

Abstract
There is good epidemiological evidence that patients with autoimmune rheumatic disease states, particularly rheumatoid arthritis, have an increased risk of cardiovascular morbidity and mortality when compared to the general population. The presence of a chronic systemic proinflammatory state in this patient group disrupts the structural and functional integrity of the endothelium and the arterial wall, favouring the onset and progression of atherosclerosis. A significant role in the detrimental effects of inflammation on endothelial function and vascular homeostasis is played by specific proatherosclerotic cytokines such as tumour necrosis factor-alpha (TNF-α), interleukin-1 (IL-1), and interleukin-6 (IL-6). Recent systematic reviews and meta-analyses have shown that treatment with methotrexate, a first-line disease-modifying antirheumatic drug (DMARD), is associated with a significant reduction in atherosclerosis-mediated cardiovascular events, such as myocardial infarction and stroke, and mortality, when compared to other DMARDs. This suggests that methotrexate might exert specific protective effects against vascular inflammation and atherosclerosis in the context of autoimmune rheumatic disease. This review discusses the available evidence regarding the potential antiatherosclerotic effects of methotrexate through the inhibition of TNF-α, IL-1, and IL-6 and provides suggestions for future experimental and human studies addressing this issue.
1. Introduction
Autoimmune rheumatic diseases such as rheumatoid arthritis (RA) are characterized by the presence of a chronic inflammatory state affecting the joints as well as a number of other organs and tissues [1]. The prevalence of RA ranges between 0.1 and 5%, depending on specific ethnic groups and geographic locations, and is higher in females than in males [1]. Patients with RA have an increased risk of death [2]. The increased mortality in this patient group, as well as in other autoimmune rheumatic conditions, is primarily due to a relatively high prevalence of cardiovascular disease and its clinical consequences, mainly acute atherosclerosis-related events such as myocardial infarction and stroke [3–5]. The risks of myocardial infarction and stroke are, respectively, 38% and 24% higher in RA patients when compared to the general population [4, 5]. This suggests that RA favours the onset of vascular damage and atherosclerosis either through conventional cardiovascular risk factors, such as diabetes, hypercholesterolaemia, or cigarette smoking, or through alternative mechanisms [6, 7]. One possible alternative mechanism is represented by the autoimmune- and inflammation-mediated disruption of the structural and/or functional integrity of the endothelium. This leads to significant alterations in vascular homeostasis, driven by an impairment of nitric oxide (NO) synthesis by endothelial NO synthase (eNOS), that include a reduced endothelium-dependent vasodilatation, an increased leukocyte and monocyte adhesion to, and deposition in, the arterial wall, an increased intima-media thickness and arterial stiffness, and a prothrombotic tendency [8–13]. Notably, these abnormalities have been reported both in animal models of autoimmune rheumatic disease and in patients with RA [8, 14–18]. The “inflammatory theory” of atherosclerosis highlights the key role of specific cytokines, particularly tumour necrosis factor-alpha (TNF-α), interleukin-1 (IL-1), and interleukin-6 (IL-6), in disrupting endothelial integrity and vascular homeostasis [19, 20]. Given the presence of a chronic inflammatory state, and therefore a sustained vascular insult, it is likely that the pathophysiological role of proatherosclerotic cytokines is further augmented in RA. This might explain the increased cardiovascular risk reported in this patient group [7].
Over the last few years, an increasing number of in vitro and in vivo studies have sought to identify pharmacological strategies targeting inflammatory pathways for cardiovascular disease prevention and management [20–22]. A number of therapeutic agents commonly prescribed to combat immune activation and inflammation in RA might also exert salutary effects on endothelial function and vascular homeostasis. Recent epidemiological evidence suggests that methotrexate, an established first-line disease-modifying antirheumatic drug (DMARD), might exert protective effects against cardiovascular disease. This review discusses the role of cytokines and inflammation in the pathophysiology of endothelial dysfunction and atherosclerosis, the pharmacology of methotrexate, the reduced cardiovascular risk associated with its use, and the available evidence regarding the effects of this DMARD on proatherosclerotic cytokines and endothelial function.
2. Cytokines, Endothelial Dysfunction, and Atherosclerosis
The endothelium plays a key role in maintaining vascular homeostasis and protecting the vascular wall from a number of endogenous and exogenous proatherosclerotic insults [23]. A key role, in this context, is played by nitric oxide (NO), an endogenous messenger synthesised by the enzyme endothelial NO synthase (eNOS) [10]. NO regulates several important physiological processes, including vasodilation of arteries and arterioles, vascular tone, arterial stiffness, wave reflection, peripheral vascular resistance, blood pressure, and platelet function (Figure 1) [10]. A reduced NO synthesis by eNOS has been shown to be associated with virtually all cardiovascular risk factors [24]. Furthermore, clinical measures of endothelial dysfunction independently predict cardiovascular morbidity and mortality in several patient groups [25–27]. Therefore, the available evidence suggests that endothelial dysfunction is a key pathophysiological step in the sequence of events linking the presence of one or more cardiovascular risk factors with the development of atherosclerosis. Furthermore, endothelial dysfunction is useful in stratifying the risk of cardiovascular events at the population level and might represent an important target of therapies designed to mitigate such risk [28].
Figure 1.

Endothelium, nitric oxide, and vascular homeostasis. NO: nitric oxide; VSMC: vascular smooth muscle cell.
A significant number of cytokines, a group of low-molecular weight proteins, are produced in several cell types, such as endothelial cells, monocytes, and vascular smooth muscle cells, that play a key role in maintaining vascular homeostasis. Amongst them, the cytokines TNF-α, IL-1, and IL-6 have been extensively investigated not only from a pathophysiological point of view but also as therapeutic targets for novel cardioprotective therapies [21, 22, 29]. Both TNF-α and IL-1 are known to stimulate the synthesis of IL-6 during the process of immune activation and inflammation [30]. This, in turn, triggers several pathways that result in endothelial dysfunction, vascular inflammation and damage, and atherosclerosis [31]. The cellular effects of TNF-α and IL-1 are primarily mediated by the p38 mitogen-activated protein kinase (p38MAPK)/nuclear factor kappa-light-chain-enhancer of the activated B-cell (NF-κB) pathways [32]. By contrast, the effects of IL-6 are mediated by the IL-6 receptor and the signal transducer protein gp130 [33].
Experimental studies have demonstrated the deleterious effects of TNF-α, IL-1, and IL-6 on endothelial function, vascular homeostasis, and cardiovascular risk and the protective effects of pharmacological agents targeting these cytokines on surrogate vascular markers (Table 1).
Table 1.
Effects of the cytokines TNF-α, IL-1, and IL-6 on endothelial function and vascular homeostasis.
| Cytokine | Reported effects |
|---|---|
| Tumour necrosis factor-alpha | Endothelial nitric oxide synthase activity ↓ |
| Inducible nitric oxide synthase activity ↑ | |
| Reactive oxygen species ↑ | |
| Endothelium-dependent vasodilation ↓ | |
| Leukocyte adhesion ↑ | |
| Endothelial cell apoptosis ↑ | |
| Asymmetric dimethylarginine ↑ | |
|
| |
| Interleukin-1 | Endothelium-dependent vasodilation ↓ |
| Vasoconstrictor response to pharmacological challenge ↑ | |
| Endothelin-1 ↑ | |
| Leukocyte adhesion ↑ | |
| Vascular smooth muscle cell growth ↑ | |
| Intima thickness ↑ | |
| Arterial stiffness ↑ | |
| Coagulation ↑ | |
|
| |
| Interleukin-6 | Expression of angiotensin II type-1 receptor ↑ |
| Endothelium-dependent vasodilation ↓ | |
| Arterial stiffness ↑ | |
| Oxidative stress ↑ | |
| Thrombosis ↑ | |
↑: Increase; ↓: decrease.
2.1. TNF-α
Treatment of endothelial cells with TNF-α has been shown to increase the expression of the inducible form of NO synthase (iNOS), decreasing at the same time the expression of the endothelial constitutive isoform eNOS [34]. While the maintenance of eNOS activity provides adequate NO synthesis for the regulation of several physiological and antatherosclerotic effects, an excessive NO synthesis by iNOS leads to the intracellular formation of reactive oxygen species with consequent development of endothelial dysfunction, apoptosis, and vascular damage [35]. Not surprisingly, several studies have also reported that TNF-α significantly impairs endothelium-dependent vasodilation, through an increase in reactive oxygen species, promotes the adhesion of leukocytes to the endothelium, and favours endothelial cell apoptosis [36–38]. Furthermore, TNF-α inhibits the activity of the enzyme dimethylarginine dimethylaminohydrolase, with consequent accumulation of the endogenous eNOS inhibitor asymmetric dimethylarginine (ADMA) [39, 40]. This effect is clinically relevant as higher plasma concentrations of ADMA have been shown to independently predict cardiovascular events in patients with a wide range of cardiovascular risk at baseline (Table 1) [41, 42].
