

Abstract
DNA glycosylases preserve genome integrity and define the specificity of the base excision repair pathway for discreet, detrimental modifications, and thus, the mechanisms by which glycosylases locate DNA damage are of particular interest. Bacterial AlkC and AlkD are specific for cationic alkylated nucleobases and have a distinctive HEAT‐like repeat (HLR) fold. AlkD uses a unique non‐base‐flipping mechanism that enables excision of bulky lesions more commonly associated with nucleotide excision repair. In contrast, AlkC has a much narrower specificity for small lesions, principally N3‐methyladenine (3mA). Here, we describe how AlkC selects for and excises 3mA using a non‐base‐flipping strategy distinct from that of AlkD. A crystal structure resembling a catalytic intermediate complex shows how AlkC uses unique HLR and immunoglobulin‐like domains to induce a sharp kink in the DNA, exposing the damaged nucleobase to active site residues that project into the DNA. This active site can accommodate and excise N3‐methylcytosine (3mC) and N1‐methyladenine (1mA), which are also repaired by AlkB‐catalyzed oxidative demethylation, providing a potential alternative mechanism for repair of these lesions in bacteria.
Keywords: 3‐methyladenine, 3‐methylcytosine, base excision repair, DNA glycosylase, DNA repair
Subject Categories: DNA Replication, Repair & Recombination; Structural Biology
Introduction
Cellular metabolites and environmental toxins damage DNA by creating a variety of covalent DNA adducts that impair normal cellular processes and lead to heritable diseases, cancer, aging, and cell death (Friedberg et al, 2006; Jackson & Bartek, 2009). Alkylation of nucleobase substituents and ring nitrogens is a major source of DNA damage that can directly inhibit replication and transcription or can degenerate to other forms of damage including abasic sites and strand breaks (Wyatt & Pittman, 2006; Krokan & Bjørås, 2013). The toxicity of these DNA lesions is the basis for the use of alkylating agents in cancer chemotherapy (Sedgwick, 2004). To avoid the toxic effects of DNA alkylation and maintain integrity of their genomes, all organisms possess multiple repair mechanisms to remove the diversity of alkyl‐DNA modifications. Nucleotide excision repair (NER) is the predominant mechanism to eliminate helix‐distorting and bulky adducts, whereas small nucleobase modifications (e.g., methyl, etheno groups) are repaired by direct reversal or base excision repair (BER) pathways (Sedgwick, 2004; Reardon & Sancar, 2005; Mishina & He, 2006). N1‐methyladenine (1mA) and N3‐methylcytosine (3mC), predominant in single‐stranded (ss) DNA and RNA, are demethylated by the AlkB family of Fe(II)/α‐ketoglutarate‐dependent dioxygenases. In contrast, N3‐ and N7‐alkylpurines, the most abundant double‐stranded DNA alkylation products, are removed from the genome by BER, whereby the damaged nucleobase is cleaved from the phosphodeoxyribose backbone by lesion‐specific DNA glycosylases that hydrolyze the N‐glycosidic bond. The resulting abasic site is nicked by apurinic/apyrimidinic (AP) endonuclease to generate a 3′‐hydroxyl substrate for gap repair synthesis by DNA polymerase.
DNA glycosylases that remove alkylation damage are essential to eukaryotes, archaea, and bacteria (Riazuddin & Lindahl, 1978; Thomas et al, 1982; Chen et al, 1989; O'Connor & Laval, 1991; Birkeland et al, 2002). Bacteria often contain paralogs that eliminate diverse types of damage resulting from endogenous versus environmental sources. For example, in Escherichia coli, the AlkA glycosylase is induced to remove a broad spectrum of alkylated and deaminated bases upon cellular exposure to alkylation agents, while the constitutively active Tag enzyme is highly specific for N3‐methyladenine (3mA) formed endogenously (McCarthy et al, 1984; Bjelland et al, 1993, 1994; Saparbaev et al, 1995; O'Brien & Ellenberger, 2004). Damage recognition and excision by these and other glycosylases rely on a base‐flipping mechanism, whereby the damaged nucleotide is rotated ~180° around the phosphate backbone and sequestered inside a nucleobase binding pocket on the protein surface (Brooks et al, 2013). This pocket contains residues that facilitate depurination by activating or stabilizing the nucleobase leaving group and/or electrostatically stabilizing an oxocarbenium intermediate that reacts with a water molecule to generate the abasic site product (Fig 1A) (Drohat & Maiti, 2014). The extrahelical orientation of the substrate in the catalytic complex is stabilized by DNA‐intercalating protein residues that fill the space generated by the missing base. Thus, the specificity of a DNA glycosylase for a particular substrate is achieved by a combination of duplex interrogation and complementarity of the nucleobase and the active site pocket.
