA and p1-mB was cut by EcoRI into two segments with the anticipated sizes of about 450 bp and 750 bp, which did not occur with the RNAs extracted from the IBDV-infected cells (Fig. 1D, lanes 2 and 3 in the RT-PCR panel). This confirms that the cotransfections of p1-mA and p1-mB were successfully transcribed into plus-sense RNAs. Thus, our data indicate that neither the purified IBDV genomic dsRNA nor the viral plus-sense RNA transcript has the ability to initiate translation to produce the viral proteins directly in host cells.
It has been postulated that IBDV genomic RNA lacks a 5′ cap, which is essential for the cap-dependent translation initiation pathway (27). However, to the best of our knowledge, the cap status of IBDV genomic RNA has not been experimentally demonstrated under a natural infection condition. To resolve this issue, total RNA extracted from the DF-1 cells either mock infected or infected with IBDV was subjected to immunoprecipitation using monoclonal antibody (MAb) H20, which specifically recognizes the cap structures of m3G or m7G of capped RNAs (28), followed by RT-PCR amplification of the viral vp1 and vp3 in parallel with that of the internal control gapdh. As shown in Fig. 1E, while the amplification of gapdh increased in the H20 precipitate compared to the level with the IgG control (Fig. 1E, gapdh, lanes 5 and 6 versus lanes 3 and 4), there was no significant difference in the amplification levels of vp1 and vp3 from the immunoprecipitation using H20 and that using the IgG control (vp1 and vp3, lanes 6 versus lanes 4). This indicates that IBDV genomic RNA is naturally uncapped.
IRES-driven translation is another essential mechanism for translation initiation of many viruses, and IRESs are often found in the RNA UTR of many viruses to functionally replace the RNA cap structure (23, 29). To explore whether the UTRs of IBDV genomic dsRNA possess a functional IRES, we generated bicistronic reporter constructs pEGFP-5′-UTR-RFP (where EGFP is enhanced green fluorescent protein and RFP is red fluorescent protein), pEGFP-3′-UTR-RFP, and pEGFP-IRES-RFP in which the IBDV 5′ UTR, 3′ UTR of segment A, and the IRES sequence from encephalomyocarditis virus (EMCV) as a positive control were inserted between the EGFP and RFP coding sequences, respectively. The reporter plasmids were transfected into HEK 293T cells, and Western blot analysis was performed to monitor the expression of EGFP and RFP. While transfection of pEGFP-IRES-RFP resulted in the simultaneous expression of both EGFP and RFP (Fig. 1F, lane 1), transfection of pEGFP-5′-UTR-RFP or pEGFP-3′-UTR-RFP led to the expression of only EGFP (Fig. 1F, lanes 2 and 3). This suggests that both the 5′ and 3′ UTRs of the IBDV genomic dsRNA were incapable of directing translation of the downstream RFP. Thus, there are no bona fide functional IRES elements within the UTR sequences of IBDV genomic dsRNA.
VP1 and VP3 are required and sufficient for rescue of the uncapped IBDV genomic dsRNA into infectious IBDV.
The plus-sense IBDV RNA transcript was noninfectious since it could not be rescued into infectious viruses by itself. We next examined whether this defect can be rescued by any viral proteins provided in trans. HEK 293T cells were cotransfected with p1-mA and p1-mB together with various combinations of the viral protein-expressing plasmids pVP1, pVP2, pVP3, pVP4, and pVP5, and the supernatant of transfected cells was collected at 72 h posttransfection and used to infect DF-1 cells. After passage three times on DF-1 cells, the whole-cell lysates of passaged cells were subjected to Western blot analysis with antibodies against various IBDV viral proteins. In the lysates of DF-1 cells infected by supernatant from cells cotransfected with p1-mA and p1-mB, there were no viral proteins detected (Fig. 2A, lane 7), suggesting that the infectious IBDV was not rescued. In contrast, in the lysates of cells inoculated with the supernatant from cells cotransfected with p1-mA and p1-mB together with plasmids expressing VP1, VP2, VP3, VP4, and VP5, the viral proteins VP1, VP3 and VP4 were detected (Fig. 2A, lane 6), suggesting that the virus was rescued.
FIG 2.

VP1 and VP3 are the minimal viral components required and sufficient for rescue of infectious virus from the uncapped IBDV RNA. (A) HEK 293T cells were cotransfected with p1-mA and p1-mB together with different combinations of pVP1, pVP2, pVP3, pVP4, and pVP5 as indicated. The supernatant of transfected cells was collected at 72 h posttransfection and passaged on DF-1 cells, and subsequently lysates of passaged cells were subjected to Western blot analysis using antibodies against VP1, VP3, and VP4. Anti-GAPDH was used as a loading control. (B) HEK 293T cells were cotransfected with p1-mA and p1-mB together with pVP1 and pVP3 or pCI-neo empty plasmid. Supernatant of the transfected cells was collected at the indicated time points posttransfection, and the viral titer was measured by plaque assay. The curve was plotted based on three independent experiments, and values are expressed as means ± standard deviations. The inset shows an example of plaques formed on a monolayer of DF-1 cells inoculated with the supernatant from HEK 293T cells cotransfected with p1-mA, p1-mB, pVP1, and pVP3 for 72 h. (C) RT-PCR analysis of vp1 and vp3 for validation of the viral RNA transcription level in the infected DF-1 cells, and discrimination of the rescued virus (IBDV/rescued) from wild-type virus (IBDV/wt) by EcoRI digestion analysis of the amplified vp1 gene fragment. (D) The plaque assay was performed to establish the one-step growth curves of the rescued viruses (IBDV/rescued) and the wild-type viruses (IBDV/wt) with the inoculation of virus at a multiplicity of infection of 0.1. (E) HEK 293T cells were cotransfected with the purified IBDV genomic dsRNA together with pVP1 and pVP3 or pCI-neo empty plasmid. The supernatant was collected at 72 h posttransfection and passaged on DF-1 cells. Western blotting was performed to verify the rescue of infectious viruses using antibodies against VP1, VP3, and VP4.
Next, we set out to determine the minimal set of viral proteins required for this rescue. A screening assay was carried out by examining the effect of withdrawal of an individual viral protein(s) on the rescue of IBDV (30). Withdrawal of either pVP1 or pVP3 from the plasmid pool for transfection resulted in no rescue of IBDV as no viral proteins were detected (Fig. 2A, lanes 1 and 3), while the deficiency of pVP2, pVP4, or pVP5 had no effect on the rescue of IBDV (Fig. 2A, lanes 2, 4, and 5). This suggests that VP1 and VP3 are the minimal viral proteins required for the rescue of infectious viruses from the viral plus-sense RNA transcripts. Further, cotransfection of only VP1 and VP3 was sufficient to rescue the uncapped plus-sense IBDV RNA transcribed from p1-mA and p1-mB into infectious viruses (Fig. 2A, lane 10).
