

ABSTRACT
Astroviruses, members of the family Astroviridae, represent an important cause of human gastroenteritis in the world. The cellular factors required for astrovirus replication have been poorly studied. In this work, we evaluated the relevance of the ubiquitin-proteasome system (UPS) in the replication of Yuc8, a human astrovirus serotype 8 strain. We found that proteasome inhibitors decrease the production of infectious viral progeny at a step in the replication cycle subsequent to virus entry. The inhibition of proteasome activity decreases viral RNA levels and viral protein synthesis; similarly, the inhibition of ubiquitination by chemical inhibitors or RNA interference (RNAi) reduces the production of viral progeny as well as viral protein synthesis. The effect on viral progeny production induced by proteasome inhibitors is not explained by a reduction in the pool of monoubiquitin or the induction of early apoptosis or autophagy. Our observations are consistent with the need of the proteolytic activity of the UPS for the efficient replication of the virus and suggest that UPS is necessary for the production of genomic and subgenomic RNA but not for antigenomic RNA.
IMPORTANCE Astroviruses are a major cause of gastroenteritis in young humans and animals, and recently, it was associated with fatal encephalitis in humans. The role of the ubiquitin-proteasome system in the replication of these viruses has not been studied previously. In this work, we present evidence that supports that the proteolytic activity of the proteasome is necessary for efficient viral progeny production and that this proteolytic system is required for the accumulation of both genomic and subgenomic viral RNAs.
KEYWORDS: RNA replication, astrovirus, proteasome, viral replication
INTRODUCTION
Astroviruses are nonenveloped, single-stranded, positive RNA viruses grouped into the family Astroviridae. Human astroviruses (HAstVs) are grouped into three clades, one comprising the classical HAstVs (8 serotypes), and the so-called novel astroviruses that include two clades, MLB and VA (1). HAstVs cause acute gastroenteritis and are associated with 2 to 9% of acute, nonbacterial diarrheal cases in children (2) and were recently associated with neurological disorders in immunocompromised individuals (3, 4).
The genome of HAstV is formed by one single-stranded, polyadenylated RNA molecule of approximately 6.8 kb. The 5′ end of the genomic RNA (gRNA) is covalently attached to a VPg protein that is required for efficient viral infection (5). The genome harbors three open reading frames (ORFs). ORF1a and ORF1b encode the nonstructural proteins, while ORF2 codes for the structural polyprotein (6). HAstV-8 enters the cell by a clathrin-dependent mechanism, and after reaching the cytoplasm, the gRNA of the incoming viral particle directs the synthesis of two polyprotein precursors, NSP1a and NSP1ab (7). The polyprotein precursors are processed by viral and cellular proteases to generate several protein products. The NSP1a precursor yields the viral protease and VPg as well as other not-well-characterized proteins. NSP1ab yields the same proteins but also the viral RNA-dependent RNA polymerase (RdRp) encoded in ORF1b. The incoming gRNA is used as the template by RdRp to synthesize the antigenomic RNA (agRNA), which in turn serves as the template for the production of two kinds of positive-stranded RNAs, the gRNA and the subgenomic RNA (sgRNA) (6). The synthesized gRNA is used as mRNA to produce more nonstructural proteins, and it is also packed as the viral genome of the newly assembled viral particles (2). The sgRNA is used as mRNA for the production of the capsid polyprotein VP90. The replication and morphogenesis of the viral particles are associated with membranes (8, 9). The viral particles are released by a nonlytic mechanism that requires processing by cellular caspases of the full-length capsid polyprotein VP90 to yield VP70 (10, 11). VP70 in the extracellular viruses is then processed by trypsin into the final three capsid proteins that constitute the mature, infectious virus.
The ubiquitin-proteasome system (UPS) is the major degradation system in the cell. Major roles of this system consist of the degradation of misfolded/damaged proteins (12, 13) and the regulation of the level of proteins related to the control of cell cycle progression, protein trafficking, transcription, the immune response, and signal transduction (14, 15). The UPS is formed by two major components: the first is the proteasome, a multiprotein complex that degrades proteins, and the second component is ubiquitin, a highly conserved protein of 76 amino acids that is covalently attached to target proteins through a three-step reaction, known as ubiquitination. Protein ubiquitination is a posttranslational modification involved in several cellular functions; one of them is the delivery of the ubiquitin-tagged proteins to the proteasome for degradation. Depending on the nature of ubiquitination, this modification can modify other aspects of protein function independently of protein degradation, such as protein-protein interactions (16) and sorting of transmembrane proteins (17). The proteasome is formed by two components, a proteolytic core particle, the 20S (720-kDa) proteasome, sandwiched between two 19S (890-kDa) “cap” regulatory complexes. The two components form the 26S proteasome, which is specialized in the degradation of ubiquitinated proteins. Versions of the proteasome, with caps other than 19S (for example, members of the activator families 11S and Blm10), can degrade nonubiquitinated proteins (14, 18).
As the UPS has a vital role in many fundamental cellular processes, many viruses have reprogrammed this machinery according to their needs. The interactions between viruses and the UPS are complex; one of the more common features is the use of the UPS to control the level of antiviral factors and to regulate innate antiviral signaling (19). In the case of viruses that depend on the cell cycle, this system is used to control the level of proteins that regulate cell cycle progression, a feature observed frequently in DNA virus infections (20). It has been reported that the UPS regulates the replication of several viruses, including rotavirus (21, 22), West Nile virus (23), circovirus (24), human hepatitis E virus (25), human immunodeficiency virus, influenza virus (26), vaccinia virus, and adenovirus (20). In the above-mentioned examples, the inhibition of the UPS had a negative impact on viral replication, but in other cases, the inhibition of the UPS favors virus replication, as in the case of papaya ringspot virus (27), or the enhancement of adeno-associated virus transduction (28). Here, we report that the function of this system is required for the efficient replication of HAstV, most likely for the synthesis of viral gRNA and sgRNA.
RESULTS
Proteasome inhibitors reduce the production of astrovirus progeny.
To determine if the proteolytic activity of the proteasome is required for HAstV replication, the effect of a set of proteasome inhibitors on the production of viral progeny was evaluated. For this, C2Bbe1 cells (29) were infected with Yuc8, a serotype 8 HAstV strain, and at the end of the adsorption period, the cells were washed and the indicated concentrations of drugs were added. The cells were then collected at 18 h postinfection (hpi), and the viral titers were evaluated by an immunoperoxidase focus-forming assay (Fig. 1A). The efficiency of drugs used was confirmed by Western blotting (Fig. 1B). As expected, proteasomal inhibitors caused an accumulation of ubiquitinated proteins and increased the expression of hsp70 (30–32). Since MG132 was the most efficient inhibitor, reducing the yield of viral progeny by more than 10-fold, its effect on virus replication was further characterized, and the dose-effect of this compound on viral replication was determined (Fig. 1C). The production of viral progeny was inhibited by MG132 in a dose-response manner with a 50% inhibitory concentration (IC50) of 26 nM. To discard the possibility that the reduction of the virus yield was due to toxicity produced by MG132, cell viability was evaluated by using a lactate dehydrogenase (LDH) release assay; at the doses and times tested, MG132 was not toxic for infected or mock-infected cells (data not shown).
FIG 1.