Figure 1. AlkCα and AlkCβ are specific for N3‐methyladenine (3mA).
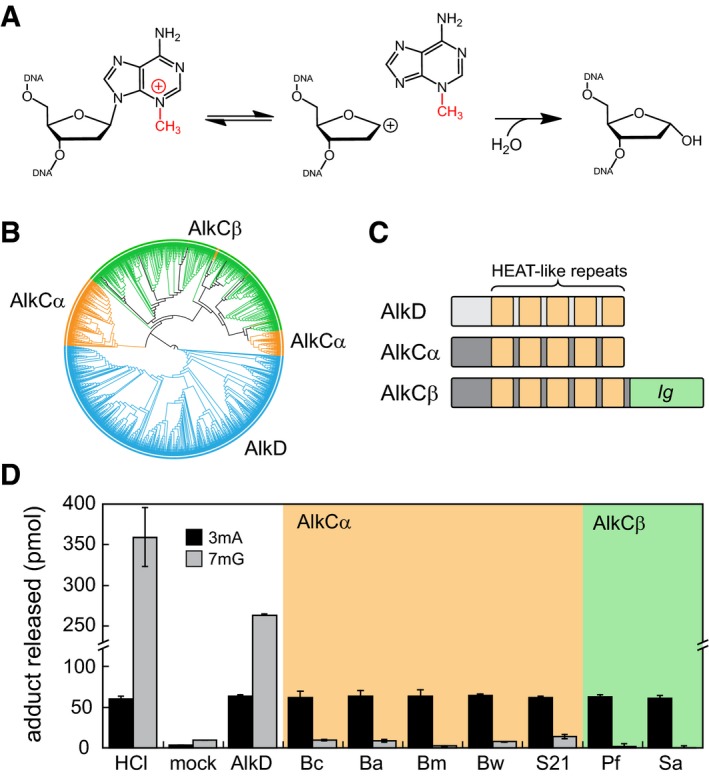
- 3mA excision reaction catalyzed by AlkC.
- Phylogenetic tree of 779 AlkC (orange, green) and 764 AlkD (blue) protein sequences visualized using the iTOL web server (Letunic & Bork, 2011).
- Schematic of AlkC and AlkD protein domains.
- Release of 3mA (black bars) and 7mG (gray bars) from methylated genomic DNA after a 1‐h incubation with either HCl, no enzyme (mock), Bacillus cereus AlkD, or one of seven AlkC orthologs. Bc, Bacillus cereus; Ba, Bacillus anthracis; Bm, Bacillus mycoides; Bw, Bacillus weihenstephanensis; S21, Sphingobacterium sp. 21; Pf, Pseudomonas fluorescens; Sa, Streptomyces albus. Values are the mean ± SD (n = 3 for each). HCl and no‐enzyme controls provide upper and lower limits of 3mA and 7mG detection.
The bacterial AlkC and AlkD glycosylases, originally identified in Bacillus cereus (Bc), comprise a distinct superfamily of DNA glycosylase selective for positively charged N3‐ and N7‐alkylpurines (Alseth et al, 2006; Dalhus et al, 2007). They specifically lack excision activity toward uncharged 1,N 6‐ethenoadenine or hypoxanthine nucleobases (Alseth et al, 2006), the common substrates of many previously characterized alkylpurine DNA glycosylases. Structures of BcAlkD revealed a new fold composed of tandem HEAT‐like repeats (HLRs) that form a left‐handed solenoid around the DNA duplex (Rubinson et al, 2008, 2010; Rubinson & Eichman, 2012). Unlike other glycosylases, AlkD does not use a base‐flipping mechanism for damage recognition or base excision (Mullins et al, 2015b). Instead, the protein traps non‐Watson–Crick base pairs in a sheared, base‐stacked conformation with catalytic active site residues in direct contact with the deoxyribose, but not the nucleobase, of the damaged nucleoside. The lack of protein–nucleobase contacts enables AlkD to excise both the major and minor groove lesions 3mA and N7‐methylguanine (7mG). Similarly, by not confining the modified nucleobase inside a binding pocket, AlkD can excise bulky lesions, including pyridyloxobutyl (POB) adducts resulting from the carcinogen NNK (nicotine‐derived nitrosamine ketone) (Rubinson et al, 2010), as well as N3‐yatakemycinyladenine (YTMA) formed from the highly genotoxic bacterial natural product yatakemycin (YTM) (Xu et al, 2012; Mullins et al, 2015b, 2017). Thus, AlkD excises a diverse spectrum of cationic lesions, including bulky lesions expected to be processed by NER.