The successful recovery of infectious IBDV was further demonstrated by the obvious cytopathic effect (CPE) induced by the supernatant of cells cotransfected with p1-mA and p1-mB together with VP1 and VP3 (Fig. 2B, inset). The rescued virus titer of supernatant harvested at 24, 48, 72, and 96 h posttransfection was determined by plaque assay, showing that the maximum rescued virus titer reached was about 6 log PFU/ml at 72 h posttransfection (Fig. 2B). To verify that the rescued virus was the transfected recombinant virus, the fragments of vp1 were amplified by RT-PCR either from the rescued virus or from the wild-type IBDV and subjected to EcoRI digestion. While the vp1 fragment amplified from the wild-type IBDV could not be cut by EcoRI, the vp1 fragment amplified from the rescued viruses was cut by EcoRI into two segments of the anticipated sizes (Fig. 2C, top panel), confirming that the recovered virus was indeed derived from the cloned cDNAs of IBDV genomic dsRNA. Furthermore, the virus recovered from cotransfection of p1-mA and p1-mB together with pVP1 and pVP3 showed growth kinetics similar to that of the wild-type IBDV (Fig. 2D, IBDV/rescued versus IBDV/wt). Moreover, the purified naked IBDV genomic dsRNA could also be rendered infectious and be rescued into infectious viruses by trans-supplementation of VP1 and VP3. As shown in Fig. 2E, inoculation of DF-1 cells with the supernatant from the HEK 293T cells transfected with the purified IBDV dsRNA alone failed to yield any viral proteins, as analyzed by Western blotting (lane 2). In contrast, as with the positive control with IBDV infection (Fig. 2E, lane 3), inoculation of the supernatant of the HEK 293T cells cotransfected with the purified IBDV dsRNA together with pVP1 and pVP3 resulted in the production of the viral proteins (lane 1).
Together, these results indicate that VP1 and VP3 are required and sufficient to render infectious the uncapped IBDV genomic dsRNA and plus-sense RNA transcripts, resulting in the rescue of IBDV.
The RdRp activity of VP1, the dsRNA binding activity of VP3, and both the 5′ and 3′ UTRs are required for the VP1/VP3-mediated translation initiation of uncapped IBDV genomic RNA.
Since the uncapped IBDV genomic RNA could not be translated by itself and since VP1 and VP3 were sufficient for allowing the uncapped IBDV dsRNA to be rescued into infectious viruses, we thus investigated whether VP1 and VP3 are required for the IBDV translation initiation of its uncapped RNA. Transfection of p1-mA alone into HEK 293T cells successfully yielded the mRNA transcript of IBDV segment A, as indicated by the RT-PCR amplification of vp3 and vp4, but the viral protein VP4, an indicator of translation of p1-mA of the viral segment A (10), was not detected by Western blotting (Fig. 3A, lane 1). This suggests that the uncapped IBDV RNA cannot be translated by itself. This defect in translation was rescued by expression of both VP1 and VP3 in trans (Fig. 3A, lane 4) but not by expression of either VP1 or VP3 (Fig. 3A, lanes 2 and 3). Thus, both VP1 and VP3 are required for translation initiation of uncapped IBDV RNA.
FIG 3.

The RdRp activity of VP1, the dsRNA binding activity of VP3, and both the 5′ and 3′ UTRs are required for VP1/VP3-mediated translation initiation of the uncapped IBDV RNA. (A) HEK 293T cells were cotransfected with the indicated plasmids, and the amount (in micrograms) of each plasmid for transfection is indicated above the lanes. Western blot analysis was performed to assess the expression of VP1, VP3, and VP4 at 72 h posttransfection. The transcription of viral mRNA from the transfected constructs was assessed by RT-PCR assay using primer pairs specific for vp1, vp3, and vp4 or for the plasmid backbone as a control. (B) HEK 293T cells were cotransfected with the indicated plasmids, and the amount (in micrograms) of each plasmid for transfection is indicated above the lanes. Western blot analysis of the cell lysate of the transfected cells was performed to assess the expression of VP1, VP3, and VP4 at 72 h posttransfection. (C) The supernatant of HEK 293T cells cotransfected with the indicated plasmids was passaged at 72 h posttransfection, and the images were taken under a phase-contrast microscope at 12 h postinfection. Scale bar, 100 μm. (D) Schematic representation of the constructs for the generation of the uncapped plus-sense RNA of segment A with deletion of the 5′ UTR (p1-mAΔ5) or 3′ UTR (p1-mAΔ3). (E) HEK 293T cells were transfected with the indicated plasmids, and the amount (in micrograms) of each plasmid for transfection is indicated above the lanes. Western blot analysis was performed to assess the expression of VP1, VP3, and VP4. The transcript level of vp4 under each condition was analyzed by RT-PCR.
VP3 interacts with IBDV genomic dsRNA and together with VP1 forms ribonucleoprotein (RNP), which is essential for the initiation of the replication cycle (17, 18). We next examined whether the RNA polymerase activity of VP1, the dsRNA binding activity of VP3, and the association between VP1 and VP3 are required for the VP1/VP3-mediated translation initiation of the viral dsRNA and for the rescue of viruses. VP4 was detected in cells cotransfected with p1-mA and VP1 together with wild-type VP3 but not with the dsRNA-binding-deficient mutant VP3/patch1 (31) or VP1-binding-deficient mutant VP3/ΔC (32) (Fig. 3B, lanes 3 and 4, respectively). Further, the supernatant of the cells cotransfected with p1-mA and p1-mB together with pVP1 plus either pVP3ΔC or pVP3/patch1 could not induce CPE in DF-1 cells (Fig. 3C, frames c and d). We also used a similar approach to assess the requirement of the polymerase activity of VP1 in the VP1/VP3-mediated translation initiation of the viral RNA and the IBDV rescue events. The viral protein VP4 was detected in the cells cotransfected with p1-mA and VP3 together with wild-type VP1 but not with the RdRp-inactive VP1 mutant of pVP1/D402A or pVP1/D416A (12, 33) (Fig. 3B, lane 6 versus lanes 7 and 8). Likely, the CPE was not observed in the DF-1 cells inoculated with the supernatant of the cells cotransfected with p1-mA, p1-mB and pVP1/D402A or pVP1/D416A (Fig. 3C, frames e and f).
In both segments, the coding ORFs of IBDV genomic dsRNA are flanked by the 5′ and 3′ UTRs, which are essential for viral replication (24). To elucidate whether the UTR plays a role in the VP1/VP3-mediated translation initiation of the uncapped IBDV RNA, we generated constructs for the generation of the plus-sense RNA of segment A with a deletion of the 5′ UTR (p1-mAΔ5) or 3′ UTR (p1-mAΔ3) (Fig. 3D). While cotransfection of p1-mA together with pVP1 and pVP3 resulted in expression of VP4 as detected by Western blotting, there was no VP4 detected in the cells cotransfected with pVP1 and pVP3 together with either p1-mA/Δ5 or p1-mA/Δ3 (Fig. 3E, lanes 4 and 6 versus lane 2 in the Western blot). This defect in translation was not caused by a defect in transcription because the RT-PCR assay showed that the deletion of the 5′ UTR or 3′ UTR had no effect on the transcription of vp4 (Fig. 3E, lanes 2, 4, and 6 in the RT-PCR panel).