Higher serum TNF-α concentrations have been associated with an increased risk of ischaemic stroke and recurrent coronary events in epidemiological studies [43, 44]. The key role of TNF-α in mediating the detrimental effects of inflammation on endothelial dysfunction and vascular homeostasis is also supported by the results of animal and human studies investigating the effects of specific TNF-α inhibitors. For example, treatment with adalimumab significantly reduced the adhesion of human leukocytes to endothelial cells and the expression of vascular cell adhesion molecule-1 (VCAM-1), intracellular adhesion molecule-1 (ICAM-1), and E-selectin [45]. Other studies have shown that treatment with adalimumab or etanercept improved clinical measures of endothelium-dependent vasodilation in patients with psoriasis, reduced the plasma concentrations of ADMA, and increased circulating endothelial progenitor cells in patients with RA (Table 1) [46–48].
2.2. IL-1
Increased production of IL-1 has been shown to induce leukocyte adhesion to the endothelium, exert procoagulant activity, and stimulate the growth and chemotaxis of vascular smooth muscle cells, key steps in the pathogenesis of atherosclerosis [49–52]. In animal studies, exposure to exogenous IL-6 causes an increase in coronary vasospastic responses to pharmacological challenge and intima thickening (Table 1) [53]. Moreover, multiple factors known to associate with atherosclerosis, such as cholesterol crystals, atheroprone oscillatory flow, hypoxia, and neutrophil extracellular traps, have recently been found to activate the critical IL-1beta producing NLRP3 inflammasome [22].
Polymorphisms in the IL-1 receptor antagonist (IL-1Ra) gene have been shown to have significant associations with the presence of single-vessel coronary artery disease, assessed by coronary angiography, in patients with ischaemic heart disease [54]. Conversely, pharmacological inhibition of IL-1 with anakinra reduces the concentrations of the potent endogenous vasoconstrictor endothelin-1 and arterial stiffness and improves clinical measures of endothelial function in patients with RA [55]. Similar effects of anakinra on endothelium-dependent vasodilation have been reported in animal models of diabetes (Table 1) [56]. In a recently completed randomized placebo-controlled trial in 10,061 participants with a previous myocardial infarction and C-reactive protein concentrations ≥2 mg/L, canakinumab, a monoclonal antibody targeting IL-1beta, significantly reduced the primary end-point of nonfatal myocardial infarction, nonfatal stroke, or cardiovascular death after a median follow-up of 3.7 years. Of the three subcutaneous doses of canakinumab studied (50 mg, 150 mg, and 300 mg every three months), only the 150 mg dose met the prespecified multiplicity adjusted threshold for statistical significance. Furthermore, there were no significant differences in all-cause mortality between treatment with canakinumab and placebo [57].
2.3. IL-6
IL-6 has been shown to upregulate the expression of the angiotensin II type-1 receptor in vascular smooth muscle cells, responsible for vasoconstriction, cell apoptosis, and proinflammatory effects, and to impair endothelium-dependent vasodilation in animal models [58]. Increased plasma IL-6 concentrations are significantly associated with a reduced endothelium-dependent vasodilation both in healthy subjects [59] and in patients with hypertension [60] or hypercholesterolaemia [61]. A significant association between plasma IL-6 concentrations and clinical markers of increased arterial stiffness has been reported in patients with hypertension (Table 1) [62]. A meta-analysis performed by the Emerging Risk Factors Collaboration has also shown that higher plasma IL-6 concentrations predict the risk of nonfatal myocardial infarction and coronary artery disease-related death, with a 25% increase in the risk of future vascular events for each increase in log IL-6 concentrations (RR 1.25, 95% CI 1.19 to 1.32) [63].
Furthermore, treatment with the IL-6 receptor inhibitor tocilizumab caused an increase in endothelium-dependent vasodilatation, and a concomitant reduction in arterial stiffness, in patients with RA [64, 65]. In another study, tocilizumab improved endothelial function and reduced markers of oxidative stress, inflammation, and thrombosis in patients with RA (Table 1) [66].
3. Methotrexate Pharmacology
Methotrexate, an analogue of the B vitamin folic acid, is a first-line synthetic DMARD for the management of RA and other autoimmune diseases that is normally administered once a week, either orally, subcutaneously, or intramuscularly, with doses ranging between 5 and 25 mg [67, 68]. Notably, methotrexate is the only DMARD that has demonstrated significant survival benefits in patients with RA [69–71]. After being transported into the cytoplasm, through the reduced folate carrier, methotrexate is converted into intracellular polyglutamates. The polyglutamate forms ensure the intracellular retention of methotrexate, allowing weekly administration despite the relatively short plasma elimination half-life (5–8 hours) [72]. The polyglutamates also mediate the immunomodulating and anti-inflammatory effects of methotrexate, by reducing the synthesis of purines, pyrimidines, and DNA through the inhibition of dihydrofolate reductase, thymidylate synthase, and aminoimidazole carboxamide ribonucleotide (AICAR) transformylase (ATIC, Figure 2) [73–75]. Furthermore, the inhibition of ATIC causes the accumulation of the substrate AICAR which, in turn, inhibits the enzymes adenosine deaminase and adenosine monophosphate (AMP) deaminase, involved in the catabolism of adenosine (Figure 2) [75]. Adenosine per se exerts significant anti-inflammatory effects, primarily through the A2A and A3 receptors [76], and mediates the anti-inflammatory effects of methotrexate. In animal models of inflammation, methotrexate has been shown to increase AICAR and adenosine concentrations and to inhibit the accumulation of leukocytes, in exudates from carrageenan-inflamed air pouches. The administration of AMP deaminase partly reversed the methotrexate-mediated reduction in leukocyte accumulation. Additionally, the administration of 3,7-dimethyl-1-propargylxanthine, an A2A receptor antagonist, but not 8-cyclopentyl-dipropylxanthine, an A1 receptor antagonist, suppressed the methotrexate-mediated reduction in leukocyte accumulation [77]. Furthermore, both AICAR and AMP activate the 5′ adenosine monophosphate-activated protein kinase (AMPK) [78]. AMPK activation is protective towards endothelial cell function and vascular homeostasis, by ensuring physiological NO synthesis, maintaining mitochondrial structure and function, and preventing oxidative stress and apoptosis [79]. Moreover, a role for AMPK in inhibiting vascular smooth muscle cell proliferation, a key proatherosclerotic event both in autoimmune rheumatic diseases and in the general population, has been proposed [80, 81]. There is experimental evidence that AMPK is a key mediator of the effects of methotrexate on inflammation and endothelial function. In vitro studies have shown that methotrexate, at concentrations between 0.1 and 0.5 μM, increased AMPK phosphorylation and AMPK activity in perivascular adipose tissue cells. In these cells, the administration of palmitic acid, a proinflammatory agent, decreased AMPK phosphorylation; however, these effects were significantly reduced by pretreatment with either methotrexate or AICAR. Furthermore, methotrexate significantly reduced the phosphorylation of NF-κB p65 and prevented the palmitic acid-mediated increase in the expression of TNF-α and IL-6, indicating anti-inflammatory effects. Silencing AMPK with AMPKa1/2-specific siRNA significantly decreased the inhibitory effects of methotrexate on palmitic acid-mediated NF-κB p65 phosphorylation and on the expression of TNF-α and IL-6. Pretreatment with methotrexate prevented the impairment of acetylcholine-induced, endothelium-dependent vasodilation in rat aorta following palmitic acid administration. Notably, the protective effects of methotrexate on endothelial function were attenuated by cotreatment with compound C, an AMPK inhibitor [82].
Figure 2.
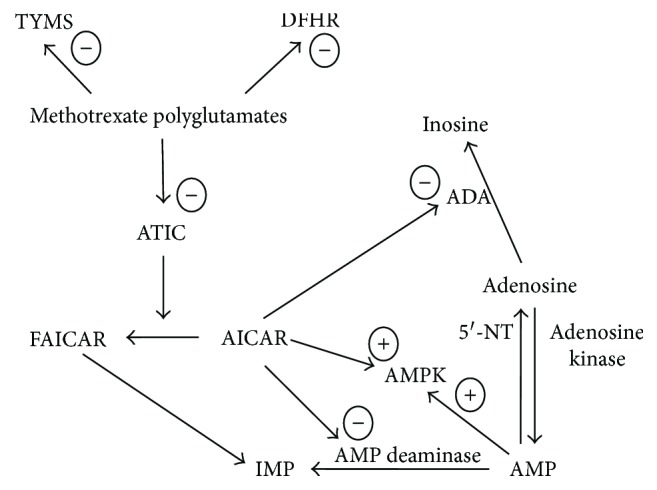
Intracellular effects of methotrexate. DHFR: dihydrofolate reductase; TYMS: thymidylate synthase; ATIC: aminoimidazole carboxamide ribonucleotide (AICAR) transformylase; FAICAR: 5-formamidoimidazole-4-carboxamide ribotide; IMP: inosine monophosphate; AMP: adenosine monophosphate; AMPK: 5′ adenosine monophosphate-activated protein kinase; ADA: adenosine deaminase; 5′-NT: 5′-nucleotidase; −: inhibition; +: activation.