Proteasome inhibitors reduce the production of viral progeny. C2Bbe1 cells were infected with HAstV Yuc8 at an MOI of 3. At the end of the adsorption period, the cells were washed, and drugs were added at the concentrations described below. At 18 hpi, cell cultures were collected and analyzed. (A) The cells and media were collected together, and the viral titer was determined by a peroxidase focus-forming assay, as described in Materials and Methods. The data are expressed as focus-forming units (ffu) per milliliter and represent the arithmetic means ± standard errors of the means of results from three independent experiments performed in triplicate. The concentrations used were 1 μM MG132, 10 μM lactacystin, and 20 μM PSI. ***, P = 0.0007; **, P = 0.0022. (B) Yuc8-infected C2Bbe1 cells were incubated for 18 h with 1 μM MG132 (MG), 10 μM lactacystin (Lact), 20 μM PSI, or dimethyl sulfoxide (DMSO) (control [C]). Total proteins were collected in Laemmli buffer, resolved by SDS-PAGE, transferred to nitrocellulose membranes, and incubated with the indicated antibodies. Ub, ubiquitin. (C) Infected cells were incubated with the indicated concentrations of MG132, and the production of viral progeny was determined as described above. The data are expressed as focus-forming units per milliliter and represent the arithmetic means ± standard errors of the means of results from four independent experiments performed in duplicate. ***, P = 0.0007; ****, P = 0.0002.
MG132 interferes with an early step during the HAstV replication cycle.
To define the step at which MG132 affects the replication cycle of HAstV, the effect of the addition of the drug at different times postadsorption on the production of viral progeny was determined. As shown in Fig. 2A, the drug inhibited the yield of viral progeny by more than 1 logarithm when added at the end of the adsorption period (time zero) or at 4 h postadsorption (hpa), while its effect was significantly reduced when MG132 was added at 8 hpa and later on. Furthermore, when MG132 was added 3 h before and during the adsorption period (−4to0 in Fig. 2A) and removed after this 4-h incubation time, viral progeny production was not affected. Taken together, these results suggest that virus entry is not affected by proteasome inhibition but that a relatively early step during viral replication requires an active proteasome.
FIG 2.
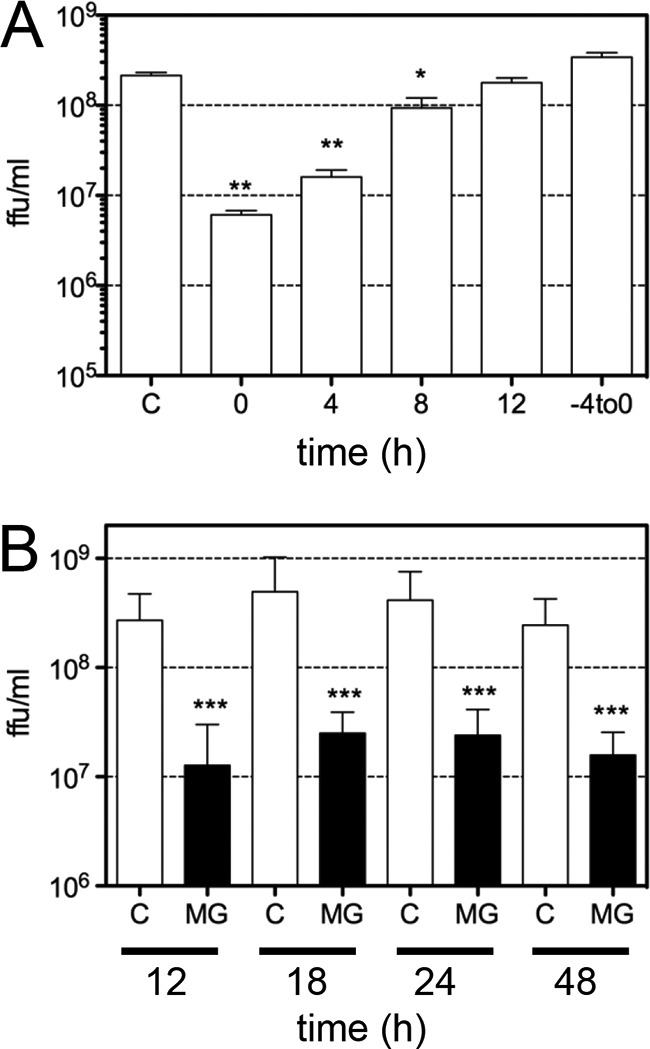
The proteasome inhibitor MG132 affects an early step of HAstV replication. C2Bbe1 cells were infected with HAstV Yuc8 at an MOI of 3, and 1 μM MG132 was added at the indicated times during infection. Viral progeny production was evaluated as described in Materials and Methods. (A) MG132 was not added (C) or was added at the indicated times postadsorption, and at 18 hpi, the cells and medium were collected together, and the amount of infectious viral particles was quantitated. “−4to0” indicates that MG132 was present for a period of only 4 h (3 h before adsorption and during the 1-h adsorption period), and after this time, the drug was washed away. Data are expressed as focus-forming units per milliliter and represent the arithmetic means ± standard errors of the means of results from three independent experiments performed in duplicate. **, P = 0.0040; *, P = 0.0162. (B) C2Bbe1 cells were infected with HAstV Yuc8 at an MOI of 3. At the end of the adsorption period, the cells were washed, 1 μM MG132 was added, the cells and media were collected together at the indicated times, and the viral titer was determined by a focus-forming assay, as described in Materials and Methods. The data are expressed as focus-forming units per milliliter and represent the arithmetic means ± standard errors of the means of results from three independent experiments performed in triplicate. ***, P = 0.0007. Open bars are control, dimethyl sulfoxide-treated cells (C), and closed bars are MG132 (MG)-treated cells.
It has been reported that MG132 can differentially affect the replication of viruses; for example, proteasome inhibitors block the replication of vesicular stomatitis virus (VSV) but only delay poliovirus growth (33). To determine its effect on astrovirus replication, we evaluated the viral progeny produced from 12 to 48 hpi in cells in which MG132 was maintained in the medium during then entire incubation period; the production of infectious virus was inhibited at similar levels at all times tested (Fig. 2B), suggesting that the replication of the virus was blocked and not delayed by MG132 treatment.
Proteasome inhibition blocks the synthesis of viral proteins.
As mentioned above, the synthesis of the capsid polyprotein precursor is directed by the sgRNA, while the nonstructural proteins are synthesized from the gRNA. In order to evaluate the effect of proteasome inhibition on the accumulation of viral proteins, C2Bbe1 cells were treated with 1 μM MG132 and analyzed by Western blotting. As shown in Fig. 3A and B, the accumulation of both the capsid and the protease decreased by 80 and 60%, respectively, suggesting that the activity of the proteasome is required for the synthesis of proteins derived from both gRNA and sgRNA. On the other hand, it was previously reported that MG132 can induce an arrest in cell translation (22, 33, 34); thus, to determine if this was the case in C2Bbe1 cells, the incorporation of [35S]Met into cellular proteins in the presence of increasing concentrations of MG132 was evaluated. As shown in Fig. 3C and D, MG132 did not significantly affect the synthesis of cellular proteins under the conditions used, suggesting that the drug specifically inhibits protein synthesis from both positive-stranded viral RNAs.
FIG 3.

Expression of viral proteins is inhibited by MG132. C2Bbe1 cells were infected with HAstV Yuc8 at an MOI of 3, and at the end of the adsorption period, MG132 was added at the indicated concentrations. The proteins were metabolically labeled with [35S]methionine-cysteine for 1 h at 17 hpi, the cells were harvested at 18 hpi in Laemmli sample buffer, and the proteins were resolved by SDS-PAGE. (A) Western blot analysis of the indicated proteins from infected or mock-infected cells in the presence or absence of 1 μM MG132. Ab, antibody. (B) Quantitation of the normalized level of expression of the indicated viral protein from infected dimethyl sulfoxide (C)- or MG132 (MG)-treated cells using tubulin as a loading control. Data are expressed as a percentage of the value for control (infected, dimethyl sulfoxide-treated) cells and represent the means ± standard errors of the means of results from five independent experiments. ***, P < 0.001. (C) Coomassie blue staining of the gel in panel D. (D) Autoradiography of metabolically labeled cellular proteins in the presence of the indicated concentrations of MG132.