Together, these data indicate that the RdRp activity of VP1, the dsRNA binding activity of VP3, and both the 5′ and 3′ UTRs are required for the VP1/VP3-mediated translation initiation of uncapped IBDV RNA.
VP1 and VP3 mediate the recovery of infectious IBDV from the minus-sense IBDV RNA.
The observation that the RdRp activity of VP1 was required for VP1/VP3-mediated translation initiation of uncapped IBDV RNA prompts us to postulate that the minus-sense strand of the viral dsRNA could be copied into the plus-sense strand and subsequently be rescued into infectious viruses in the presence of VP1 and VP3. To test this hypothesis, we generated constructs p1-RmA and p1-RmB in which the cDNAs of IBDV segment A and segment B, respectively, were cloned in reverse orientation under the control of Pol I for production of authentic minus-sense IBDV RNA transcripts of either segment A or segment B (Fig. 4A). Transfection of p1-RmA without the cotransfection of pVP1 and pVP3 failed to generate the viral proteins. In contrast, cotransfection of p1-RmA together with pVP1 and pVP3 led to the synthesis of VP4 (Fig. 4B, lane 2 versus lane 1 in the Western blot). The levels of transcription from p1-RmA or p1-mA with and without in trans expression of VP1 and VP3 were comparable, as indicated by RT-PCR analysis of the relative level of vp4 mRNA in the transfected cells (Fig. 4B, lanes 1, 2, 3, and 4 in the RT-PCR panel). Further, similar to the results with p1-mA and p1-mB (Fig. 4C, frame b), cotransfection of p1-RmA and p1-RmB together with pVP1 and pVP3 resulted in a full recovery of infectious IBDV, which causes apparent CPE in DF-1 cells (Fig. 4C, frame d). However, the plaque assay showed that viruses recovered from cotransfection of p1-RmA and p1-RmB together with pVP1 and pVP3 had a slightly delayed growth kinetics relative to that of viruses rescued from cotransfection of p1-mA and p1-mB together with pVP1 and pVP3 (Fig. 4D). Thus, these data demonstrate that VP1 and VP3 can mediate the recovery of infectious viruses from not only the plus-sense RNA but also the minus-sense RNA of IBDV genomic dsRNA.
FIG 4.

VP1 and VP3 mediate the recovery of the minus-sense IBDV RNA into infectious viruses. (A) Schematic of the constructs for the generation of the uncapped authentic minus-sense RNAs of IBDV genome. The construct contains the cDNA of the minus strand of segment A (p1-RmA) or segment B (p1-RmB), flanked immediately by Pol I and Ter I. (B) HEK 293T cells were transfected with the indicated plasmids, and the amount (in micrograms) of each plasmid for transfection is indicated above the lanes. Western blotting was used to analyze the expression of the viral proteins VP1, VP3, and VP4 at 72 h posttransfection. Alternatively, RT-PCR was used to detect the relative level of vp4 transcript in the transfected cells as indicated. (C) The supernatant of HEK 293T cells cotransfected with the indicated plasmids was passaged at 72 h posttransfection, and the images were taken under a phase-contrast microscope at 12 h postinfection. Scale bar, 100 μm. (D) A plaque assay was performed to measure viral titer in the supernatant at the indicated times from HEK 293T cells cotransfected with the indicated plasmids. The histogram was plotted based on three independent experiments. Values are expressed as means ± standard deviations.
The cap-binding protein eIF4E is not required for VP1/VP3-mediated translation initiation of uncapped IBDV RNA.
The requirement of VP1 and VP3 for translation initiation of the viral uncapped dsRNA prompts us to speculate that VP1 and VP3 might act as a substitute for the cap structure for dsRNA translation. To test this hypothesis, we set out to examine whether the defect in the translation initiation of IBDV RNA can be rescued by artificially adding a cap at the 5′ end of the viral mRNA. To achieve this, the cDNA of either segment A or segment B was inserted downstream of a T7 promoter, and this was followed by in vitro transcription of mRNA of the IBDV dsRNA. With or without an addition of the m7G cap analog in the in vitro transcription mixture, the 5′ end of the in vitro-transcribed mRNA from the T7 promoter can be either capped or uncapped. In the absence of VP1 and VP3, transfection of in vitro-transcribed and capped IBDV RNA transcripts into HEK 293T cells gave rise to the expression of VP1, VP3, and VP4 while transfection of in vitro-transcribed but uncapped IBDV RNA transcripts did not (Fig. 5A, lane 3 versus lane 2). Moreover, inoculation with the supernatant from the cells transfected with capped segments A and B rather than with that from cells transfected with uncapped A and B led to the obvious CPE in DF-1 cells (Fig. 5A, panel b). Thus, the in vitro-transcribed and artificially capped IBDV RNA gains the ability to initiate translation without a requirement for VP1 and VP3. This result further suggests that the VP1 and VP3 are responsible for the translation of IBDV RNA by acting as a substitute for the 5′ cap required for viral mRNA translation.
FIG 5.

VP1- and VP3-mediated translation initiation of uncapped plus-sense IBDV RNA transcripts is independent of the 5′ cap and the cap-binding protein eIF4E. (A) Western blot analysis of the cell lysate from HEK 293T cells transfected with the in vitro-transcribed IBDV RNAs of both A and B segments with or without an artificial addition of a 5′ cap, using antibodies against VP1, VP3, and VP4 (panel a). The supernatant was passaged at 72 h posttransfection, and the images were taken under a phase-contrast microscope at 12 h postinfection (panel b). Scale bar, 100 μm. (B) HEK 293T cells were transfected with the indicated plasmids, and the amount (in micrograms) of each plasmid for transfection is indicated above the lanes. The transfected cells were either untreated, mock (DMSO) treated (4 μl/well), or treated with wortmannin (800 nmol in 4 μl of DMSO/well) for 72 h. Western blot analysis was performed to analyze the expression of VP4, VP1, VP3, phospho-Akt (Ser473) (p-Akt), and total Akt. (C) HEK 293T cells were transfected with the shRNA plasmids for 24 h, and the resultant cells were cotransfected with p1-mA, pIRES-VP1, and pIRES-VP3 for another 72 h; Western blotting was performed to analyze the levels of VP4, VP1, VP3, and eIF4E.