4. Methotrexate and Cardiovascular Risk
Recent systematic reviews and meta-analyses have investigated the associations between methotrexate treatment and cardiovascular risk. Micha et al. identified observational studies in 66,334 patients with either RA (9 studies), psoriasis (1 study), or polyarthritis (1 study), that reported 6235 cardiovascular events. The median duration of follow-up in these studies was 5.8 years. Six studies compared methotrexate users versus never users, three compared current versus noncurrent users, and two compared initiators versus noninitiators. Cardiovascular endpoints included total or fatal cardiovascular disease (7 studies; two studies also provided separate estimates for myocardial infarction and ischaemic/haemorrhagic stroke), myocardial infarction (3 studies), and ischaemic stroke (1 study). Combined assessment of the included studies showed that methotrexate treatment was associated with a significant reduction in cardiovascular events (RR 0.79, 95% CI 0.73 to 0.87). Assessment of specific cardiovascular endpoints showed a similar effect size (overall cardiovascular disease events: RR 0.76, 95% CI 0.69 to 0.84; myocardial infarction: RR 0.82, 95% CI 0.71 to 0.96; stroke: RR 0.70, 95% CI 0.56 to 0.87) [83]. In a subsequent systematic review and meta-analysis, Roubille et al. identified 34 studies (28 studies in 236,525 RA patients, reporting 5410 cardiovascular events, and 6 studies in 220,209 patients with either psoriasis or psoriatic arthritis, reporting 2701 cardiovascular events). In studies conducted in RA patients, methotrexate use was associated with a reduced risk of all cardiovascular events (RR 0.72, 95% CI 0.57 to 0.91, P = 0.007) and myocardial infarction (RR 0.81, 95% CI 0.68 to 0.86, P = 0.01) when compared to other synthetic DMARDs. Although there was no significant effect of methotrexate on the risk of either stroke (RR 0.78, 95% CI 0.40 to 1.50) or major adverse cardiovascular events (RR 0.38, 95% CI 0.05 to 2.84), the number of studies assessing these endpoints was relatively low (one study for stroke and two studies for major adverse cardiovascular events, resp.) [84]. Therefore, the results of observational studies support the hypothesis that methotrexate exhibits specific protective cardiovascular effects when compared to other DMARDs.
5. Methotrexate and Proatherosclerotic Cytokines
In order to review the available evidence on the direct or indirect, mediated by adenosine, AICAR or AMPK activation, effects of methotrexate on proatherosclerotic cytokines, a PubMed literature search was conducted from inception to June 2017, using the following terms: methotrexate, adenosine, AICAR, AMPK, endothelium, inflammation, atherosclerosis, TNF-α, IL-1, and IL-6.
5.1. TNF-α
In in vitro studies, clinical concentrations of methotrexate (2 × 10−8 M and 2 × 10−7 M) have been shown to increase the release of soluble TNF receptor p75 from the cell surface [85]. This phenomenon is able to significantly inhibit the proinflammatory effects of TNF-α [86]. Adenosine is a known inhibitor of TNF-α expression through stimulation of the A3 receptor (Table 2) [87, 88]. There is recent evidence that methotrexate stimulates AMPK phosphorylation and activity, induces manganese superoxide dismutase mRNA and protein, and increases the expression of the cytoprotective genes haem oxygenase-1 and Bcl-2-related protein in human umbilical vein endothelial cells (HUVECs) and arterial endothelial cells (HAECs). The pretreatment of endothelial cells with TNF-α did not affect the methotrexate-mediated upregulation of manganese superoxide dismutase and haem oxygenase-1 mRNA [89]. Methotrexate (10−8 M to 10−6 M) and/or adenosine treatment has also been shown to prevent the TNF-α-induced (a) expression of ICAM-1 and VCAM-1 in human umbilical vein endothelial cells (HUVEC) [90] and (b) reduction of mitochondrial mass, membrane potential, and intracellular ATP concentrations, in endothelial cells through increased activity of eNOS [91]. Furthermore, adenosine has been shown to reduce the circulating concentrations of TNF-α and IL-6 in a mice model of sepsis (Table 2) [92]. By contrast, therapeutic concentrations of methotrexate, 0.1, 0.25, and 0.5 μM, significantly increased the expression of the TNF-α receptor-associated factor 1 gene (~threefold change) and of the TNF-α receptor superfamily member 9 gene (~threefold change) in EA.hy 926 cells, derived from the fusion of primary endothelial cells with an epithelial tumour cell line [93].
Table 2.
Effects of methotrexate, adenosine, AICAR, and AMPK activation on endothelial function and vascular homeostasis.
| Mediator | Reported effects |
|---|---|
| Methotrexate | Release of soluble TNF-α receptor p75 ↑ |
| TNF-α expression/concentrations ↓ | |
| IL-6 expression/concentrations ↓ | |
| ICAM-1 expression ↓ | |
| VCAM-1 expression ↓ | |
| eNOS activity ↑ | |
| Endothelium-dependent vasodilatation ↑ | |
| Mitochondrial mass, membrane potential, and intracellular ATP concentrations ↑ | |
|
| |
| Adenosine | TNF-α expression/concentrations ↓ |
| IL-6 expression/concentrations ↓ | |
| ICAM-1 expression ↓ | |
| VCAM-1 expression ↓ | |
| E-selectin expression ↓ | |
| eNOS activity ↑ | |
| Blood pressure ↓ | |
| Mitochondrial mass, membrane potential, and intracellular ATP concentrations ↑ | |
| Formation of atherosclerotic lesions ↓ | |
| Cholesterol concentrations ↓ | |
| Triglyceride concentrations ↓ | |
|
| |
| AICAR/AMPK | IL-1 expression/concentrations ↓ |
| IL-6 expression/concentrations ↓ | |
| ICAM-1 expression ↓ | |
| VCAM-1 expression ↓ | |
| NO synthesis ↑ | |
| Endothelium-dependent vasodilation ↑ | |
| Endothelium-independent vasodilation ↑ | |
| Blood pressure ↓ | |
| Oxidative stress ↓ | |
| Endoplasmic reticulum stress ↓ | |
| Manganese superoxide dismutase induction ↑ | |
| NF-κB ↓ | |
| Monocyte adhesion to endothelial cells ↓ | |
| Restenosis ↓ | |
| Cholesterol efflux capacity ↑ | |
| Cellular glucose uptake ↑ | |
| Glycolysis ↑ | |
AICAR: aminoimidazole carboxamide ribonucleotide; AMPK: 5′ adenosine monophosphate-activated protein kinase; TNF-α: tumour necrosis factor-alpha; IL-1: interleukin-1; IL-6: interleukin-6; ICAM-1: intercellular adhesion molecule-1; VCAM-1: vascular cell adhesion molecule-1; ATP: adenosine triphosphate; eNOS: endothelial nitric oxide synthase; NF-κB: nuclear factor kappa-light-chain-enhancer of the activated B-cell; NO: nitric oxide; ↑: increase; ↓: decrease.
5.2. IL-1
In an in vitro model of palmitate-induced endothelial dysfunction, AICAR-mediated AMPK activation has been shown to significantly reduce IL-1, IL-6, and VCAM-1 synthesis, by preventing the activation of the NLRP3 inflammasome [94]. Similarly, pharmacological activation of AMPK with oestradiol prevented the IL-1-induced expression of VCAM-1 and ICAM-1 in cultured human endothelial cells (Table 2) [95]. By contrast, therapeutic concentrations of methotrexate, 0.1, 0.25, and 0.5 μM, significantly increased the expression of the IL-1 alpha gene (~threefold change) and of the IL-1 receptor-like 1 gene (~fourfold change) in EA.hy 926 cells [93].