Ubiquitination is required for HAstV replication.
The proteasome degrades ubiquitinated proteins, but several proteasome substrates can be degraded in a ubiquitin-independent way (14). If the activity of the proteasome during HAstV replication depends on ubiquitination, it is expected that the inhibition of ubiquitination will affect the replication of HAstV. To evaluate this possibility, the effect of Pyr-41, an inhibitor of the E1 ubiquitin-activating enzyme, on the generation of viral progeny and viral protein synthesis was tested. In the presence of this inhibitor, de novo ubiquitination does not occur (35). Figure 4A shows that treatment of the cells with Pyr-41 reduced the viral progeny titer in a dose-response manner, with an IC50 of 24 μM. At the doses and under the conditions tested, this drug was not toxic to the cells, as judged by an LDH assay (data not shown). The efficiency of Pyr-41 was confirmed by Western blotting in which ubiquitinated proteins and the accumulation of hsp70 were detected (Fig. 4B), since it has been reported that Pyr-41 causes an accumulation of ubiquitinated proteins and an increase in the expression of hsp70 (36). As in the case of MG132, the syntheses of the capsid protein and the viral protease were reduced by about 80% in Pyr-41-treated cells (Fig. 4C and D).
FIG 4.

Pyr-41, an inhibitor of E1-activating enzymes, reduces viral progeny production and the synthesis of viral proteins. C2Bbe1 cells were infected with HAstV Yuc8 at an MOI of 3. At the end of the adsorption period, Pyr-41 was added at the indicated concentrations, and cells were then processed at 18 hpi. (A) Cells and medium were collected together, and the titer of the viral progeny produced was evaluated. The data are expressed as focus-forming units per milliliter and represent the means of results from three independent experiments performed in duplicate ± standard errors of the means. *, P ≤ 0.05; **, P ≤ 0.01. (B) Yuc8-infected C2Bbe1 cells were incubated for 18 h with 100 μM Pyr-41 (Pyr) or dimethyl sulfoxide (C). Total proteins collected in Laemmli buffer were resolved by SDS-PAGE and transferred onto nitrocellulose membranes for detection of the indicated proteins. (C) Western blot analysis of the indicated proteins from infected or mock-infected cells in the presence or absence of 100 μM Pyr-41. The proteins were collected in Laemmli sample buffer, resolved by SDS-PAGE, transferred onto a nitrocellulose membrane, and stained with the indicated antibodies. Immunodetection was performed by using Alexa 647-conjugated secondary antibodies. (D) Quantification of normalized values (using tubulin as a loading control) of expression for the indicated viral proteins in infected dimethyl sulfoxide (C)- or Pyr-41-treated cells. Data are expressed as a percentage of values for the control (dimethyl sulfoxide-treated cells) and represent the means ± standard errors of the means of results from five independent experiments. ***, P < 0.001.
As an alternative approach to evaluate the role of ubiquitination in HAstV replication, the expression of the ubc gene, one of the four genes that encode ubiquitin (37), was silenced by RNA interference. A small interfering RNA (siRNA) pool directed against ubc was transfected into cells, and viral replication was evaluated. When the viral progeny produced in control and ubiquitin-silenced cells were compared, a reduction of more than 95% was observed, a level similar to that obtained when cells were treated with MG132 or Pyr-41 (Fig. 1A and 4A). Under these conditions, the synthesis of the viral protease and the capsid was also noticeably reduced (Fig. 5B and C). The efficiency of interference was evaluated by Western blotting (Fig. 5D). Under these conditions, a reduction of more than 95% in the accumulation of ubiquitin was observed, as judged by the level of monoubiquitin molecules detected, i.e., the pool of free ubiquitin plus the ubiquitin associated with E1 and E2 enzymes. The reduction in the amount of ubiquitin was not toxic to the cells, as judged by an LDH release assay (data not shown), nor did it affect the synthesis of cellular proteins (Fig. 5D). Taken together, these results suggest that ubiquitination is necessary for the efficient replication of HAstV.
FIG 5.

Interference of ubiquitin (Ub) synthesis reduces the amount of viral progeny produced and viral protein synthesis. C2Bbe1 cells were transfected with either an siRNA directed to the ubc gene or an irrelevant control (NT) or were untransfected (UT), and at 48 h posttransfection, cells were infected with HAstV Yuc8 at an MOI of 3. (A) Cells and media were collected together at 18 hpi, and the viral titer was determined by a peroxidase focus-forming assay, as described in Materials and Methods. Data are expressed as focus-forming units per milliliter and represent the arithmetic means ± standard errors of the means of results from three independent experiments performed in duplicate. **, P = 0.004. (B) Proteins collected at 18 hpi in Laemmli sample buffer were resolved by SDS-PAGE and processed for immunodetection using the indicated antibodies. (C) Quantification of the normalized value (using tubulin as a loading control) of expression of the indicated viral proteins of infected cells transfected with an siRNA with no target in the human genome (NT) or ubiquitin-targeting siRNA. Data are expressed as a percentage of the value for control cells (transfected with NT siRNA) and represent the means ± standard errors of the means of results from three independent experiments. *, P < 0.02; ***, P < 0.001. (D) Cells transfected with the indicated siRNAs were metabolically labeled with [35S]methionine-cysteine for 1 h at 17 hpi and then collected in Laemmli sample buffer and resolved by SDS-PAGE. Data from Coomassie blue staining, autoradiography, and immunodetection of ubiquitin (anti-Ub) are shown. In order to detect monoubiquitin, the proteins were resolved on Tris-Tricine gels. (E) Incubation of cells with MG132 does not decreases the pool of monomeric ubiquitin. C2Bbe1 cells were infected with HAstV Yuc8 at an MOI of 3. At the end of the adsorption period, 1 μM MG132 was added or not, and the cells were incubated for 18 h at 37°C, lysed in Laemmli sample buffer, and resolved in Tris-Tricine gels for immunodetection of viral capsid or ubiquitin by Western blotting. The position of the migration of monomeric ubiquitin is indicated with an arrow.
When cells are incubated with proteasome inhibitors for long periods of time, ubiquitin is not recycled, and it is has been shown that the pool of ubiquitin available for ubiquitination can be reduced (22, 38). Under these conditions, proteasome inhibitors could affect viral replication not by a direct effect on proteolysis but by an indirect effect on the ubiquitination of proteins required for viral replication. To explore this possibility, the level of monoubiquitin in control and MG132-treated cells was evaluated by Western blotting. In MG132-treated cells, the pool of monoubiquitin was not reduced but instead was augmented, and, as expected, the level of ubiquitinated proteins was increased (Fig. 5E). These data suggest that the effect of proteasome inhibitors on HAstV replication is more likely due to the proteolytic component of the UPS. However, the possibility that the ubiquitination of proteins, not related to proteolysis, could be necessary for virus replication cannot be ruled out.
The reduction in the amount of viral progeny caused by proteasome inhibition is not related to the induction of autophagy or apoptosis.