The cap structure at the 5′ end of eukaryotic mRNA interacts with eIF4E, which is essential for cap-dependent initiation of protein synthesis (34). Next, we sought to determine whether VP1 and VP3 could also replace eIF4E in the translation initiation of the uncapped IBDV RNA. Wortmannin inhibits phosphatidylinositol 3-kinase (PI3K), resulting in hypophosphorylation of 4E-binding protein 1 (4E-BP1), which in turn sequestrates eIF4E from eIF4F and thus inhibits cap-dependent translation (35). HEK 293T cells were cotransfected with p1-mA, pIRES-VP1, and pIRES-VP3, followed by mock (dimethyl sulfoxide [DMSO]) or wortmannin treatment. The IRES-driven translation of VP1 and VP3 from pIRES-VP1 and pIRES-VP3 was not affected upon inhibition of the cap-dependent synthesis pathway by treatment with wortmannin. As expected, wortmannin treatment effectively inhibited the activity of PI3K, which was reflected in a significant reduction of the phosphorylation of Akt at S473 (Fig. 5B, lane 4 versus lane 2). Interestingly, upon wortmannin treatment, the VP1/VP3-mediated translation of p1-mA, which was indicated by the expression level of VP4, was not decreased but, rather, was remarkably enhanced up to 4-fold compared to that with the mock (DMSO) treatment (Fig. 5B, lane 4 versus lane 3). Further, more specifically, we used a short hairpin RNA (shRNA) approach to knock down expression of eIF4E. Western blotting confirmed that the expression of eIF4E was successfully reduced by 80% upon expression of either of two independent eIF4E-specific shRNA clones (Fig. 5C, lanes 3 and 4). Similarly, knockdown of eIF4E using an shRNA did not decrease but, rather, increased the expression of the VP1/VP3-mediated translation initiation of uncapped IBDV RNA (p1-mA), which was indicated by the expression level of VP4 (Fig. 5C, lanes 3 and 4). Together, these data suggest that VP1/VP3-mediated translation is independent of the cap-binding protein eIF4E.
DISCUSSION
IBDV, a virus belonging to the Birnaviridae family, contains a bisegmented dsRNA. IBDV genomic dsRNA lacks a 5′ cap. While the initiation of viral translation is a key point of control for virus replication, the means by which the uncapped IBDV RNA is translated effectively remains unclear. In this study, we demonstrated that the naked IBDV genomic dsRNA or the plus-sense strand RNA with an authentic terminus could not be translated by itself and rescued into infectious viruses. This defect in translation and virus production was rescued by VP1 and VP3 provided in trans. VP1 and VP3 were required and sufficient to initiate translation of the uncapped IBDV RNA and to rescue infectious viruses from both the plus-sense and minus-sense strands of the IBDV genomic dsRNA. The VP1/VP3-mediated translation initiation of uncapped IBDV RNA is independent of the cap-interacting initiation factor eIF4E.
Our immunoprecipitation with H20, an antibody specific against m3G and m7G, in combination with RT-PCR analysis revealed that under the naturally occurring infection condition, the IBDV genomic dsRNA and the viral RNA products generated during replication are uncapped (Fig. 1E). There was no functional IRES element in the UTRs (Fig. 1F). This explains why the plus-sense strand RNA of IBDV cannot directly be translated into viral proteins by itself and is thus noninfectious. This is, however, in direct contradiction to a previous report that the in vitro-transcribed plus-sense RNA copy of the IBDV genome was recovered in infectious virus (36). Our explanation for the difference was that the in vitro synthetic plus-sense transcripts of IBDV dsRNA used in that study were actually capped according to the experimental procedure described in the paper (36). In support of this explanation, we also showed that the in vitro-transcribed and artificially 5′ capped IBDV RNA could successfully be translated into viral proteins and rescued into infectious IBDV by itself (Fig. 5A) and that IBDV could also be efficiently rescued from the capped viral RNAs generated by the RNA polymerase II promoter (37).
The defect in virus production and translation of IBDV uncapped RNA imposed by the lack of a 5′ cap was rescued by the viral proteins VP1 and VP3 provided in trans, which is in line with previous observations (38). Significantly, VP1 and VP3 are sufficient for translation initiation and recovery of infectious viruses not only for the plus-sense RNA but also for the minus-sense RNA. Similar to the rescued viruses from IBDV plus-sense RNA, the viruses recovered from the minus-sense RNA could replicate to high titers and form plaques in DF-1 cells. The only difference is that the latter exhibited slightly delayed replication kinetics in the cell culture relative to that of the viruses rescued from the plus-sense transcripts. This suggests that VP1 and VP3 play a role similar to that of the 5′ cap or may function directly as a substitute for the 5′ cap structure in IBDV translation initiation. The importance of VP1 and VP3 as a substitute for the 5′ cap structure in IBDV translation initiation was further demonstrated by finding that the in vitro artificial addition of a 5′ cap to the plus-sense IBDV RNA could restore and complement the translation of IBDV uncapped RNA and rescue infectious IBDV in the absence of VP1 and VP3 (Fig. 5A).
Interestingly, the RNA-dependent RNA polymerase (RdRp) activity of VP1 was required for the VP1/VP3-dependent translation initiation of the uncapped IBDV plus-sense RNA and rescue of the infectious viruses (Fig. 3B and C), suggesting that a primary transcription mediated by VP1 is required prior to the translation initiation of IBDV RNA in the viral life cycle. Further, the dsRNA binding ability of VP3 was also required for VP1/VP3-mediated uncapped IBDV RNA translation initiation (Fig. 3B and C), suggesting that assembly of VP1 and VP3 together with the viral dsRNA into RNP complexes is a prerequisite for priming the translation of IBDV uncapped RNA. This suggests that apart from its role in viral RNA transcription, the RNP complex has an additional role in translation initiation of the viral RNA during IBDV replication (12).
In contrast to cap-dependent translation initiation where a single-strand RNA with a 5′ cap is used as a template, the VP1/VP3-mediated translation initiation of IBDV utilizes at least a short stretch of dsRNA formed by UTRs, given that the binding of VP3 to dsRNA and the presence of both the 5′ and 3′ UTRs are required (Fig. 3B and E). There is thus a need for VP1-dependent synthesis of complementary-strand RNA to form the dsRNA, which can be derived from both the plus- and minus-sense strands. This also explains why the RdRp activity of VP1 is required for the VP1/VP3-mediated translation initiation and the recovery of infectious viruses from both plus- and minus-sense RNAs of the IBDV genome. This unique prerequisite for dsRNA instead of ssRNA in the translation initiation stage could confer more specificity to IBDV translation. Thus, IBDV uses a novel cap-independent mechanism of protein synthesis initiation that relies on the viral proteins VP1 and VP3, which act as a substitute for the 5′ cap structure. Interestingly, caliciviruses employ a similar translation initiation mechanism to translate their genes that is mediated by the interaction of the VPg with the cellular eIF4E (39), and hantavirus nucleocapsid protein (N) replaces the cellular cap-binding complex to mediate translation initiation (40).
Since the UTRs interact with VP1 and VP3 (19, 41), it is not a surprise that both the 5′ and 3′ UTRs are essential for VP1/VP3-mediated translation initiation of IBDV RNA. In line with this, our data showed that deletion of the 5′ UTR or 3′ UTR of segment A abolished VP1/VP3-mediated translation initiation of IBDV uncapped RNA (Fig. 3E). Additionally, the coding sequences may also be involved in this process because the gfp sequence flanked by the 5′ and 3′ UTRs could not be encapsidated into IBDV (42). Thus, the specific RNA sequences and secondary structures are likely prerequisites for this process.