5.3. IL-6
In db/db mice fed with Western diet, methotrexate treatment (4 mg/kg) caused a significant reduction in the circulating concentrations of IL-6 and TNF-α. These effects were associated with a significant increase in endothelium-dependent vasodilatation and a reduction in VCAM-1 [96]. In another study, methotrexate treatment (0.1–0.5 μM) significantly reduced the expression of IL-6 and TNF-α. These effects were mediated by the activation of AMPK and were associated with an improvement in eNOS activity and endothelium-dependent vasodilation in rat aorta (Table 2) [82]. Adenosine has been shown to prevent the thrombin-mediated increased expression of IL-6, VCAM-1, ICAM-1, and E-selectin in HUVECs. These effects are mediated by the activation of the adenosine receptor A2A [97]. Similarly, adenosine inhibited, in a dose-dependent fashion, the release of IL-6, VCAM-1, and ICAM-1 in HUVECs pretreated with either IL-1, TNF-α, or lipopolysaccharide (Table 2) [98].
6. Additional Vascular Effects of Adenosine, AICAR, and AMPK Activation
A number of studies have shown that adenosine, AICAR, and AMPK activation provide beneficial effects on endothelial function and vascular homeostasis that are independent of those on TNF-α, IL-1, or IL-6.
6.1. Adenosine Accumulation
There is good evidence that adenosine, through the activation of the A2A and A2B receptors, lowers blood pressure as a result of increased NO synthesis in the endothelium and direct vasodilation [99, 100]. Studies have also demonstrated the presence of central nervous system-mediated hypotensive effects through the activation of the A3 receptor [101]. Furthermore, the pharmacological activation of the A2B receptor prevents the formation of atherosclerotic lesions and reduces the plasma concentrations of cholesterol and triglycerides, possibly through the reduced activation of the transcription factor sterol regulatory element-binding protein 1 in the liver [102, 103]. Activation of adenosine receptors seems also to modulate glucose homeostasis. However, the pathophysiological and clinical relevance of this finding is yet to be determined (Table 2) [104].
6.2. AICAR and Activation of 5′ Adenosine Monophosphate-Activated Protein Kinase (AMPK)
There is increasing evidence that AICAR and or/AMPK activation stimulates NO synthesis in the endothelium, enhances endothelium-dependent and endothelium-independent vasodilation, reduces blood pressure, prevents vessel restenosis, and increases cholesterol efflux capacity [105–110]. Furthermore, AMPK stimulates cellular glucose uptake, through GLUT-1 and GLUT-4 transporters, and glycolysis, through phosphorylation of two isoforms of the enzyme 6-phoshofructo-2-kinase: fructose-2,6-biphosphatase, with beneficial effects on glucose homeostasis [111–114]. There is also evidence that AMPK activation protects endothelial cells from the deleterious effects of chronic exposure to high concentrations of glucose and fatty acids and consequently reduces oxidative stress, inflammation, and endoplasmic reticulum stress (Table 2) [115, 116].
7. Discussion
The available evidence from systematic reviews and meta-analyses of observational studies in patients with either RA or other autoimmune rheumatic disease states suggests that the use of methotrexate is associated with a significant reduction in cardiovascular morbidity and mortality when compared to other DMARDs. Furthermore, a relatively small number of experimental studies have shown that methotrexate can exert beneficial effects on endothelial function and vascular homeostasis by preventing or blocking the effects of key proatherosclerotic cytokines such as TNF-α, IL-1, and IL-6. The reported effects are mediated either by methotrexate directly or through the activation of adenosine receptors, AICAR, or AMPK (Table 2). Pending further in vitro and in vivo studies investigating the exact mechanisms involved in such effects, a number of limitations need to be considered when interpreting the available data:
The dose of methotrexate used in some studies [96] and its consequent local concentrations in target cells and tissues are quite different from those normally observed in patients with autoimmune disorders.
No study has assessed the intracellular concentrations of methotrexate polyglutamates, as a factor mediating the effects of the drug on the study end-points.
The effects of methotrexate, adenosine, AICAR, or AMPK activation on proatherosclerotic cytokines were not compared to those of other synthetic or biologic DMARDs.
Other studies, not specifically investigating TNF-α, IL-1, and IL-6, have shown that methotrexate can also exert antiproliferative effects in human umbilical vein endothelial cells and EA.hy 926 cells [93, 117, 118] and reduce NO synthesis, possibly as a result of reduced availability of tetrahydrobiopterin, an essential cofactor for endothelial nitric oxide synthase, secondary to dihydrofolate reductase inhibition [119].
There is no direct evidence that the methotrexate-induced inhibition of TNF-α, IL-1, or IL-6 leads to sustained beneficial effects on endothelial function, atherosclerosis, arterial structure and function, and cardiovascular risk in human studies.
These issues should be accounted for in future studies investigating the effects of methotrexate on proatherosclerotic cytokines. In particular, the use of other DMARDs as comparator should help to determine whether methotrexate treatment exerts specific antiatherosclerotic effects that might help to explain its superiority, in terms of cardiovascular risk reduction, reported in observational studies. Furthermore, a comprehensive assessment of proatherosclerotic and antiatherosclerotic cytokines, such as transforming growth factor-β, IL-10, and IL-35 [120], as well as measures of endothelial cell proliferation, apoptosis, and nitric oxide synthesis, might provide additional mechanistic insights regarding the possible vasculoprotective effects of methotrexate and the potential rationale for combining methotrexate treatment with other interventional strategies targeting specific cytokines. In this context, the results of the Cardiovascular Inflammation Reduction Trial (CIRT, clinicaltrials.gov identifier NCT01594333), investigating the effects of methotrexate on myocardial infarction, stroke, and cardiovascular death in patients with type 2 diabetes or metabolic syndrome and stable coronary artery disease, will provide additional knowledge regarding the potential repurposing of methotrexate for cardiovascular risk management and prevention in different patient populations [121].
8. Conclusions
The evidence recently generated from experimental studies suggests that methotrexate, an anchor drug in the treatment of RA and other autoimmune disorders, might exert significant beneficial effects on endothelial function and vascular homeostasis by targeting key cytokines regulating the immune responses and inflammation pathways responsible for the development of atherosclerosis and thrombosis. However, further studies are required to fully establish the role of this DMARD as an antiatherosclerotic and cardioprotective agent.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
References
- 1.Smolen J. S., Aletaha D., McInnes I. B. Rheumatoid arthritis. The Lancet. 2016;388(10055):2023–2038. doi: 10.1016/S0140-6736(16)30173-8. [DOI] [PubMed] [Google Scholar]
- 2.Gonzalez A., Maradit Kremers H., Crowson C. S., et al. The widening mortality gap between rheumatoid arthritis patients and the general population. Arthritis & Rheumatism. 2007;56(11):3583–3587. doi: 10.1002/art.22979. [DOI] [PubMed] [Google Scholar]
- 3.Piga M., Casula L., Perra D., et al. Population-based analysis of hospitalizations in a West-European region revealed major changes in hospital utilization for patients with systemic lupus erythematosus over the period 2001-2012. Lupus. 2016;25(1):28–37. doi: 10.1177/0961203315596597. [DOI] [PubMed] [Google Scholar]
- 4.Chung W. S., Lin C. L., Peng C. L., et al. Rheumatoid arthritis and risk of acute myocardial infarction–a nationwide retrospective cohort study. International Journal of Cardiology. 2013;168(5):4750–4754. doi: 10.1016/j.ijcard.2013.07.233. [DOI] [PubMed] [Google Scholar]