Long exposure of cells to proteasomal inhibitors can induce apoptosis or autophagy, and autophagy proteins can target viral components or virions for lysosomal degradation, a process termed xenophagy (39). In addition, HAstV infection induces apoptosis, an event related to virus release (10, 11, 40), but at early times during infection, apoptosis could reduce viral production. To evaluate if the reduction in the amount of viral progeny observed in the presence of MG132 was a consequence of the induction of apoptosis or autophagy, the effects of bafilomycin, an inhibitor of the vacuolar proton ATPase, and of carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]-fluoromethylketone (Z-VAD-fmk), a caspase inhibitor, on the virus yield were evaluated. If autophagy or apoptosis was responsible for the inhibition of viral replication induced by MG132, it would be expected that these drugs would revert the inhibition observed. As shown in Fig. 6, neither bafilomycin nor Z-VAD-fmk reversed the reduction in the amount of viral progeny. The drugs were confirmed to efficiently exert their actions by evaluation of the effect of bafilomycin on the infectivity of HAstV and of Z-VAD-fmk on the processing of the capsid protein, as reported previously (7, 10, 11) (data not shown). These results support the conclusion that proteasomal activity is required for HAstV replication.
FIG 6.
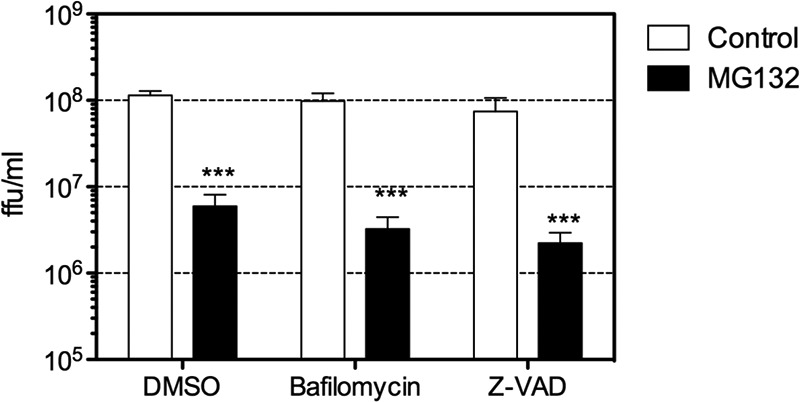
The decrease in the amount of viral progeny caused by MG132 is not related to the induction of autophagy or apoptosis. Cells were infected with Yuc8 at an MOI of 3, and at the end of the adsorption period, the indicated drugs were added. At 20 hpi, the cells were collected and lysed by three freeze-thaw cycles, and the viral titer was determined by a focus-forming assay, as described in Materials and Methods. The data are expressed as focus-forming units per milliliter and represent the arithmetic means ± standard errors of the means of data from three independent experiments performed in duplicate. ***, P = 0.0007. Drug concentrations used were 1 μM MG132, 50 nM bafilomycin, and 50 μM Z-VAD.
The proteasome inhibitor MG132 reduces the production of genomic and subgenomic RNAs.
The reduction in the synthesis of viral proteins observed in the presence of MG132 could be due to an effect on the efficiency of translation of viral gRNA and sgRNA or to a reduction in the level of these RNAs, with the consequent reduction of viral protein synthesis. To discern between these two alternatives, we evaluated the levels of gRNA and agRNAs in control and MG132-treated cells by real-time reverse transcription-quantitative PCR (qRT-PCR). As shown in Fig. 7, the level of gRNA was reduced to about 10% in MG132-treated cells at 12 hpi, and it was increased to about 28% at 24 hpi, with respect to the levels of gRNA observed in mock-treated control cells. In contrast, the level of agRNA did not change at 12 hpi and was reduced by only about 60% at 24 hpi. The reduction in the amount of agRNA observed at 24 hpi could be explained given that the template for its synthesis, the gRNA, appears to be diminished at 12 hpi, and a drop in the gRNA level will eventually result in decreased agRNA synthesis. These data suggest that the step inhibited by MG132 could be the production of gRNA and, as a consequence, the level of agRNA at late times during infection.
FIG 7.
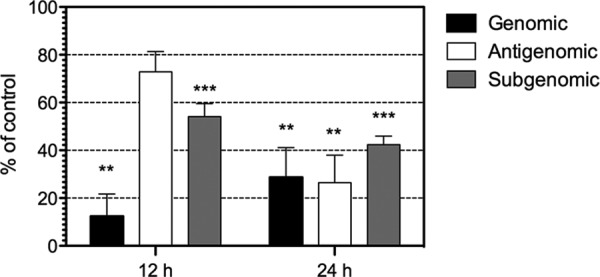
MG132 reduces the amount of viral RNAs. C2Bbe1 cells were infected with HAstV Yuc8 at an MOI of 3. At the end of the adsorption period, 1 μM MG132 or dimethyl sulfoxide was added, the cells were incubated for the indicated periods of time, and total RNA was isolated. The levels of genomic, antigenomic, and subgenomic (ORF2) RNAs were determined by a qRT-PCR assay, as described in Materials and Methods. Data are expressed as a percentage of the value for dimethyl sulfoxide-treated cells as a control, which was taken as 100%, and represent the means ± standard errors of the means of data from three (genomic and antigenomic) or five (subgenomic) independent experiments. *, P < 0.05.
Since the synthesis of the capsid protein was reduced by MG132 (Fig. 3A and B), it is possible that, as observed for the gRNA, the level of sgRNA could also be affected in MG132-treated cells. To evaluate the level of sgRNA, we performed qRT-PCR using primers designed to amplify the ORF2 region of the two positive-stranded viral RNAs, as opposed to the primers designed to measure the level of gRNA, which amplify a region located in ORF1b. As shown in Fig. 7, the level of RNA detected was reduced by about 50% at both 12 and 24 hpi in MG132-treated cells. Given that the sequence of ORF2 is present in both the sgRNA and the gRNA, it is not possible to differentiate between the two RNAs by qRT-PCR. However, based on the differences between the threshold cycle (CT) values for the amplification of the ORF1b (gRNA) and the ORF2 (gRNA plus sgRNA) regions, we determined that the quantity of sgRNA represents about 90% of the RNA evaluated with the ORF2 primers. Thus, this indicates that the level of the ORF2 region detected by qRT-PCR represents mostly the level of sgRNA.
The inhibition of the proteasome does not affect the expression of the viral protease at early times postinfection.
The data presented above suggest that the production of positive-sense RNAs but not of negative-sense RNAs is inhibited by MG132. In this regard, it is important to take into consideration that the first round of nonstructural viral protein synthesis is directed by the incoming genomic RNA present in the original viral particle that infected the cell. Thus, one prediction from our data is that at early times of infection, the level of expression of nonstructural proteins (produced by the translation of the original gRNA) is not affected by drug treatment. To evaluate this hypothesis, we used a multiplicity of infection (MOI) of 50 to infect C2Bbe1 cells and detected the level of viral protease at 4, 6, 8, and 10 hpi. Although difficult to detect due to the small amount of viral protein produced, at 4 hpi, the level of viral protease was not observed to be reduced by MG132, while a reduction in its accumulation was found at later times (Fig. 8A and B). These results suggest that the reduction in the synthesis of gRNA and sgRNA observed in the presence of MG132 is not related, at least at early times of infection, to a failure in the production of the viral nonstructural proteins, including the RdRp. Furthermore, the abundance of the VP90 structural protein, which is translated from the sgRNA, is reduced from the initial time when it can be detected, at 6 hpi (Fig. 8A and C). Altogether, these data suggest that the inhibition of the proteasome activity does not affect directly the translation of viral mRNAs but one step related to the accumulation of the viral positive-sense RNAs.