AlkC from B. cereus (BcAlkC) shares 15.8% identity and 33.7% similarity with BcAlkD and has been predicted to adopt the HLR architecture (Dalhus et al, 2007). However, in contrast to AlkD, AlkC has a strong preference for 3mA and N3‐methylguanine (3mG) and only weak activity for 7mG (Alseth et al, 2006; Mullins et al, 2013). Given AlkD's unique non‐base‐flipping mechanism and activity for bulky lesions, we were interested to expand our understanding of this new DNA repair superfamily by determining the basis for AlkC's apparent limited substrate specificity. We carried out a comprehensive phylogenetic, biochemical, and structural comparison of AlkC and AlkD proteins and found that the majority of AlkC proteins contain an immunoglobulin (Ig)‐like domain not yet observed in a DNA repair enzyme and that is essential for base excision activity in those enzymes. We also found that unlike AlkD, AlkC does not remove YTMA adducts either in vitro or in vivo. The crystal structure of Pseudomonas fluorescens (Pf) AlkC bound to damaged DNA revealed that the HLR and Ig‐like domains wrap almost completely around the DNA duplex to recognize the damaged base pair from opposite major and minor grooves. Like AlkD, AlkC does not extrude the damaged nucleobase from the DNA helix. However, unlike AlkD, AlkC induces a sharp kink in the DNA and accesses the damage by inserting active site residues into the exposed base stack. The structure and supporting mutational analysis of base excision by AlkC provide a mechanistic basis for how AlkC selectively excises 3mA from DNA. Additionally, we show that AlkC's unique active site is also capable of robust excision activity for 3mC, which is normally repaired by AlkB‐catalyzed oxidative demethylation. This work provides a molecular basis for how a non‐base‐flipping glycosylase selects for discrete methylated bases, and describes an alternative mechanism for removal of 3mC in bacteria.
Results and Discussion
Proteins within two AlkC subgroups are functionally distinct from AlkD
Despite a predicted similarity in HLR secondary structure, AlkC and AlkD from B. cereus were previously shown to be phylogenetically and functionally distinct (Alseth et al, 2006; Mullins et al, 2013). To better understand the AlkC family of proteins and how they differ from AlkD, we obtained 779 AlkC and 764 AlkD orthologous sequences by performing an iterative PSI‐BLAST search against the NCBI non‐redundant protein database and then used them to reconstruct a phylogenetic tree. 93% of AlkC sequences were widespread among actinobacteria, bacteroidetes, firmicutes, and proteobacteria, and only rarely found in archaea and eukaryotes, largely consistent with a previous analysis (Backe et al, 2013). We found that AlkC proteins clustered into two distinct clades that we denote AlkCα and AlkCβ (Fig 1B), both of which contain an HLR domain distinct from AlkD mainly within the N‐terminal 60–80 residues. The AlkCβ clade, which constitutes the majority (70%) of the total AlkC sequences, contains an additional ~100‐residue putative immunoglobulin (Ig)‐like fold at the C‐terminus (Fig 1C, Appendix Fig S1).
The specificity of AlkC for 3mA has been characterized only from the B. cereus enzyme, which belongs to the AlkCα class (Alseth et al, 2006; Mullins et al, 2013). To determine whether this 3mA selectivity is a general characteristic of both AlkCα and AlkCβ proteins, we cloned and purified five AlkCα and two AlkCβ orthologs and measured their ability to excise 3mA and 7mG nucleobases from N‐methyl‐N‐nitrosourea (MNU)‐treated genomic DNA, the major methylated products of which are 7mG (66%) and 3mA (8%) (Lawley, 1976). All seven AlkC proteins released the maximum amount of 3mA after 1 h as judged by treatment of substrate with HCl, but exhibited only very low activity toward 7mG as compared to an AlkD control (Fig 1D). Consistent with this, both BcAlkC (α) and PfAlkC (β) excised 7mG from a defined oligodeoxynucleotide substrate after 24 h, albeit with much slower kinetics than BcAlkD (Appendix Fig S2A). Thus, 3mA specificity seems to be a general property of all AlkC proteins, independent of the presence of the putative Ig‐like domain.