- 5.Liou T. H., Huang S. W., Lin J. W., Chang Y. S., Wu C. W., Lin H. W. Risk of stroke in patients with rheumatism: a nationwide longitudinal population-based study. Scientific Reports. 2014;4:p. 5110. doi: 10.1038/srep05110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baghdadi L. R., Woodman R. J., Shanahan E. M., Mangoni A. A. The impact of traditional cardiovascular risk factors on cardiovascular outcomes in patients with rheumatoid arthritis: a systematic review and meta-analysis. PLoS One. 2015;10(2, article e0117952) doi: 10.1371/journal.pone.0117952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lauper K., Gabay C. Cardiovascular risk in patients with rheumatoid arthritis. Seminars in Immunopathology. 2017;39(4):447–459. doi: 10.1007/s00281-017-0632-2. [DOI] [PubMed] [Google Scholar]
- 8.Murdaca G., Colombo B. M., Cagnati P., Gulli R., Spano F., Puppo F. Endothelial dysfunction in rheumatic autoimmune diseases. Atherosclerosis. 2012;224(2):309–317. doi: 10.1016/j.atherosclerosis.2012.05.013. [DOI] [PubMed] [Google Scholar]
- 9.Cardaropoli S., Silvagno F., Morra E., Pescarmona G. P., Todros T. Infectious and inflammatory stimuli decrease endothelial nitric oxide synthase activity in vitro. Journal of Hypertension. 2003;21(11):2103–2110. doi: 10.1097/00004872-200311000-00020. [DOI] [PubMed] [Google Scholar]
- 10.Napoli C., de Nigris F., Williams-Ignarro S., Pignalosa O., Sica V., Ignarro L. J. Nitric oxide and atherosclerosis: an update. Nitric Oxide. 2006;15(4):265–279. doi: 10.1016/j.niox.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 11.Loscalzo J. Nitric oxide insufficiency, platelet activation, and arterial thrombosis. Circulation Research. 2001;88(8):756–762. doi: 10.1161/hh0801.089861. [DOI] [PubMed] [Google Scholar]
- 12.Rudic R. D., Sessa W. C. Nitric oxide in endothelial dysfunction and vascular remodeling: clinical correlates and experimental links. American Journal of Human Genetics. 1999;64(3):673–677. doi: 10.1086/302304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wilkinson I. B., Franklin S. S., Cockcroft J. R. Nitric oxide and the regulation of large artery stiffness: from physiology to pharmacology. Hypertension. 2004;44(2):112–116. doi: 10.1161/01.HYP.0000138068.03893.40. [DOI] [PubMed] [Google Scholar]
- 14.Haruna Y., Morita Y., Komai N., et al. Endothelial dysfunction in rat adjuvant-induced arthritis: vascular superoxide production by NAD(P)H oxidase and uncoupled endothelial nitric oxide synthase. Arthritis & Rheumatism. 2006;54(6):1847–1855. doi: 10.1002/art.21891. [DOI] [PubMed] [Google Scholar]
- 15.Abbot S. E., Whish W. J., Jennison C., Blake D. R., Stevens C. R. Tumour necrosis factor α stimulated rheumatoid synovial microvascular endothelial cells exhibit increased shear rate dependent leucocyte adhesion in vitro. Annals of the Rheumatic Diseases. 1999;58(9):573–581. doi: 10.1136/ard.58.9.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang P., Guan S. Y., Xu S. Z., et al. Increased carotid intima-media thickness in rheumatoid arthritis: an update meta-analysis. Clinical Rheumatology. 2016;35(2):315–323. doi: 10.1007/s10067-015-3130-8. [DOI] [PubMed] [Google Scholar]
- 17.Roman M. J., Devereux R. B., Schwartz J. E., et al. Arterial stiffness in chronic inflammatory diseases. Hypertension. 2005;46(1):194–199. doi: 10.1161/01.HYP.0000168055.89955.db. [DOI] [PubMed] [Google Scholar]
- 18.Beinsberger J., Heemskerk J. W., Cosemans J. M. Chronic arthritis and cardiovascular disease: altered blood parameters give rise to a prothrombotic propensity. Seminars in Arthritis & Rheumatism. 2014;44(3):345–352. doi: 10.1016/j.semarthrit.2014.06.006. [DOI] [PubMed] [Google Scholar]
- 19.Libby P. Inflammation in atherosclerosis. Arteriosclerosis, Thrombosis, and Vascular Biology. 2012;32(9):2045–2051. doi: 10.1161/ATVBAHA.108.179705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tousoulis D., Oikonomou E., Economou E. K., Crea F., Kaski J. C. Inflammatory cytokines in atherosclerosis: current therapeutic approaches. European Heart Journal. 2016;37(22):1723–1732. doi: 10.1093/eurheartj/ehv759. [DOI] [PubMed] [Google Scholar]
- 21.Ramji D. P., Davies T. S. Cytokines in atherosclerosis: key players in all stages of disease and promising therapeutic targets. Cytokine & Growth Factor Reviews. 2015;26(6):673–685. doi: 10.1016/j.cytogfr.2015.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ridker P. M. From C-reactive protein to interleukin-6 to interleukin-1: moving upstream to identify novel targets for atheroprotection. Circulation Research. 2016;118(1):145–156. doi: 10.1161/CIRCRESAHA.115.306656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boulanger C. M. Endothelium. Arteriosclerosis, Thrombosis, and Vascular Biology. 2016;36(4):e26–e31. doi: 10.1161/ATVBAHA.116.306940. [DOI] [PubMed] [Google Scholar]
- 24.Reriani M. K., Lerman L. O., Lerman A. Endothelial function as a functional expression of cardiovascular risk factors. Biomarkers in Medicine. 2010;4(3):351–360. doi: 10.2217/bmm.10.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matsuzawa Y., Kwon T. G., Lennon R. J., Lerman L. O., Lerman A. Prognostic value of flow-mediated vasodilation in brachial artery and fingertip artery for cardiovascular events: a systematic review and meta-analysis. Journal of the American Heart Association. 2015;4(11, article e002270) doi: 10.1161/JAHA.115.002270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu Y., Arora R. C., Hiebert B. M., et al. Non-invasive endothelial function testing and the risk of adverse outcomes: a systematic review and meta-analysis. European Heart Journal Cardiovascular Imaging. 2014;15(7):736–746. doi: 10.1093/ehjci/jet256. [DOI] [PubMed] [Google Scholar]
- 27.Ras R. T., Streppel M. T., Draijer R., Zock P. L. Flow-mediated dilation and cardiovascular risk prediction: a systematic review with meta-analysis. International Journal of Cardiology. 2013;168(1):344–351. doi: 10.1016/j.ijcard.2012.09.047. [DOI] [PubMed] [Google Scholar]
- 28.Jensen H. A., Mehta J. L. Endothelial cell dysfunction as a novel therapeutic target in atherosclerosis. Expert Review of Cardiovascular Therapy. 2016;14(9):1021–1033. doi: 10.1080/14779072.2016.1207527. [DOI] [PubMed] [Google Scholar]
- 29.Tedgui A., Mallat Z. Cytokines in atherosclerosis: pathogenic and regulatory pathways. Physiological Reviews. 2006;86(2):515–581. doi: 10.1152/physrev.00024.2005. [DOI] [PubMed] [Google Scholar]
- 30.Feldmann M., Brennan F. M., Maini R. N. Role of cytokines in rheumatoid arthritis. Annual Review of Immunology. 1996;14(1):397–440. doi: 10.1146/annurev.immunol.14.1.397. [DOI] [PubMed] [Google Scholar]
- 31.Libby P., Ridker P. M. Novel inflammatory markers of coronary risk: theory versus practice. Circulation. 1999;100(11):1148–1150. doi: 10.1161/01.CIR.100.11.1148. [DOI] [PubMed] [Google Scholar]
- 32.Chan K. F., Siegel M. R., Lenardo J. M. Signaling by the TNF receptor superfamily and T cell homeostasis. Immunity. 2000;13(4):419–422. doi: 10.1016/S1074-7613(00)00041-8. [DOI] [PubMed] [Google Scholar]
- 33.Ait-Oufella H., Taleb S., Mallat Z., Tedgui A. Recent advances on the role of cytokines in atherosclerosis. Arteriosclerosis, Thrombosis, and Vascular Biology. 2011;31(5):969–979. doi: 10.1161/ATVBAHA.110.207415. [DOI] [PubMed] [Google Scholar]
- 34.MacNaul K. L., Hutchinson N. I. Differential expression of iNOS and cNOS mRNA in human vascular smooth muscle cells and endothelial cells under normal and inflammatory conditions. Biochemical and Biophysical Research Communications. 1993;196(3):1330–1334. doi: 10.1006/bbrc.1993.2398. [DOI] [PubMed] [Google Scholar]
- 35.Nowak W. N., Deng J., Ruan X. Z., Xu Q. Reactive oxygen species generation and atherosclerosis. Arteriosclerosis, Thrombosis, and Vascular Biology. 2017;37(5):e41–e52. doi: 10.1161/ATVBAHA.117.309228. [DOI] [PubMed] [Google Scholar]
- 36.Gao X., Belmadani S., Picchi A., et al. Tumor necrosis factor-α induces endothelial dysfunction in Leprdb mice. Circulation. 2007;115(2):245–254. doi: 10.1161/CIRCULATIONAHA.106.650671. [DOI] [PubMed] [Google Scholar]