FIG 8.

The level of expression of the viral protease is not reduced by MG132 at early times of infection. C2Bbe1 cells were infected with HAstV Yuc8 at an MOI of 50. At the end of the adsorption period, 1 μM MG132 or dimethyl sulfoxide was added, the cells were incubated for the indicated periods of time, and cells were then collected in Laemmli sample buffer and resolved by SDS-PAGE. (A) Representative Western blot analyses using the indicated antibodies and controls of uninfected cells (MG132 or dimethyl sulfoxide treated) are included. (B and C) Densitometric analysis of Western blots. Graphs show the normalized values (using tubulin as a loading control) of expression of the viral protease (B) and the capsid protein (C) compared to control (dimethyl sulfoxide-treated) cells. Data are expressed as a percentage of values for the control (nontreated cells) and represent the means ± standard errors of the means of results from three independent experiments. *, P < 0.05; **, P < 0.01.
DISCUSSION
The proteasome is the major protein degradative system in the cytoplasm. In this work, we show that proteasomal inhibitors and the inhibition of cellular ubiquitination result in the reduction of the production of astroviral progeny. Inhibition of proteasomal activity may lead to a reduction in the pool of free ubiquitin, which in turn may affect the de novo ubiquitination of proteins. For instance, long exposures of QT6 cells to MG132 reduced the cell egress of the retrovirus respiratory syncytial virus (RSV) by decreasing the pool of ubiquitin available for ubiquitination, which is required for virus budding (38). In our case, the fact that the pool of monoubiquitin was not decreased in the presence of MG132 (Fig. 5E) strongly suggests that the proteolytic activity of the proteasome is necessary for HAstV replication. This conclusion is supported by the fact that three different proteasomal inhibitors (MG132, lactacystin, and N-[(phenylmethoxy)carbonyl]-l-isoleucyl-l-α-glutamyl-tert-butyl ester-N-[(1S)-1-formyl-3-methylbutyl]-l-alaninamide [PSI]), with different mechanisms of action (41–43), reduced the production of viral progeny. Thus, the effect of Pyr-41 and the ubiquitin siRNA on virus replication could be explained because protein ubiquitination is important for protein degradation, although a role of ubiquitination in the replication cycle of the virus, independent of protein degradation, cannot be discarded. Preliminary results in our laboratory suggest that the replication of other classical astroviruses is affected in a manner similar to that of Yuc8.
Exposure of cells to proteasome inhibitors can induce autophagy or apoptosis (44–46). As these mechanisms could have antiviral effects, one possibility is that MG132 reduces HAstV replication by an indirect mechanism, inducing the degradation of viral components by autophagy or by early cell death. However, the fact that the effect of MG132 was not relieved by treatment with autophagy or apoptosis inhibitors suggests that these processes are not involved in the reduction of HAstV progeny production caused by MG132 (Fig. 6).
The replication cycle of HAstV begins with the translation of the incoming viral gRNA that directs the synthesis of the viral nonstructural proteins, followed by the formation of a polymerase complex that copies the gRNA into an agRNA. The agRNA is then used as the template for the synthesis of additional gRNA molecules and of sgRNA, which encodes the viral capsid polyprotein (6). In this work, we found that the synthesis of both nonstructural and structural viral proteins is inhibited by MG132. In principle, the effect of the drug could be due to the inhibition of any of the steps described above; i.e., it could block either the synthesis of the negative- or the positive-strand viral RNAs or the synthesis of the viral proteins necessary for the transcription and replication of the gRNA. In this context, it is hard to pinpoint the virus replication step inhibited by MG132.
There are, however, some indications of the stage of virus replication that could be affected by MG132. First, the inhibitory effect of MG132 is maintained when the drug is added to the cells at 4 hpi (Fig. 2A), indicating that the entry process of the virus is not affected by the drug, since the half-time for the release of HAstV RNA into the cytoplasm is about 130 min (7). Second, at early times of infection, the expression of the protease (a nonstructural protein) does not seem to be reduced, suggesting that the translation of the incoming gRNA into nonstructural proteins is not affected. Third, at 12 hpi, the level of gRNA is decreased by about 10-fold, while that of the agRNA is not reduced, and at this time, the level of the sgRNA is also reduced by 50%, indicating that the synthesis of the agRNA is not affected by MG132 at early times of infection. The reduction in the level of agRNA at 24 hpi, to about 50% of that of the control, could be the consequence of the lower levels of the template gRNA available at earlier times. Fourth, the fact that the level of sgRNA was diminished at both 12 and 24 hpi suggests that the primary effect of MG132 is on the accumulation of the two positive-sense viral RNAs, which in turn is reflected in the reduced synthesis of viral proteins. Altogether, these findings suggest that the RdRp complexes involved in the synthesis of positive- and negative-sense RNAs could be different, although an effect of MG132 on RNA stability rather than on its synthesis could not be discarded. A similar observation has been reported for influenza virus, where proteasome activity is necessary for efficient polymerase function (26).
Why the proteolytic activity of the proteasome is required for the efficient production of positive-sense viral RNAs during HAstV replication is not clear; however, there are a number of possible mechanisms. First, one of the more common features of the interaction between the UPS and viruses is the degradation of antiviral factors (47, 48). It is possible that a cellular factor that inhibits the RdRp complex in charge of the synthesis of positive-sense RNAs needs to be degraded for efficient gRNA and sgRNA production. A second alternative is that the stability and not the production of gRNA and sgRNA is affected by a cellular factor (for example, an RNase); thus, it is possible that the agRNA is present only in virus replication centers associated with membranes, where it might be protected from cellular RNases, but positive RNAs (gRNA and sgRNA), present in the cytoplasm, could be available for RNase degradation.
The role of the innate immune response in astrovirus infection has been poorly studied so far. It has been reported that it contributes to the control of murine astrovirus (MuAstV) (49), and recent publications reported that HAstV-1 induces a mild and delayed interferon (IFN) response and that its replication is IFN sensitive (50, 51). Thus, it is possible that factors of the innate immune system that restrict astrovirus replication could be degraded by the proteasome during virus infection. Finally, it is possible that the proteasome could regulate the degradation or processing of proteins that modulate the production of positive-sense RNAs directly or indirectly. For example, its activity could be required for activating a signaling pathway involved in the formation of replication centers, as shown for brome mosaic virus; in this case, the formation of these centers depends on the activation of lipid metabolism in a process regulated by the proteasome (52, 53). In the case of HAstV, it was reported that cellular proteins related to lipid metabolism are expressed in subcellular fractions where the viral RdRp is present, and RNA interference in the synthesis of these proteins reduced viral RNA production (54). Proteasome activity could be necessary for the stability, processing, and intracellular distribution of the viral proteins. It has been shown for turnip yellow mosaic virus that the UPS regulates the temporal synthesis of positive- and negative-sense viral RNAs by regulating the RdRp abundance (55, 56). Also, in the case of rotavirus, it has been shown that the inhibition of the UPS results in the delocalization of the viral polymerase (21, 22). One interesting idea derived from the results presented in this report is that if the proteolytic activity of the proteasome is necessary for the synthesis of the HAstV positive-sense RNAs, the replication complexes that produce the positive- and negative-sense RNAs could be different.
MATERIALS AND METHODS
Cells, virus, and reagents.