The cap structure at the 5′ end of the eukaryotic mRNA and its interaction with eIF4E, a cap-binding factor, are essential for recruitment of the host translation machinery and for mRNA recognition in the initiation of protein synthesis (43). Importantly, functional inhibition or downregulation of eIF4E did not block or attenuate but, rather, increased VP1/VP3-mediated translation initiation of the uncapped IBDV RNA (Fig. 5B and C), suggesting that VP1 and VP3 cannot only compensate the deficiency of the 5′ cap but also replace the role of eIF4E in the recruitment of the host translation machinery, such as the 43S preinitiation complex, to the suitable start codon, AUG, of the uncapped IBDV RNA. One possible explanation for the enhanced translation of IBDV RNA upon wortmannin inhibition or shRNA downregulation of eIF4E is that inhibition of the cap-dependent synthesis pathway by eIF4E suppression will supply more resources, such as availability of ribosomes, for the VP1/VP3-mediated translation, which is cap independent. However, it is not yet clear how VP1 and VP3 function to replace eIF4E in the translation initiation of uncapped IBDV RNAs in host cells. It is likely that the IBDV VP1 or/and VP3 may interact with one or more components of the cellular translation initiation factors in the translation initiation complex, thus facilitating the entry of the ribosome to the translational sequences. For example, it was reported that VP1 of IBDV interacts with the carboxy-terminal domain of eIF4AII (44, 45), an essential player in the initiation of translation of both capped and uncapped mRNAs. Further investigation of the host-specific factors or pathways implicated in the VP1/VP3-mediated translation initiation of the uncapped IBDV RNA will provide detailed insights into this process.
Together, our results support a model of translation initiation of IBDV uncapped dsRNA wherein VP1 and VP3 are required and sufficient to compensate the deficiency of a 5′ cap and replace eIF4E to mediate the recruitment and assembly of an active translation initiation complex, including factors such as eIF4A/G and eIF3, subsequently allowing for access to the start codon AUG by 40S ribosomes (Fig. 6). Importantly, VP1/VP3 mediates translation initiation of the uncapped IBDV RNA that can be derived from both the plus- and minus-sense strands. This reconfigures the existing model that the plus-sense viral RNA is used to translate the viral proteins and the minus-sense viral RNA is used for the synthesis of the viral genome. These findings will provide not only new insights into the molecular mechanisms of the life cycle of IBDV but also a new tool for an efficient alternative strategy for the recovery of IBDV from the cDNA of the viral genomic dsRNA.
FIG 6.
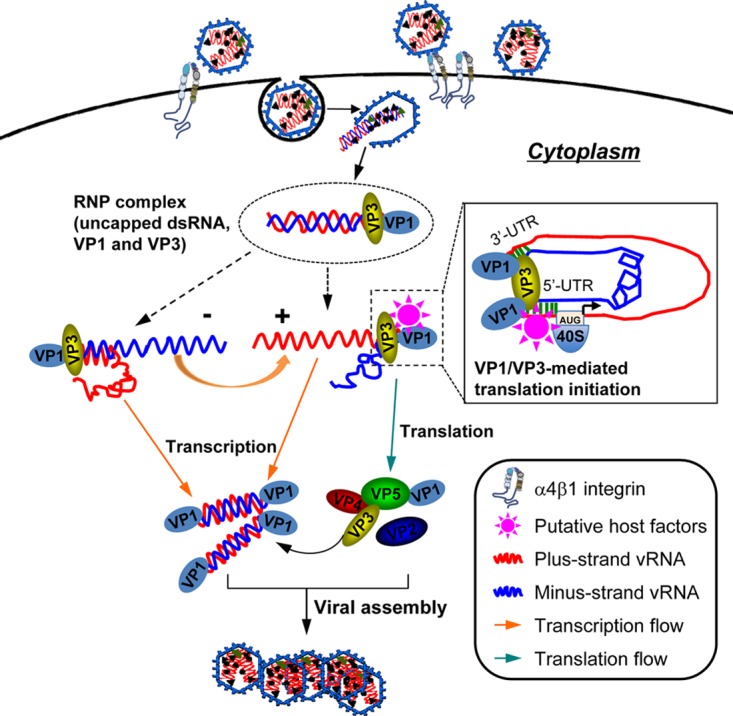
Suggested model of the VP1/VP3-mediated translation of uncapped IBDV RNA by functionally compensating for the deficiency of the 5′ cap and replacing the cap-binding protein eIF4E during viral replication. Upon cell entry, the IBDV genomic dsRNA is released from the endosome into the cytoplasm. While the IBDV genomic dsRNA is uncapped and does not contain an IRES element, it associates with the viral proteins VP1 and VP3 in RNP complexes that function as transcription engines in the viral replication (17). Our current model supports the observation that VP1 and VP3 in the RNP complex are required and sufficient to initiate translation of the uncapped viral RNA, which is dependent on both the RdRp activity of VP1 and dsRNA binding activity of VP3. The VP1/VP3-mediated translation initiation of the viral uncapped RNA requires the binding of VP3 to the dsRNA, e.g., a short stretch of dsRNA in the UTRs and the presence of both the 5′ and 3′ UTRs. The VP1 and VP3 can mediate translation initiation of not only the plus-sense strand but also the minus-sense strand of the viral dsRNA. Thus, VP1 and VP3 in the RNP complex have a dual role in IBDV replication contributing not only to the viral RNA transcription but also to translation initiation of the associated uncapped viral genomic RNA by compensating for the deficiency of the 5′ cap and replacing eIF4E.
MATERIALS AND METHODS
Cells, virus, and reagents.
HEK 293T cells (ATCC; CRL-11268) and the chicken fibroblast cell line DF-1 (ATCC; CRL-12203) were routinely maintained in Dulbecco's modified Eagle's medium (Gibco, Carlsbad, CA) supplemented with 10% fetal bovine serum (Gibco). The IBDV attenuated strain HZ2 that was adapted to growth in DF-1 cells was a gift from Yaowei Huang (46). The MAb H20 that recognizes m3G and m7G was purchased from Merck Millipore (Billerica, MA). Mouse anti-VP1, mouse anti-VP3, and mouse anti-VP4 polyclonal antibodies were generated from the sera of mice by immunization with purified recombinant VP1, VP3, and VP4, respectively. Monoclonal mouse anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was purchased from Kangchen Biotechnology (Shanghai, China). Monoclonal mouse antibodies against GFP and RFP were purchased from ThermoFisher Scientific (Waltham, MA). The chemical inhibitor wortmannin was purchased from Selleck (Houston, TX). The rabbit monoclonal antibodies against Akt, phospho-Akt (S473), and eIF4E were purchased from Cell Signaling Technology (Danvers, MA).
Purification of the naked IBDV genomic dsRNA.
IBDV was propagated and concentrated as described previously (47). Briefly, IBDV-containing virus fluid was concentrated to 1/100 of the original volume by ultracentrifugation at 50,000 × g for 3 h and redissolved in phosphate-buffered saline (PBS). The concentrated IBDV was subjected to digestion with proteinase K in a 55°C water bath for 1 h with occasional shaking. The IBDV genomic dsRNA was purified by phenol-chloroform extraction and ethanol precipitation and dissolved in the diethyl pyrocarbonate (DEPC)-treated water. The purified IBDV genomic dsRNAs were analyzed by electrophoresis on a 0.8% agarose gel.
Construction of plasmids.