- 37.Picchi A., Gao X., Belmadani S., et al. Tumor necrosis factor-α induces endothelial dysfunction in the prediabetic metabolic syndrome. Circulation Research. 2006;99(1):69–77. doi: 10.1161/01.RES.0000229685.37402.80. [DOI] [PubMed] [Google Scholar]
- 38.Madge L. A., Pober J. S. TNF signaling in vascular endothelial cells. Experimental and Molecular Pathology. 2001;70(3):317–325. doi: 10.1006/exmp.2001.2368. [DOI] [PubMed] [Google Scholar]
- 39.Ito A., Tsao P. S., Adimoolam S., Kimoto M., Ogawa T., Cooke J. P. Novel mechanism for endothelial dysfunction: dysregulation of dimethylarginine dimethylaminohydrolase. Circulation. 1999;99(24):3092–3095. doi: 10.1161/01.CIR.99.24.3092. [DOI] [PubMed] [Google Scholar]
- 40.Wadham C., Mangoni A. A. Dimethylarginine dimethylaminohydrolase regulation: a novel therapeutic target in cardiovascular disease. Expert Opinion on Drug Metabolism & Toxicology. 2009;5(3):303–319. doi: 10.1517/17425250902785172. [DOI] [PubMed] [Google Scholar]
- 41.Willeit P., Freitag D. F., Laukkanen J. A., et al. Asymmetric dimethylarginine and cardiovascular risk: systematic review and meta-analysis of 22 prospective studies. Journal of the American Heart Association. 2015;4(6, article e001833) doi: 10.1161/JAHA.115.001833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xuan C., Tian Q. W., Li H., Zhang B. B., He G. W., Lun L. M. Levels of asymmetric dimethylarginine (ADMA), an endogenous nitric oxide synthase inhibitor, and risk of coronary artery disease: a meta-analysis based on 4713 participants. European Journal of Preventive Cardiology. 2016;23(5):502–510. doi: 10.1177/2047487315586094. [DOI] [PubMed] [Google Scholar]
- 43.Cui G., Wang H., Li R., et al. Polymorphism of tumor necrosis factor alpha (TNF-alpha) gene promoter, circulating TNF-alpha level, and cardiovascular risk factor for ischemic stroke. Journal of Neuroinflammation. 2012;9:p. 235. doi: 10.1186/1742-2094-9-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ridker P. M., Rifai N., Pfeffer M., Sacks F., Lepage S., Braunwald E. Elevation of tumor necrosis factor-α and increased risk of recurrent coronary events after myocardial infarction. Circulation. 2000;101(18):2149–2153. doi: 10.1161/01.CIR.101.18.2149. [DOI] [PubMed] [Google Scholar]
- 45.Rios-Navarro C., de Pablo C., Collado-Diaz V., et al. Differential effects of anti-TNF-α and anti-IL-12/23 agents on human leukocyte-endothelial cell interactions. European Journal of Pharmacology. 2015;765:355–365. doi: 10.1016/j.ejphar.2015.08.054. [DOI] [PubMed] [Google Scholar]
- 46.Avgerinou G., Tousoulis D., Siasos G., et al. Anti-tumor necrosis factor alpha treatment with adalimumab improves significantly endothelial function and decreases inflammatory process in patients with chronic psoriasis. International Journal of Cardiology. 2011;151(3):382–383. doi: 10.1016/j.ijcard.2011.06.112. [DOI] [PubMed] [Google Scholar]
- 47.Spinelli F. R., Di Franco M., Metere A., et al. Decrease of asymmetric dimethyl arginine after anti-TNF therapy in patients with rheumatoid arthritis. Drug Development Research. 2014;75(Supplement 1):S67–S69. doi: 10.1002/ddr.21200. [DOI] [PubMed] [Google Scholar]
- 48.Spinelli F. R., Metere A., Barbati C., et al. Effect of therapeutic inhibition of TNF on circulating endothelial progenitor cells in patients with rheumatoid arthritis. Mediators of Inflammation. 2013;2013:8. doi: 10.1155/2013/537539.537539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bevilacqua M. P., Pober J. S., Wheeler M. E., Cotran R. S., Gimbrone M. A., Jr. Interleukin 1 acts on cultured human vascular endothelium to increase the adhesion of polymorphonuclear leukocytes, monocytes, and related leukocyte cell lines. The Journal of Clinical Investigation. 1985;76(5):2003–2011. doi: 10.1172/JCI112200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bevilacqua M. P., Pober J. S., Majeau G. R., Cotran R. S., Gimbrone M. A., Jr Interleukin 1 (IL-1) induces biosynthesis and cell surface expression of procoagulant activity in human vascular endothelial cells. The Journal of Experimental Medicine. 1984;160(2):618–623. doi: 10.1084/jem.160.2.618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ikeda U., Ikeda M., Oohara T., Kano S., Yaginuma T. Mitogenic action of interleukin-1α on vascular smooth muscle cells mediated by PDGF. Atherosclerosis. 1990;84(2-3):183–188. doi: 10.1016/0021-9150(90)90089-2. [DOI] [PubMed] [Google Scholar]
- 52.Nomoto A., Mutoh S., Hagihara H., Yamaguchi I. Smooth muscle cell migration induced by inflammatory cell products and its inhibition by a potent calcium antagonist, nilvadipine. Atherosclerosis. 1988;72(2-3):213–219. doi: 10.1016/0021-9150(88)90083-4. [DOI] [PubMed] [Google Scholar]
- 53.Shimokawa H., Ito A., Fukumoto Y., et al. Chronic treatment with interleukin-1 beta induces coronary intimal lesions and vasospastic responses in pigs in vivo. The role of platelet-derived growth factor. The Journal of Clinical Investigation. 1996;97(3):769–776. doi: 10.1172/JCI118476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Francis S. E., Camp N. J., Dewberry R. M., et al. Interleukin-1 receptor antagonist gene polymorphism and coronary artery disease. Circulation. 1999;99(7):861–866. doi: 10.1161/01.CIR.99.7.861. [DOI] [PubMed] [Google Scholar]
- 55.Ikonomidis I., Lekakis J. P., Nikolaou M., et al. Inhibition of interleukin-1 by anakinra improves vascular and left ventricular function in patients with rheumatoid arthritis. Circulation. 2008;117(20):2662–2669. doi: 10.1161/CIRCULATIONAHA.107.731877. [DOI] [PubMed] [Google Scholar]
- 56.Vallejo S., Palacios E., Romacho T., Villalobos L., Peiro C., Sanchez-Ferrer C. F. The interleukin-1 receptor antagonist anakinra improves endothelial dysfunction in streptozotocin-induced diabetic rats. Cardiovascular Diabetology. 2014;13(1):p. 158. doi: 10.1186/s12933-014-0158-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ridker P. M., Everett B. M., Thuren T., et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. The New England Journal of Medicine. 2017;377(12):1119–1131. doi: 10.1056/NEJMoa1707914. [DOI] [PubMed] [Google Scholar]
- 58.Wassmann S., Stumpf M., Strehlow K., et al. Interleukin-6 induces oxidative stress and endothelial dysfunction by overexpression of the angiotensin II type 1 receptor. Circulation Research. 2004;94(4):534–541. doi: 10.1161/01.RES.0000115557.25127.8D. [DOI] [PubMed] [Google Scholar]
- 59.Esteve E., Castro A., Lopez-Bermejo A., Vendrell J., Ricart W., Fernandez-Real J. M. Serum interleukin-6 correlates with endothelial dysfunction in healthy men independently of insulin sensitivity. Diabetes Care. 2007;30(4):939–945. doi: 10.2337/dc06-1793. [DOI] [PubMed] [Google Scholar]
- 60.Naya M., Tsukamoto T., Morita K., et al. Plasma interleukin-6 and tumor necrosis factor-α can predict coronary endothelial dysfunction in hypertensive patients. Hypertension Research. 2007;30(6):541–548. doi: 10.1291/hypres.30.541. [DOI] [PubMed] [Google Scholar]
- 61.Nawawi H., Osman N. S., Annuar R., Khalid B. A., Yusoff K. Soluble intercellular adhesion molecule-1 and interleukin-6 levels reflect endothelial dysfunction in patients with primary hypercholesterolaemia treated with atorvastatin. Atherosclerosis. 2003;169(2):283–291. doi: 10.1016/S0021-9150(03)00193-X. [DOI] [PubMed] [Google Scholar]
- 62.Mahmud A., Feely J. Arterial stiffness is related to systemic inflammation in essential hypertension. Hypertension. 2005;46(5):1118–1122. doi: 10.1161/01.HYP.0000185463.27209.b0. [DOI] [PubMed] [Google Scholar]
- 63.Kaptoge S., Seshasai S. R., Gao P., et al. Inflammatory cytokines and risk of coronary heart disease: new prospective study and updated meta-analysis. European Heart Journal. 2014;35(9):578–589. doi: 10.1093/eurheartj/eht367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Protogerou A. D., Zampeli E., Fragiadaki K., Stamatelopoulos K., Papamichael C., Sfikakis P. P. A pilot study of endothelial dysfunction and aortic stiffness after interleukin-6 receptor inhibition in rheumatoid arthritis. Atherosclerosis. 2011;219(2):734–736. doi: 10.1016/j.atherosclerosis.2011.09.015. [DOI] [PubMed] [Google Scholar]