C2Bbe1 cells, derived from the colon adenocarcinoma Caco-2 cell line, were obtained from the American Type Culture Collection and propagated in a 10% CO2 atmosphere at 37°C in high-glucose Dulbecco's modified Eagle's medium (DMEM-HG) (Sigma Chemical Co., St. Louis, MO) supplemented with nonessential amino acids (catalog number 1140; Gibco Life Technologies, Carlsbad, CA) and 10% fetal bovine serum (FBS) (Cansera, Ontario, Canada). HAstV serotype 8 strain Yuc8 was isolated in our laboratory, and it was grown as described previously (57, 58). The siRNA UBC (smart pool) against the ubiquitin gene was purchased from Dharmacon Research (Lafayette, CO). Horseradish peroxidase-conjugated goat anti-rabbit polyclonal and anti-mouse antibodies were purchased from PerkinElmer Life Sciences (Boston, MA), and an antitubulin monoclonal antibody (clone TU-01) was purchased from Invitrogen. Rabbit polyclonal antibody to ubiquitin was purchased from Cell Signaling. Rabbit polyclonal sera to the capsid protein of HAstV (Yuc8 strain), to the viral protease, and to the viral RdRp were previously described (59). The proteasome inhibitors PSI, MG132, and lactacystin (proteasome inhibitor set I) and the E1 inhibitor Pyr-41 were purchased from Calbiochem.
Determination of yields of infectious virus.
Confluent C2Bbe1 cells grown in 96-multiwell plates were infected with Yuc8 at an MOI of 3 for 1 h at 37°C, and unbound virus was removed by washing. When used, the drugs were present at the indicated times, added just at the end of the adsorption period, and maintained in medium until harvest, unless otherwise indicated. In all the experiments, the medium was supplemented with nonessential amino acids (NEAAs). The cells were collected at the indicated times and lysed by three cycles of freeze-thawing, and the virus present in the lysates was then activated by incubation with trypsin at 200 μg/ml for 1 h at 37°C. Viral titers were determined by an immunoperoxidase focus determination assay, as described previously (11).
Western blots.
Cells were infected with Yuc8 as described above. At the indicated times, the cells were lysed with Laemmli sample buffer, and the proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto nitrocellulose membranes (Millipore, Bedford, MA). Membranes were blocked with 5% nonfat dried milk in phosphate-buffered saline (PBS)–0.1% Tween and incubated at 4°C with the indicated primary antibodies diluted in PBS–0.1% Tween, followed by incubation with secondary, species-specific, horseradish peroxidase-conjugated or Alexa 647-conjugated antibodies (as indicated). For monoubiquitin detection, Tris-Tricine gels were used as described previously (22).
siRNA transfection.
To transfect siRNAs into C2Bbe1 cells, a modified reverse transfection method was used (60). Briefly, the siRNAs (32 nM) were diluted in minimal essential medium (MEM) in a final volume of 15 μl and mixed with 90 μl of MEM containing 1.34 μl of Oligofectamine (Invitrogen, Carlsbad, CA) directly in the wells of a 48-well plate. The siRNA-Oligofectamine mix was incubated for 20 min at room temperature, and 200 μl of a suspension of 100,000 cells/ml in DMEM supplemented with 15% of FBS was then added to each well. The final concentration of siRNA under these conditions was 1.5 nM. The cells were incubated for 48 h at 37°C.
Metabolic labeling.
Cells were infected at an MOI of 3 and incubated with or without MG132 at the indicated concentrations, or siRNA-transfected cells were infected as previously described. At 17 hpi, the medium was replaced with Express-35S labeling mix (Dupont, NEN), maintaining the same concentration of MG132, and the mixture was incubated for 1 h. Cells were then washed and lysed with Laemmli sample buffer. The proteins were separated by SDS-PAGE, and gels were stained with Coomassie blue and subjected to autoradiography.
RNA extraction.
C2Bbe1 cells grown in 6-well plates were infected with Yuc8 at an MOI of 3 and incubated for 12 or 24 h in the presence or absence of 1 μM MG132. Total RNA was extracted by using TRIzol (Invitrogen) according to the manufacturer's instructions. RNA was resuspended in RNase-free water and quantified by spectrometry using a NanoDrop ND-1000 instrument. RNA from mock-infected cells was also obtained as a control.
qRT-PCR analysis.
qRT-PCR was performed according to protocols for the standard First Strand cDNA synthesis kit with Maxima SYBR green qPCR master mix (Thermo Scientific, Waltham, MA). Quantitative analysis of data was performed by using the Prism 7000 analysis software program (Applied Biosystems, Life Technologies, Carlsbad, CA). The primers used to amplify ORF1b astrovirus Yuc8 RNA for the gRNA and agRNA were described previously. Briefly, in these experiments, the primer used for cDNA synthesis determines the polarity of the strand analyzed, as was reported previously by us and others (51, 54, 61). To ensure that only the strand selected is reverse transcribed, after the cDNA synthesis step, the reaction mix is heated to 90°C for 15 min and immediately transferred to ice to prevent the renaturation of the reverse transcriptase. After this step, PCR with both primers is set up. The primers used to amplify the sgRNA were forward primer 5′-TGGAACACTGCCTATCACGG-3′ and reverse primer 5′-GAAGGCCAGAGTCACGAAGCT-3′. The results were normalized to the level of total glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA detected in each RNA sample. The relative fold changes in gene expression levels were calculated by the 2ΔCT method, where CT is the threshold cycle, as described previously (54).
ACKNOWLEDGMENTS
This work was partially supported by grants IN212211, IN207714, and IN208317 from DGAPA-UNAM. Luis A. Casorla-Pérez was a recipient of a scholarship from CONACyT.
We thank Rafaela Espinosa and Pedro Romero for their valuable technical assistance and Andrea Murillo and Noemí Flores for their advice on qPCR assays.