The cDNAs of both segment A and segment B of the IBDV genome (GenBank accession numbers AF321054.1 and AF493979.1, respectively) were amplified by reverse transcription-PCR (RT-PCR) using the extracted IBDV genomic dsRNA as the template with specific primers (48). p1-mA and p1-mB were generated by precisely embedding the viral cDNA of segment A and segment B, respectively, between Pol I and Ter I based on the backbone of pTA2 (Toyobo, Osaka, Japan). p1-RmA and p1-RmB for the generation of minus-sense RNA transcripts were generated by reversing the direction of the cDNAs of segment A and segment B in p1-mA and p1-mB, respectively. Site-directed mutagenesis was carried out as previously described (21) to introduce the EcoRI restriction site in segment B as the molecular marker. pEGFP-5′-UTR-RFP, pEGFP-3′-UTR-RFP, and pEGFP-IRES-RFP were generated by inserting the sequence of the segment A 5′ UTR, segment A 3′ UTR, and the IRES from EMCV between the EGFP and RFP coding sequences, respectively. pVP1, pVP2, pVP3, pVP4, and pVP5 were generated by inserting the corresponding coding sequences into the vector of pCI-neo (Promega, Madison, WI). pIRES-VP1 and pIRES-VP3 were generated by embedding the sequences of VP1 and VP3, respectively, into the vector of pIRES (Clontech, Mountain View, CA). All plasmids were confirmed by Sanger sequencing, and all the primer sequences used for plasmid construction are available on request.
In vitro transcription.
The full-length sequence of segment A or segment B flanked by the upstream T7 promoter sequence was generated by standard PCR. The primer sequences are available on the request. In vitro transcription was performed with a Riboprobe In Vitro Transcription System (Promega). Briefly, the template DNA was recovered by cleanup and added to a transcription reaction mixture (2 μg/100 μl) containing 1× transcription buffer, 10 mM dithiothreitol (DTT), 100 U of RNasin, 0.5 mM nucleoside triphosphate (NTP), and 80 U of T7 RNA polymerase, with or without 0.5 mM m7G cap analog, and incubated at 37°C for 1 h. The reaction mixture was treated with DNase I to remove the template, and the synthetic RNA transcripts were purified by phenol-chloroform extraction and ethanol precipitation.
Transfection.
Transfection of DNA plasmids was performed by Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's instructions, and the total amount of DNA in each transfection was kept constant by adding empty pCI-neo plasmid DNA (vector). Transfection of the purified IBDV genomic dsRNA or the in vitro-transcribed RNA of IBDV was performed as for the transfection of DNA.
RNA interference.
Two tested target of eIF4E (GGATGGTATTGAGCCTATG and AAGCAAACCTGCGGCTGATCT) (49) were selected to generate the shRNA plasmids (sheIF4E-1 and sheIF4E-2) as described previously (50). The knockdown of expression of eIF4E in HEK 293T cells was performed by transient transfection of shRNA plasmids, and the shRNA targeting EGFP (shEGFP) was used as a negative control.
Virus passage.
After transfected cells were subjected to three freeze-thaw cycles, the supernatant was filtered through a 0.22-μm-pore-size filter unit (Millipore) and used as an inoculum in the monolayer of DF-1 cells (1:50) for 72 h; the supernatant was then passaged serially by this method an additional two times on DF-1 cells. After the final passage, the samples used for CPE imaging, Western blot analysis, or total RNA extraction were prepared by incubating the supernatant on the monolayer of DF-1 cells (1:100) for 12 h.
RT-PCR and EcoRI digestion.
The total RNA of the cells in six-well plates was prepared with TRIzol reagent (Invitrogen) according to the manufacturer's instructions. Reverse transcription of 1 μg of total RNA was carried out using a SuperScript first-strand synthesis system (Fermentas, Pittsburgh, PA) according to the manufacturer's protocol using specific primers. The amplifications of vp1, vp3, vp4, and gapdh as the internal control were carried out according to the standard protocol. The amplification product of vp1 was recovered by a cleanup kit (TaKaRa, Dalian, China) and subjected to EcoRI digestion at 37°C in a water bath for 1 h. The products were subjected to 1% agarose gel electrophoresis analysis. The primer sequences used for RT-PCR are available on request.
Immunoprecipitation of RNAs with anti-cap MAb.
Immunoprecipitation of RNAs with anti-cap MAb was performed as previously described (28). Briefly, H20, a specific anti-cap MAb, was incubated with protein G-Sepharose beads for 1 h at 4°C, and then the beads were washed three times with binding buffer (10 mM Tris base, 150 mM NaCl, 10 mM EDTA, pH 7.5). An irrelevant MAb coupled to the beads under the same conditions was used as the control. For the reaction, 100 μl of a 50% slurry of MAb-coupled beads was mixed with the total RNA (10 μg), diluted with the binding buffer to a final volume of 300 μl, and incubated at 4°C for 2 h. After centrifugation, the beads were washed three times in 1,000 μl of binding buffer. The RNA bound to the beads was recovered by proteinase K digestion, phenol-chloroform extraction, and ethanol precipitation. The RNA pellets were suspended in 11 μl of DEPC-treated water. The RT-PCR amplification of gapdh was used as the internal control, and the amplification of vp1 and vp3 was used to assess the capped viral RNA of IBDV.
Western blotting.
Western blot analysis was performed as described previously (50). Briefly, whole-cell lysate was extracted by using lysis buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% Triton X-100, and 1% sodium deoxycholate) at 4°C for 30 min, followed by centrifugation at 12,000 × g at 4°C for 30 min. Equivalent amounts of cell lysate were subjected to 12% SDS-PAGE and transferred to nitrocellulose membranes. After being blocked with 5% bovine serum albumin in PBS containing 0.1% Tween 20 (PBST) at room temperature for 1 h, the membranes were incubated with primary antibodies at 4°C overnight, followed by horseradish peroxidase-conjugated secondary antibody incubation at 37°C for 1 h. The blots were developed with enhanced chemiluminescence (ECL) reagent, and GAPDH was probed as the loading control.
Plaque assay.
A plaque assay for IBDV on DF-1 cells was performed as described previously (50). Briefly, a 10-fold dilution series of a virus-containing sample (0.1-ml aliquots of each dilution) was inoculated onto the monolayer of DF-1 cells in six-well plates. The cells were incubated at 37°C for 1 h for virus internalization, and then the cells were covered with 1% low-melting-point agarose after three washes with PBS. The plates were stained with 1% crystal violet after another 72 h of incubation. The titer was determined by counting the plaques induced.
ACKNOWLEDGMENTS
This work was supported by Zhejiang A&F University (ZAFU) research and development grants (2014FR007 and 2015FR008), the China Postdoctoral Science Foundation (2017M621977), the National Natural Science Foundation of China (31502061 and 31470840), the Natural Science Foundation of Zhejiang province (LY18C180002), and the Science and Technology Foundation of Jiangsu province (BK20131181).