- 65.Bacchiega B. C., Bacchiega A. B., Usnayo M. J., Bedirian R., Singh G., Pinheiro G. D. Interleukin 6 inhibition and coronary artery disease in a high-risk population: a prospective community-based clinical study. Journal of the American Heart Association. 2017;6(3, article e005038) doi: 10.1161/JAHA.116.005038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ruiz-Limon P., Ortega R., Arias de la Rosa I., et al. Tocilizumab improves the proatherothrombotic profile of rheumatoid arthritis patients modulating endothelial dysfunction, NETosis, and inflammation. Translational Research. 2017;183:87–103. doi: 10.1016/j.trsl.2016.12.003. [DOI] [PubMed] [Google Scholar]
- 67.Floris A., Piga M., Cauli A., Mathieu A. Predictors of flares in systemic lupus erythematosus: preventive therapeutic intervention based on serial anti-dsDNA antibodies assessment. Analysis of a monocentric cohort and literature review. Autoimmunity Reviews. 2016;15(7):656–663. doi: 10.1016/j.autrev.2016.02.019. [DOI] [PubMed] [Google Scholar]
- 68.Braun J. Methotrexate: optimizing the efficacy in rheumatoid arthritis. Therapeutic Advances in Musculoskeletal Disease. 2011;3(3):151–158. doi: 10.1177/1759720X11408635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Choi H. K., Hernan M. A., Seeger J. D., Robins J. M., Wolfe F. Methotrexate and mortality in patients with rheumatoid arthritis: a prospective study. The Lancet. 2002;359(9313):1173–1177. doi: 10.1016/S0140-6736(02)08213-2. [DOI] [PubMed] [Google Scholar]
- 70.Krause D., Schleusser B., Herborn G., Rau R. Response to methotrexate treatment is associated with reduced mortality in patients with severe rheumatoid arthritis. Arthritis & Rheumatism. 2000;43(1):14–21. doi: 10.1002/1529-0131(200001)43:1<14::AID-ANR3>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 71.Wasko M. C., Dasgupta A., Hubert H., Fries J. F., Ward M. M. Propensity-adjusted association of methotrexate with overall survival in rheumatoid arthritis. Arthritis & Rheumatism. 2013;65(2):334–342. doi: 10.1002/art.37723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bannwarth B., Pehourcq F., Schaeverbeke T., Dehais J. Clinical pharmacokinetics of low-dose pulse methotrexate in rheumatoid arthritis. Clinical Pharmacokinetics. 1996;30(3):194–210. doi: 10.2165/00003088-199630030-00002. [DOI] [PubMed] [Google Scholar]
- 73.Pan S., Stamp L. K., Duffull S. B., et al. Assessment of the relationship between methotrexate polyglutamates in red blood cells and clinical response in patients commencing methotrexate for rheumatoid arthritis. Clinical Pharmacokinetics. 2014;53(12):1161–1170. doi: 10.1007/s40262-014-0179-5. [DOI] [PubMed] [Google Scholar]
- 74.de Rotte M. C., den Boer E., de Jong P. H., et al. Methotrexate polyglutamates in erythrocytes are associated with lower disease activity in patients with rheumatoid arthritis. Annals of the Rheumatic Diseases. 2015;74(2):408–414. doi: 10.1136/annrheumdis-2013-203725. [DOI] [PubMed] [Google Scholar]
- 75.Inoue K., Yuasa H. Molecular basis for pharmacokinetics and pharmacodynamics of methotrexate in rheumatoid arthritis therapy. Drug Metabolism and Pharmacokinetics. 2014;29(1):12–19. doi: 10.2133/dmpk.DMPK-13-RV-119. [DOI] [PubMed] [Google Scholar]
- 76.Hasko G., Cronstein B. Regulation of inflammation by adenosine. Frontiers in Immunology. 2013;4:p. 85. doi: 10.3389/fimmu.2013.00085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cronstein B. N., Naime D., Ostad E. The antiinflammatory mechanism of methotrexate. Increased adenosine release at inflamed sites diminishes leukocyte accumulation in an in vivo model of inflammation. The Journal of Clinical Investigation. 1993;92(6):2675–2682. doi: 10.1172/JCI116884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hardie D. G. AMP-activated protein kinase-an energy sensor that regulates all aspects of cell function. Genes & Development. 2011;25(18):1895–1908. doi: 10.1101/gad.17420111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Antonioli L., Colucci R., Pellegrini C., et al. The AMPK enzyme-complex: from the regulation of cellular energy homeostasis to a possible new molecular target in the management of chronic inflammatory disorders. Expert Opinion on Therapeutic Targets. 2016;20(2):179–191. doi: 10.1517/14728222.2016.1086752. [DOI] [PubMed] [Google Scholar]
- 80.Boin F., Erre G. L., Posadino A. M., et al. Oxidative stress-dependent activation of collagen synthesis is induced in human pulmonary smooth muscle cells by sera from patients with scleroderma-associated pulmonary hypertension. Orphanet Journal of Rare Diseases. 2014;9(1):p. 123. doi: 10.1186/s13023-014-0123-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Igata M., Motoshima H., Tsuruzoe K., et al. Adenosine monophosphate-activated protein kinase suppresses vascular smooth muscle cell proliferation through the inhibition of cell cycle progression. Circulation Research. 2005;97(8):837–844. doi: 10.1161/01.RES.0000185823.73556.06. [DOI] [PubMed] [Google Scholar]
- 82.Ma Y., Li L., Shao Y., Bai X., Bai T., Huang X. Methotrexate improves perivascular adipose tissue/endothelial dysfunction via activation of AMPK/eNOS pathway. Molecular Medicine Reports. 2017;15(4):2353–2359. doi: 10.3892/mmr.2017.6225. [DOI] [PubMed] [Google Scholar]
- 83.Micha R., Imamura F., Wyler von Ballmoos M., et al. Systematic review and meta-analysis of methotrexate use and risk of cardiovascular disease. The American Journal of Cardiology. 2011;108(9):1362–1370. doi: 10.1016/j.amjcard.2011.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Roubille C., Richer V., Starnino T., et al. The effects of tumour necrosis factor inhibitors, methotrexate, non-steroidal anti-inflammatory drugs and corticosteroids on cardiovascular events in rheumatoid arthritis, psoriasis and psoriatic arthritis: a systematic review and meta-analysis. Annals of the Rheumatic Diseases. 2015;74(3):480–489. doi: 10.1136/annrheumdis-2014-206624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Seitz M., Zwicker M., Loetscher P. Effects of methotrexate on differentiation of monocytes and production of cytokine inhibitors by monocytes. Arthritis & Rheumatism. 1998;41(11):2032–2038. doi: 10.1002/1529-0131(199811)41:11<2032::AID-ART19>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 86.Edwards C. K., 3rd, Bendele A. M., Reznikov L. I., et al. Soluble human p55 and p75 tumor necrosis factor receptors reverse spontaneous arthritis in transgenic mice expressing transmembrane tumor necrosis factor α. Arthritis & Rheumatism. 2006;54(9):2872–2885. doi: 10.1002/art.22077. [DOI] [PubMed] [Google Scholar]
- 87.Sajjadi F. G., Takabayashi K., Foster A. C., Domingo R. C., Firestein G. S. Inhibition of TNF-alpha expression by adenosine: role of A3 adenosine receptors. Journal of Immunology. 1996;156(9):3435–3442. [PubMed] [Google Scholar]
- 88.Bulgarelli A., Martins Dias A. A., Caramelli B., Maranhao R. C. Treatment with methotrexate inhibits atherogenesis in cholesterol-fed rabbits. Journal of Cardiovascular Pharmacology. 2012;59(4):308–314. doi: 10.1097/FJC.0b013e318241c385. [DOI] [PubMed] [Google Scholar]
- 89.Thornton C. C., Al-Rashed F., Calay D., et al. Methotrexate-mediated activation of an AMPK-CREB-dependent pathway: a novel mechanism for vascular protection in chronic systemic inflammation. Annals of the Rheumatic Diseases. 2016;75(2):439–448. doi: 10.1136/annrheumdis-2014-206305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yamasaki E., Soma Y., Kawa Y., Mizoguchi M. Methotrexate inhibits proliferation and regulation of the expression of intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 by cultured human umbilical vein endothelial cells. The British Journal of Dermatology. 2003;149(1):30–38. doi: 10.1046/j.1365-2133.2003.05407.x. [DOI] [PubMed] [Google Scholar]
- 91.Kalogeris T. J., Baines C., Korthuis R. J. Adenosine prevents TNFα-induced decrease in endothelial mitochondrial mass via activation of eNOS-PGC-1α regulatory axis. PLoS One. 2014;9(6, article e98459) doi: 10.1371/journal.pone.0098459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cohen E. S., Law W. R., Easington C. R., et al. Adenosine deaminase inhibition attenuates microvascular dysfunction and improves survival in sepsis. American Journal of Respiratory and Critical Care Medicine. 2002;166(1):16–20. doi: 10.1164/rccm.200109-014OC. [DOI] [PubMed] [Google Scholar]