REFERENCES
- 1.De Benedictis P, Schultz-Cherry S, Burnham A, Cattoli G. 2011. Astrovirus infections in humans and animals—molecular biology, genetic diversity, and interspecies transmissions. Infect Genet Evol 11:1529–1544. doi: 10.1016/j.meegid.2011.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bosch A, Pinto RM, Guix S. 2014. Human astroviruses. Clin Microbiol Rev 27:1048–1074. doi: 10.1128/CMR.00013-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brown JR, Morfopoulou S, Hubb J, Emmett WA, Ip W, Shah D, Brooks T, Paine SM, Anderson G, Virasami A, Tong CY, Clark DA, Plagnol V, Jacques TS, Qasim W, Hubank M, Breuer J. 2015. Astrovirus VA1/HMO-C: an increasingly recognized neurotropic pathogen in immunocompromised patients. Clin Infect Dis 60:881–888. doi: 10.1093/cid/ciu940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Naccache SN, Peggs KS, Mattes FM, Phadke R, Garson JA, Grant P, Samayoa E, Federman S, Miller S, Lunn MP, Gant V, Chiu CY. 2015. Diagnosis of neuroinvasive astrovirus infection in an immunocompromised adult with encephalitis by unbiased next-generation sequencing. Clin Infect Dis 60:919–923. doi: 10.1093/cid/ciu912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fuentes C, Bosch A, Pinto RM, Guix S. 2012. Identification of human astrovirus genome-linked protein (VPg) essential for virus infectivity. J Virol 86:10070–10078. doi: 10.1128/JVI.00797-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mendez E, Arias CF. 2013. Astroviruses, p 981–1000. In Knipe DE, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 6th ed, vol 1 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 7.Mendez E, Munoz-Yanez C, Sanchez-San Martin C, Aguirre-Crespo G, Banos-Lara MDR, Gutierrez M, Espinosa R, Acevedo Y, Arias CF, Lopez S. 2014. Characterization of human astrovirus cell entry. J Virol 88:2452–2460. doi: 10.1128/JVI.02908-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mendez E, Aguirre-Crespo G, Zavala G, Arias CF. 2007. Association of the astrovirus structural protein VP90 with membranes plays a role in virus morphogenesis. J Virol 81:10649–10658. doi: 10.1128/JVI.00785-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guix S, Caballero S, Bosch A, Pinto RM. 2004. C-terminal nsP1a protein of human astrovirus colocalizes with the endoplasmic reticulum and viral RNA. J Virol 78:13627–13636. doi: 10.1128/JVI.78.24.13627-13636.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Banos-Lara MDR, Mendez E. 2010. Role of individual caspases induced by astrovirus on the processing of its structural protein and its release from the cell through a non-lytic mechanism. Virology 401:322–332. doi: 10.1016/j.virol.2010.02.028. [DOI] [PubMed] [Google Scholar]
- 11.Mendez E, Salas-Ocampo E, Arias CF. 2004. Caspases mediate processing of the capsid precursor and cell release of human astroviruses. J Virol 78:8601–8608. doi: 10.1128/JVI.78.16.8601-8608.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goldberg AL. 2003. Protein degradation and protection against misfolded or damaged proteins. Nature 426:895–899. doi: 10.1038/nature02263. [DOI] [PubMed] [Google Scholar]
- 13.Schwartz AL, Ciechanover A. 2009. Targeting proteins for destruction by the ubiquitin system: implications for human pathobiology. Annu Rev Pharmacol Toxicol 49:73–96. doi: 10.1146/annurev.pharmtox.051208.165340. [DOI] [PubMed] [Google Scholar]
- 14.Jariel-Encontre I, Bossis G, Piechaczyk M. 2008. Ubiquitin-independent degradation of proteins by the proteasome. Biochim Biophys Acta 1786:153–177. doi: 10.1016/j.bbcan.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 15.Goldberg AL. 2007. Functions of the proteasome: from protein degradation and immune surveillance to cancer therapy. Biochem Soc Trans 35:12–17. doi: 10.1042/BST0350012. [DOI] [PubMed] [Google Scholar]
- 16.Welchman RL, Gordon C, Mayer RJ. 2005. Ubiquitin and ubiquitin-like proteins as multifunctional signals. Nat Rev Mol Cell Biol 6:599–609. doi: 10.1038/nrm1700. [DOI] [PubMed] [Google Scholar]
- 17.Hanson PI, Cashikar A. 2012. Multivesicular body morphogenesis. Annu Rev Cell Dev Biol 28:337–362. doi: 10.1146/annurev-cellbio-092910-154152. [DOI] [PubMed] [Google Scholar]
- 18.Stadtmueller BM, Hill CP. 2011. Proteasome activators. Mol Cell 41:8–19. doi: 10.1016/j.molcel.2010.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin D, Zhong B. 2015. Regulation of cellular innate antiviral signaling by ubiquitin modification. Acta Biochim Biophys Sin (Shanghai) 47:149–155. doi: 10.1093/abbs/gmu133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Calistri A, Munegato D, Carli I, Parolin C, Palu G. 2014. The ubiquitin-conjugating system: multiple roles in viral replication and infection. Cells 3:386–417. doi: 10.3390/cells3020386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Contin R, Arnoldi F, Mano M, Burrone OR. 2011. Rotavirus replication requires a functional proteasome for effective assembly of viroplasms. J Virol 85:2781–2792. doi: 10.1128/JVI.01631-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lopez T, Silva-Ayala D, Lopez S, Arias CF. 2011. Replication of the rotavirus genome requires an active ubiquitin-proteasome system. J Virol 85:11964–11971. doi: 10.1128/JVI.05286-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gilfoy F, Fayzulin R, Mason PW. 2009. West Nile virus genome amplification requires the functional activities of the proteasome. Virology 385:74–84. doi: 10.1016/j.virol.2008.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheng S, Yan W, Gu W, He Q. 2014. The ubiquitin-proteasome system is required for the early stages of porcine circovirus type 2 replication. Virology 456–457:198–204. doi: 10.1016/j.virol.2014.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karpe YA, Meng XJ. 2012. Hepatitis E virus replication requires an active ubiquitin-proteasome system. J Virol 86:5948–5952. doi: 10.1128/JVI.07039-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kirui J, Mondal A, Mehle A. 28 September 2016. Ubiquitination up-regulates influenza virus polymerase function. J Virol doi: 10.1128/JVI.01829-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sahana N, Kaur H, Basavaraj, Tena F, Jain RK, Palukaitis P, Canto T, Praveen S. 2012. Inhibition of the host proteasome facilitates papaya ringspot virus accumulation and proteosomal catalytic activity is modulated by viral factor HcPro. PLoS One 7:e52546. doi: 10.1371/journal.pone.0052546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mitchell AM, Samulski RJ. 2013. Mechanistic insights into the enhancement of adeno-associated virus transduction by proteasome inhibitors. J Virol 87:13035–13041. doi: 10.1128/JVI.01826-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peterson MD, Mooseker MS. 1992. Characterization of the enterocyte-like brush border cytoskeleton of the C2BBe clones of the human intestinal cell line, Caco-2. J Cell Sci 102(Part 3):581–600. [DOI] [PubMed] [Google Scholar]
- 30.Bush KT, Goldberg AL, Nigam SK. 1997. Proteasome inhibition leads to a heat-shock response, induction of endoplasmic reticulum chaperones, and thermotolerance. J Biol Chem 272:9086–9092. doi: 10.1074/jbc.272.14.9086. [DOI] [PubMed] [Google Scholar]
- 31.Grossin L, Etienne S, Gaborit N, Pinzano A, Cournil-Henrionnet C, Gerard C, Payan E, Netter P, Terlain B, Gillet P. 2004. Induction of heat shock protein 70 (Hsp70) by proteasome inhibitor MG 132 protects articular chondrocytes from cellular death in vitro and in vivo. Biorheology 41:521–534. [PubMed] [Google Scholar]
- 32.Kim D, Kim SH, Li GC. 1999. Proteasome inhibitors MG132 and lactacystin hyperphosphorylate HSF1 and induce hsp70 and hsp27 expression. Biochem Biophys Res Commun 254:264–268. doi: 10.1006/bbrc.1998.9840. [DOI] [PubMed] [Google Scholar]