REFERENCES
- 1.Hirai K, Kunihiro K, Shimakura S. 1979. Characterization of immunosuppression in chickens by infectious bursal disease virus. Avian Dis 23:950–965. doi: 10.2307/1589611. [DOI] [PubMed] [Google Scholar]
- 2.Sharma JM, Kim I-J, Rautenschlein S, Yeh H-Y. 2000. Infectious bursal disease virus of chickens: pathogenesis and immunosuppression. Dev Comp Immunol 24:223–235. doi: 10.1016/S0145-305X(99)00074-9. [DOI] [PubMed] [Google Scholar]
- 3.Jackwood DJ, Saif YM, Hughes JH. 1982. Characteristics and serologic studies of two serotypes of infectious bursal disease virus in turkeys. Avian Dis 26:871–882. doi: 10.2307/1589875. [DOI] [PubMed] [Google Scholar]
- 4.Ismail N, Saif Y, Moorhead P. 1988. Lack of pathogenicity of five serotype 2 infectious bursal disease viruses in chickens. Avian Dis 32:757–759. doi: 10.2307/1590995. [DOI] [PubMed] [Google Scholar]
- 5.Wu Y, Hong L, Ye J, Huang Z, Zhou J. 2009. The VP5 protein of infectious bursal disease virus promotes virion release from infected cells and is not involved in cell death. Arch Virol 154:1873–1882. doi: 10.1007/s00705-009-0524-4. [DOI] [PubMed] [Google Scholar]
- 6.Lombardo E, Maraver A, Espinosa I, Fernández-Arias A, Rodriguez JF. 2000. VP5, the nonstructural polypeptide of infectious bursal disease virus, accumulates within the host plasma membrane and induces cell lysis. Virology 277:345–357. doi: 10.1006/viro.2000.0595. [DOI] [PubMed] [Google Scholar]
- 7.Méndez F, de Garay T, Rodríguez D, Rodríguez JF. 2015. Infectious bursal disease virus VP5 polypeptide: a phosphoinositide-binding protein required for efficient cell-to-cell virus dissemination. PLoS One 10:e0123470. doi: 10.1371/journal.pone.0123470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spies U, Müller H, Becht H. 1989. Nucleotide sequence of infectious bursal disease virus genome segment A delineates two major open reading frames. Nucleic Acids Res 17:7982–7982. doi: 10.1093/nar/17.19.7982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kibenge FS, Jackwood DJ, Mercado CC. 1990. Nucleotide sequence analysis of genome segment A of infectious bursal disease virus. J Gen Virol 71:569–577. doi: 10.1099/0022-1317-71-3-569. [DOI] [PubMed] [Google Scholar]
- 10.Lejal N, Da Costa B, Huet J-C, Delmas B. 2000. Role of Ser-652 and Lys-692 in the protease activity of infectious bursal disease virus VP4 and identification of its substrate cleavage sites. J Gen Virol 81:983–992. doi: 10.1099/0022-1317-81-4-983. [DOI] [PubMed] [Google Scholar]
- 11.Lee CC, Ko TP, Chou CC, Yoshimura M, Doong SR, Wang MY, Wang AH. 2006. Crystal structure of infectious bursal disease virus VP2 subviral particle at 2.6 Å resolution: implications in virion assembly and immunogenicity. J Struct Biol 155:74–86. doi: 10.1016/j.jsb.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 12.Ursula I, Gorbalenya AE, Schirrmeier H, Behrens S-E, Letzel T, Mundt E. 2004. VP1 of infectious bursal disease virus is an RNA-dependent RNA polymerase. J Gen Virol 85:2221–2229. doi: 10.1099/vir.0.19772-0. [DOI] [PubMed] [Google Scholar]
- 13.Müller H, Nitschke R. 1987. The two segments of the infectious bursal disease virus genome are circularized by a 90,000-Da protein. Virology 159:174–177. doi: 10.1016/0042-6822(87)90363-1. [DOI] [PubMed] [Google Scholar]
- 14.Calvert JG, Nagy E, Soler M, Dobos P. 1991. Characterization of the VPg-dsRNA linkage of infectious pancreatic necrosis virus. J Gen Virol 72:2563–2567. doi: 10.1099/0022-1317-72-10-2563. [DOI] [PubMed] [Google Scholar]
- 15.Spies U, Muller H. 1990. Demonstration of enzyme activities required for cap structure formation in infectious bursal disease virus, a member of the birnavirus group. J Gen Virol 71:977–981. doi: 10.1099/0022-1317-71-4-977. [DOI] [PubMed] [Google Scholar]
- 16.Dobos P. 1993. In vitro guanylylation of infectious pancreatic necrosis virus polypeptide VP1. Virology 193:403–413. doi: 10.1006/viro.1993.1137. [DOI] [PubMed] [Google Scholar]
- 17.Tacken MG, Peeters BP, Thomas AA, Rottier PJ, Boot HJ. 2002. Infectious bursal disease virus capsid protein VP3 interacts both with VP1, the RNA-dependent RNA polymerase, and with viral double-stranded RNA. J Virol 76:11301–11311. doi: 10.1128/JVI.76.22.11301-11311.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lombardo E, Maraver A, Castón JR, Rivera J, Fernández-Arias A, Serrano A, Carrascosa JL, Rodriguez JF. 1999. VP1, the putative RNA-dependent RNA polymerase of infectious bursal disease virus, forms complexes with the capsid protein VP3, leading to efficient encapsidation into virus-like particles. J Virol 73:6973–6983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ferrero D, Garriga D, Navarro A, Rodríguez JF, Verdaguer N. 2015. Infectious bursal disease virus VP3 upregulates VP1-mediated RNA-dependent RNA replication. J Virol 89:11165–11168. doi: 10.1128/JVI.00218-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Busnadiego I, Maestre AM, Rodríguez D, Rodríguez JF. 2012. The infectious bursal disease virus RNA-binding VP3 polypeptide inhibits PKR-mediated apoptosis. PLoS One 7:e46768. doi: 10.1371/journal.pone.0046768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ye C, Jia L, Sun Y, Hu B, Wang L, Lu X, Zhou J. 2014. Inhibition of antiviral innate immunity by birnavirus VP3 protein via blockage of viral double-stranded RNA binding to the host cytoplasmic RNA detector MDA5. J Virol 88:11154–11165. doi: 10.1128/JVI.01115-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Richter JD, Sonenberg N. 2005. Regulation of cap-dependent translation by elF4E inhibitory proteins. Nature 433:477. doi: 10.1038/nature03205. [DOI] [PubMed] [Google Scholar]
- 23.Lee KM, Chen CJ, Shih SR. 2017. Regulation mechanisms of viral IRES-driven translation. Trends Microbiol 25:546–561. doi: 10.1016/j.tim.2017.01.010. [DOI] [PubMed] [Google Scholar]
- 24.Boot HJ, Pritz-Verschuren SB. 2004. Modifications of the 3′-UTR stem–loop of infectious bursal disease virus are allowed without influencing replication or virulence. Nucleic Acids Res 32:211–222. doi: 10.1093/nar/gkh177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Palmer TD, Miller AD, Reeder RH, McStay B. 1993. Efficient expression of a protein coding gene under the control of an RNA polymerase I promoter. Nucleic Acids Res 21:3451–3457. doi: 10.1093/nar/21.15.3451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rehwinkel J, Tan CP, Goubau D, Schulz O, Pichlmair A, Bier K, Robb N, Vreede F, Barclay W, Fodor E, Reis e Sousa C. 2010. RIG-I detects viral genomic RNA during negative-strand RNA virus infection. Cell 140:397–408. doi: 10.1016/j.cell.2010.01.020. [DOI] [PubMed] [Google Scholar]