- 93.Summers C. M., Hammons A. L., Arora J., et al. Methotrexate modulates folate phenotype and inflammatory profile in EA.hy 926 cells. European Journal of Pharmacology. 2014;732:60–67. doi: 10.1016/j.ejphar.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Li J., Wang Y., Wang Y., et al. Pharmacological activation of AMPK prevents Drp1-mediated mitochondrial fission and alleviates endoplasmic reticulum stress-associated endothelial dysfunction. Journal of Molecular and Cellular Cardiology. 2015;86:62–74. doi: 10.1016/j.yjmcc.2015.07.010. [DOI] [PubMed] [Google Scholar]
- 95.Hou X., Pei F. Estradiol inhibits cytokine-induced expression of VCAM-1 and ICAM-1 in cultured human endothelial cells via AMPK/PPARα activation. Cell Biochemistry and Biophysics. 2015;72(3):709–717. doi: 10.1007/s12013-015-0522-y. [DOI] [PubMed] [Google Scholar]
- 96.Quan A., Pan Y., Singh K. K., et al. Cardiovascular inflammation is reduced with methotrexate in diabetes. Molecular and Cellular Biochemistry. 2017;432(1-2):159–167. doi: 10.1007/s11010-017-3006-0. [DOI] [PubMed] [Google Scholar]
- 97.Hassanian S. M., Dinarvand P., Rezaie A. R. Adenosine regulates the proinflammatory signaling function of thrombin in endothelial cells. Journal of Cellular Physiology. 2014;229(9):1292–1300. doi: 10.1002/jcp.24568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bouma M. G., van den Wildenberg F. A., Buurman W. A. Adenosine inhibits cytokine release and expression of adhesion molecules by activated human endothelial cells. The American Journal of Physiology. 1996;270, 2 Part 1:C522–C529. doi: 10.1152/ajpcell.1996.270.2.C522. [DOI] [PubMed] [Google Scholar]
- 99.Shryock J. C., Belardinelli L. Adenosine and adenosine receptors in the cardiovascular system: biochemistry, physiology, and pharmacology. The American Journal of Cardiology. 1997;79(12) Supplement 1:2–10. doi: 10.1016/S0002-9149(97)00256-7. [DOI] [PubMed] [Google Scholar]
- 100.Olsson R. A., Pearson J. D. Cardiovascular purinoceptors. Physiological Reviews. 1990;70(3):761–845. doi: 10.1152/physrev.1990.70.3.761. [DOI] [PubMed] [Google Scholar]
- 101.Stella L., de Novellis V., Marabese I., et al. The role of A3 adenosine receptors in central regulation of arterial blood pressure. British Journal of Pharmacology. 1998;125(3):437–440. doi: 10.1038/sj.bjp.0702126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Koupenova M., Johnston-Cox H., Ravid K. Regulation of atherosclerosis and associated risk factors by adenosine and adenosine receptors. Current Atherosclerosis Reports. 2012;14(5):460–468. doi: 10.1007/s11883-012-0263-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Varani K., Portaluppi F., Merighi S., Ongini E., Belardinelli L., Borea P. A. Caffeine alters A2A adenosine receptors and their function in human platelets. Circulation. 1999;99(19):2499–2502. doi: 10.1161/01.CIR.99.19.2499. [DOI] [PubMed] [Google Scholar]
- 104.Antonioli L., Blandizzi C., Csoka B., Pacher P., Hasko G. Adenosine signalling in diabetes mellitus--pathophysiology and therapeutic considerations. Nature Reviews. Endocrinology. 2015;11(4):228–241. doi: 10.1038/nrendo.2015.10. [DOI] [PubMed] [Google Scholar]
- 105.Li D., Wang D., Wang Y., Ling W., Feng X., Xia M. Adenosine monophosphate-activated protein kinase induces cholesterol efflux from macrophage-derived foam cells and alleviates atherosclerosis in apolipoprotein E-deficient mice. The Journal of Biological Chemistry. 2010;285(43):33499–33509. doi: 10.1074/jbc.M110.159772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chen Z., Peng I. C., Sun W., et al. AMP-activated protein kinase functionally phosphorylates endothelial nitric oxide synthase Ser633. Circulation Research. 2009;104(4):496–505. doi: 10.1161/CIRCRESAHA.108.187567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Goirand F., Solar M., Athea Y., et al. Activation of AMP kinase α1 subunit induces aortic vasorelaxation in mice. The Journal of Physiology. 2007;581(3):1163–1171. doi: 10.1113/jphysiol.2007.132589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nagata D., Takeda R., Sata M., et al. AMP-activated protein kinase inhibits angiotensin II-stimulated vascular smooth muscle cell proliferation. Circulation. 2004;110(4):444–451. doi: 10.1161/01.CIR.0000136025.96811.76. [DOI] [PubMed] [Google Scholar]
- 109.Ford R. J., Teschke S. R., Reid E. B., Durham K. K., Kroetsch J. T., Rush J. W. AMP-activated protein kinase activator AICAR acutely lowers blood pressure and relaxes isolated resistance arteries of hypertensive rats. Journal of Hypertension. 2012;30(4):725–733. doi: 10.1097/HJH.0b013e32835050ca. [DOI] [PubMed] [Google Scholar]
- 110.Buhl E. S., Jessen N., Pold R., et al. Long-term AICAR administration reduces metabolic disturbances and lowers blood pressure in rats displaying features of the insulin resistance syndrome. Diabetes. 2002;51(7):2199–2206. doi: 10.2337/diabetes.51.7.2199. [DOI] [PubMed] [Google Scholar]
- 111.Wu N., Zheng B., Shaywitz A., et al. AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Molecular Cell. 2013;49(6):1167–1175. doi: 10.1016/j.molcel.2013.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.McGee S. L., van Denderen B. J., Howlett K. F., et al. AMP-activated protein kinase regulates GLUT4 transcription by phosphorylating histone deacetylase 5. Diabetes. 2008;57(4):860–867. doi: 10.2337/db07-0843. [DOI] [PubMed] [Google Scholar]
- 113.Marsin A. S., Bertrand L., Rider M. H., et al. Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Current Biology. 2000;10(20):1247–1255. doi: 10.1016/S0960-9822(00)00742-9. [DOI] [PubMed] [Google Scholar]
- 114.Marsin A. S., Bouzin C., Bertrand L., Hue L. The stimulation of glycolysis by hypoxia in activated monocytes is mediated by AMP-activated protein kinase and inducible 6-phosphofructo-2-kinase. The Journal of Biological Chemistry. 2002;277(34):30778–30783. doi: 10.1074/jbc.M205213200. [DOI] [PubMed] [Google Scholar]
- 115.Xie Z., Zhang J., Wu J., Viollet B., Zou M. H. Upregulation of mitochondrial uncoupling protein-2 by the AMP-activated protein kinase in endothelial cells attenuates oxidative stress in diabetes. Diabetes. 2008;57(12):3222–3230. doi: 10.2337/db08-0610. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 116.Li X. N., Song J., Zhang L., et al. Activation of the AMPK-FOXO3 pathway reduces fatty acid-induced increase in intracellular reactive oxygen species by upregulating thioredoxin. Diabetes. 2009;58(10):2246–2257. doi: 10.2337/db08-1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hirata S., Matsubara T., Saura R., Tateishi H., Hirohata K. Inhibition of in vitro vascular endothelial cell proliferation and in vivo neovascularization by low-dose methotrexate. Arthritis & Rheumatism. 1989;32(9):1065–1073. doi: 10.1002/anr.1780320903. [DOI] [PubMed] [Google Scholar]
- 118.Annussek T., Szuwart T., Kleinheinz J., Koiky C., Wermker K. In vitro inhibition of HUVECs by low dose methotrexate - insights into oral adverse events. Head & Face Medicine. 2014;10(1):p. 19. doi: 10.1186/1746-160X-10-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gao L., Chalupsky K., Stefani E., Cai H. Mechanistic insights into folic acid-dependent vascular protection: dihydrofolate reductase (DHFR)-mediated reduction in oxidant stress in endothelial cells and angiotensin II-infused mice: a novel HPLC-based fluorescent assay for DHFR activity. Journal of Molecular and Cellular Cardiology. 2009;47(6):752–760. doi: 10.1016/j.yjmcc.2009.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Pastrana J. L., Sha X., Virtue A., et al. Regulatory T cells and atherosclerosis. Journal of Clinical & Experimental Cardiology. 2013;1(Supplement 12):p. 2. doi: 10.4172/2155-9880.S12-002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Everett B. M., Pradhan A. D., Solomon D. H., et al. Rationale and design of the cardiovascular inflammation reduction trial: a test of the inflammatory hypothesis of atherothrombosis. American Heart Journal. 2013;166(2):199–207.e15. doi: 10.1016/j.ahj.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]