- 33.Neznanov N, Dragunsky EM, Chumakov KM, Neznanova L, Wek RC, Gudkov AV, Banerjee AK. 2008. Different effect of proteasome inhibition on vesicular stomatitis virus and poliovirus replication. PLoS One 3:e1887. doi: 10.1371/journal.pone.0001887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiang HY, Wek RC. 2005. Phosphorylation of the alpha-subunit of the eukaryotic initiation factor-2 (eIF2alpha) reduces protein synthesis and enhances apoptosis in response to proteasome inhibition. J Biol Chem 280:14189–14202. doi: 10.1074/jbc.M413660200. [DOI] [PubMed] [Google Scholar]
- 35.Yang Y, Kitagaki J, Dai RM, Tsai YC, Lorick KL, Ludwig RL, Pierre SA, Jensen JP, Davydov IV, Oberoi P, Li CC, Kenten JH, Beutler JA, Vousden KH, Weissman AM. 2007. Inhibitors of ubiquitin-activating enzyme (E1), a new class of potential cancer therapeutics. Cancer Res 67:9472–9481. doi: 10.1158/0008-5472.CAN-07-0568. [DOI] [PubMed] [Google Scholar]
- 36.Kapuria V, Peterson LF, Showalter HD, Kirchhoff PD, Talpaz M, Donato NJ. 2011. Protein cross-linking as a novel mechanism of action of a ubiquitin-activating enzyme inhibitor with anti-tumor activity. Biochem Pharmacol 82:341–349. doi: 10.1016/j.bcp.2011.05.012. [DOI] [PubMed] [Google Scholar]
- 37.Oh C, Park S, Lee EK, Yoo YJ. 2013. Downregulation of ubiquitin level via knockdown of polyubiquitin gene Ubb as potential cancer therapeutic intervention. Sci Rep 3:2623. doi: 10.1038/srep02623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Patnaik A, Chau V, Wills JW. 2000. Ubiquitin is part of the retrovirus budding machinery. Proc Natl Acad Sci U S A 97:13069–13074. doi: 10.1073/pnas.97.24.13069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bauckman KA, Owusu-Boaitey N, Mysorekar IU. 2015. Selective autophagy: xenophagy. Methods 75:120–127. doi: 10.1016/j.ymeth.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guix S, Bosch A, Ribes E, Martinez LD, Pinto RM. 2004. Apoptosis in astrovirus-infected CaCo-2 cells. Virology 319:249–261. doi: 10.1016/j.virol.2003.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rock KL, Gramm C, Rothstein L, Clark K, Stein R, Dick L, Hwang D, Goldberg AL. 1994. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell 78:761–771. doi: 10.1016/S0092-8674(94)90462-6. [DOI] [PubMed] [Google Scholar]
- 42.Figueiredo-Pereira ME, Berg KA, Wilk S. 1994. A new inhibitor of the chymotrypsin-like activity of the multicatalytic proteinase complex (20S proteasome) induces accumulation of ubiquitin-protein conjugates in a neuronal cell. J Neurochem 63:1578–1581. doi: 10.1046/j.1471-4159.1994.63041578.x. [DOI] [PubMed] [Google Scholar]
- 43.Fenteany G, Standaert RF, Lane WS, Choi S, Corey EJ, Schreiber SL. 1995. Inhibition of proteasome activities and subunit-specific amino-terminal threonine modification by lactacystin. Science 268:726–731. doi: 10.1126/science.7732382. [DOI] [PubMed] [Google Scholar]
- 44.Saji C, Higashi C, Niinaka Y, Yamada K, Noguchi K, Fujimuro M. 2011. Proteasome inhibitors induce apoptosis and reduce viral replication in primary effusion lymphoma cells. Biochem Biophys Res Commun 415:573–578. doi: 10.1016/j.bbrc.2011.10.107. [DOI] [PubMed] [Google Scholar]
- 45.Bao W, Gu Y, Ta L, Wang K, Xu Z. 2016. Induction of autophagy by the MG132 proteasome inhibitor is associated with endoplasmic reticulum stress in MCF7 cells. Mol Med Rep 13:796–804. doi: 10.3892/mmr.2015.4599. [DOI] [PubMed] [Google Scholar]
- 46.Lan D, Wang W, Zhuang J, Zhao Z. 2015. Proteasome inhibitor-induced autophagy in PC12 cells overexpressing A53T mutant alpha-synuclein. Mol Med Rep 11:1655–1660. doi: 10.3892/mmr.2014.3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bhoj VG, Chen ZJ. 2009. Ubiquitylation in innate and adaptive immunity. Nature 458:430–437. doi: 10.1038/nature07959. [DOI] [PubMed] [Google Scholar]
- 48.Oudshoorn D, Versteeg GA, Kikkert M. 2012. Regulation of the innate immune system by ubiquitin and ubiquitin-like modifiers. Cytokine Growth Factor Rev 23:273–282. doi: 10.1016/j.cytogfr.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yokoyama CC, Loh J, Zhao G, Stappenbeck TS, Wang D, Huang HV, Virgin HW, Thackray LB. 2012. Adaptive immunity restricts replication of novel murine astroviruses. J Virol 86:12262–12270. doi: 10.1128/JVI.02018-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guix S, Perez-Bosque A, Miro L, Moreto M, Bosch A, Pinto RM. 2015. Type I interferon response is delayed in human astrovirus infections. PLoS One 10:e0123087. doi: 10.1371/journal.pone.0123087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marvin SA, Huerta CT, Sharp B, Freiden P, Cline TD, Schultz-Cherry S. 2015. Type I interferon response limits astrovirus replication and protects against increased barrier permeability in vitro and in vivo. J Virol 90:1988–1996. doi: 10.1128/JVI.02367-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee WM, Ahlquist P. 2003. Membrane synthesis, specific lipid requirements, and localized lipid composition changes associated with a positive-strand RNA virus RNA replication protein. J Virol 77:12819–12828. doi: 10.1128/JVI.77.23.12819-12828.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang X, Diaz A, Hao L, Gancarz B, den Boon JA, Ahlquist P. 2011. Intersection of the multivesicular body pathway and lipid homeostasis in RNA replication by a positive-strand RNA virus. J Virol 85:5494–5503. doi: 10.1128/JVI.02031-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Murillo A, Vera-Estrella R, Barkla BJ, Mendez E, Arias CF. 2015. Identification of host cell factors associated with astrovirus replication in Caco-2 cells. J Virol 89:10359–10370. doi: 10.1128/JVI.01225-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chenon M, Camborde L, Cheminant S, Jupin I. 2012. A viral deubiquitylating enzyme targets viral RNA-dependent RNA polymerase and affects viral infectivity. EMBO J 31:741–753. doi: 10.1038/emboj.2011.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Camborde L, Planchais S, Tournier V, Jakubiec A, Drugeon G, Lacassagne E, Pflieger S, Chenon M, Jupin I. 2010. The ubiquitin-proteasome system regulates the accumulation of Turnip yellow mosaic virus RNA-dependent RNA polymerase during viral infection. Plant Cell 22:3142–3152. doi: 10.1105/tpc.109.072090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mendez-Toss M, Romero-Guido P, Munguia ME, Mendez E, Arias CF. 2000. Molecular analysis of a serotype 8 human astrovirus genome. J Gen Virol 81:2891–2897. doi: 10.1099/0022-1317-81-12-2891. [DOI] [PubMed] [Google Scholar]
- 58.Mendez E, Fernandez-Luna T, Lopez S, Mendez-Toss M, Arias CF. 2002. Proteolytic processing of a serotype 8 human astrovirus ORF2 polyprotein. J Virol 76:7996–8002. doi: 10.1128/JVI.76.16.7996-8002.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mendez E, Salas-Ocampo MP, Munguia ME, Arias CF. 2003. Protein products of the open reading frames encoding nonstructural proteins of human astrovirus serotype 8. J Virol 77:11378–11384. doi: 10.1128/JVI.77.21.11378-11384.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lopez T, Silva-Ayala D, Lopez S, Arias CF. 2012. Methods suitable for high-throughput screening of siRNAs and other chemical compounds with the potential to inhibit rotavirus replication. J Virol Methods 179:242–249. doi: 10.1016/j.jviromet.2011.11.010. [DOI] [PubMed] [Google Scholar]
- 61.Ayala-Breton C, Arias M, Espinosa R, Romero P, Arias CF, Lopez S. 2009. Analysis of the kinetics of transcription and replication of the rotavirus genome by RNA interference. J Virol 83:8819–8831. doi: 10.1128/JVI.02308-08. [DOI] [PMC free article] [PubMed] [Google Scholar]