- 27.Jackson RJ, Hellen CU, Pestova TV. 2010. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol 11:113. doi: 10.1038/nrm2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kabrane-Lazizi Y, Meng X-J, Purcell RH, Emerson SU. 1999. Evidence that the genomic RNA of hepatitis E virus is capped. J Virol 73:8848–8850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brown EA, Zajac AJ, Lemon SM. 1994. In vitro characterization of an internal ribosomal entry site (IRES) present within the 5′ nontranslated region of hepatitis A virus RNA: comparison with the IRES of encephalomyocarditis virus. J Virol 68:1066–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takahashi K, Yamanaka S. 2006. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 31.Valli A, Busnadiego I, Maliogka V, Ferrero D, Castón JR, Rodríguez JF, García JA. 2012. The VP3 factor from viruses of Birnaviridae family suppresses RNA silencing by binding both long and small RNA duplexes. PLoS One 7:e45957. doi: 10.1371/journal.pone.0045957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maraver A, Clemente R, Rodríguez JF, Lombardo E. 2003. Identification and molecular characterization of the RNA polymerase-binding motif of infectious bursal disease virus inner capsid protein VP3. J Virol 77:2459–2468. doi: 10.1128/JVI.77.4.2459-2468.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pan J, Lin L, Tao YJ. 2009. Self-guanylylation of birnavirus VP1 does not require an intact polymerase activity site. Virology 395:87–96. doi: 10.1016/j.virol.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Niedzwiecka A, Marcotrigiano J, Stepinski J, Jankowska-Anyszka M, Wyslouch-Cieszynska A, Dadlez M, Gingras A-C, Mak P, Darzynkiewicz E, Sonenberg N. 2002. Biophysical studies of eIF4E cap-binding protein: recognition of mRNA 5′ cap structure and synthetic fragments of eIF4G and 4E-BP1 proteins. J Mol Biol 319:615–635. doi: 10.1016/S0022-2836(02)00328-5. [DOI] [PubMed] [Google Scholar]
- 35.Vanhaesebroeck B, Leevers SJ, Ahmadi K, Timms J, Katso R, Driscoll PC, Woscholski R, Parker PJ, Waterfield MD. 2001. Synthesis and function of 3-phosphorylated inositol lipids. Annu Rev Biochem 70:535–602. doi: 10.1146/annurev.biochem.70.1.535. [DOI] [PubMed] [Google Scholar]
- 36.Mundt E, Vakharia VN. 1996. Synthetic transcripts of double-stranded Birnavirus genome are infectious. Proc Natl Acad Sci U S A 93:11131–11136. doi: 10.1073/pnas.93.20.11131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abdeljelil NB, Khabouchi N, Mardassi H. 2008. Efficient rescue of infectious bursal disease virus using a simplified RNA polymerase II-based reverse genetics strategy. Arch Virol 153:1131–1137. doi: 10.1007/s00705-008-0080-3. [DOI] [PubMed] [Google Scholar]
- 38.Dalton RM, Rodríguez JF. 2014. Rescue of Infectious Birnavirus from recombinant ribonucleoprotein complexes. PLoS One 9:e87790. doi: 10.1371/journal.pone.0087790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goodfellow I, Chaudhry Y, Gioldasi I, Gerondopoulos A, Natoni A, Labrie L, Laliberté JF, Roberts L. 2005. Calicivirus translation initiation requires an interaction between VPg and eIF4E. EMBO Rep 6:968–972. doi: 10.1038/sj.embor.7400510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mir MA, Sheema S, Haseeb A, Haque A. 2010. Hantavirus nucleocapsid protein has distinct m7G cap-and RNA-binding sites. J Biol Chem 285:11357–11368. doi: 10.1074/jbc.M110.102459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Garriga D, Navarro A, Querol-Audí J, Abaitua F, Rodríguez JF, Verdaguer N. 2007. Activation mechanism of a noncanonical RNA-dependent RNA polymerase. Proc Natl Acad Sci U S A 104:20540–20545. doi: 10.1073/pnas.0704447104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mosley YY, Wu CC, Lin TL. 2016. IBDV particles packaged with only segment A dsRNA. Virology 488:68–72. doi: 10.1016/j.virol.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 43.Hinnebusch AG. 2012. Translational homeostasis via eIF4E and 4E-BP1. Mol Cell 46:717–719. doi: 10.1016/j.molcel.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 44.Gao L, Li K, Zhong L, Zhang L, Qi X, Wang Y, Gao Y, Wang X. 2017. Eukaryotic translational initiation factor 4AII reduces the replication of infectious bursal disease virus by inhibiting VP1 polymerase activity. Antiviral Res 139:102–111. doi: 10.1016/j.antiviral.2016.11.022. [DOI] [PubMed] [Google Scholar]
- 45.Tacken MG, Thomas AA, Peeters B, Rottier PJ, Boot HJ. 2004. VP1, the RNA-dependent RNA polymerase and genome-linked protein of infectious bursal disease virus, interacts with the carboxy-terminal domain of translational eukaryotic initiation factor 4AII. Arch Virol 149:2245–2260. doi: 10.1007/s00705-004-0365-0. [DOI] [PubMed] [Google Scholar]
- 46.Yu L, Li J, Huang Y, Dikki J, Deng R. 2001. Molecular characteristics of full-length genomic segment A of three infectious bursal disease viruses in China: two attenuated strains and one virulent field strain. Avian Dis 45:862–874. doi: 10.2307/1592866. [DOI] [PubMed] [Google Scholar]
- 47.Pattison M, Alexander DJ, Harkness JW. 1975. Purification and preliminary characterisation of a pathogenic strain of infectious bursal disease virus. Avian Pathol 4:175–187. doi: 10.1080/03079457509353864. [DOI] [PubMed] [Google Scholar]
- 48.Boot HJ, ter Huurne AHM, Peeters BPH. 2000. Generation of full-length cDNA of the two genomic dsRNA segments of infectious bursal disease virus. J Virol Methods 84:49–58. doi: 10.1016/S0166-0934(99)00132-9. [DOI] [PubMed] [Google Scholar]
- 49.Soni A, Akcakanat A, Singh G, Luyimbazi D, Zheng Y, Kim D, Gonzalez-Angulo A, Meric-Bernstam F. 2008. eIF4E knockdown decreases breast cancer cell growth without activating Akt signaling. Mol Cancer Ther 7:1782–1788. doi: 10.1158/1535-7163.MCT-07-2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ye C, Han X, Yu Z, Zhang E, Wang L, Liu H. 2017. Infectious bursal disease virus activates c-Src to promote α4β1 integrin-dependent viral entry via modulating downstream Akt-RhoA GTPase-actin rearrangement cascade. J Virol 91:e01891-. doi: 10.1128/JVI.01891-16. [DOI] [PMC free article] [PubMed] [Google Scholar]