We also found that AlkC's specificity for N3‐alkyladenine adducts is limited to small adducts. Neither BcAlkC nor PfAlkC was able to cleave YTMA lesions for which AlkD exhibits robust activity (Mullins et al, 2017) (Appendix Fig S2B). We previously showed that in addition to AlkD excision of YTMA in vitro, Bacillus anthracis cells lacking AlkD exhibited a sensitivity to YTM (Mullins et al, 2015b). We therefore tested growth of AlkC‐deficient B. anthracis strains in the presence of YTM. Consistent with our in vitro results, YTM sensitivity of a ΔalkC strain did not differ from that of wild‐type B. anthracis, nor did a ΔalkCΔalkD double mutant show additional sensitivity compared to ΔalkD (Appendix Fig S2C). Thus, substrate specificities of both α‐ and β‐subgroups of AlkC are distinct from AlkD, suggesting that AlkC utilizes a different strategy to recognize damaged DNA despite its predicted structural similarity to AlkD.
AlkC encircles and bends damaged DNA
To determine the structural basis for AlkC's preference for 3mA and whether a non‐base‐flipping mechanism is a common feature among HLR glycosylases, we determined a crystal structure of PfAlkC in complex with DNA containing 1′‐aza‐2′,4′‐dideoxyribose (1aR), which mimics the oxocarbenium ion intermediate formed upon nucleobase dissociation (Fig 1A) (Hollis et al, 2000; Chu et al, 2011; Schramm, 2011). The PfAlkC/1aR‐DNA model was refined against X‐ray diffraction data extending to 1.8 Å resolution to a crystallographic residual of 14.1% (R free = 16.4%) (Appendix Table S1) and contained two crystallographically unique protein–DNA complexes, each with 361 of 369 amino acids and all 22 nucleotides clearly defined by the electron density. The structures of the two complexes in the asymmetric unit (asu) were virtually identical (r.m.s.d. = 0.42 Å for all atoms), differing only in the location of a statically disordered 5ʹ‐overhanging adenosine A1 on the undamaged strand that forms a crystal packing contact with the same nucleotide in an adjacent protein/DNA complex.
The PfAlkC crystal structure confirmed the presence of distinct HLR and Ig‐like domains, which together wrap almost completely around a highly bent DNA duplex (Fig 2). As a consequence of its interactions with the HLR and Ig‐like domains, the DNA is sharply bent at the 1aR lesion by 60° away from the minor groove and the minor groove widened by 7 Å. The HLR domain (helices αA′‐αN) is similar to that of AlkD in that it contains an N‐terminal α‐helical bundle (αA′‐αC) followed by five HLRs—αD‐αE, αF‐αG, αH‐αI, αJ‐αK, and αL‐αM—that together form a left‐handed solenoid that wraps around one‐half of the DNA duplex from the minor groove side (Fig 2A and B). The N‐terminal α‐helical bundle contacts the backbone of the undamaged strand from the minor groove using polar side chains and the helix dipoles of helices αA′ and αC, which point directly at thymines T6 (opposite the 1aR) and T7, respectively (Fig 2A). All HLRs except HLR1 directly contact the DNA, primarily along the phosphate backbones. The C‐terminal helices of each HLR adorn basic and polar side chains that bind either strand from the minor (HLR2, HLR3) or major (HLR4, HLR5) groove sides (Fig 2C). HLR2 and HLR3 also contain conserved glutamate side chains that directly contact the 1aR. Between HLR3 and HLR4, an 11‐residue loop/α‐helix insertion (αIJ‐loop) not found in AlkD penetrates the minor groove at the lesion. The HLR domain is tethered by a 9‐residue linker to the Ig‐like fold, which is composed of nine β‐strands (βA1‐βG) and an extended loop that contacts the DNA from the major groove side (Fig 2A). Both domains form a positively charged, highly conserved concave surface that engulfs the DNA (Fig 2B). Extensive polar and van der Waals contacts are formed with the DNA backbone 5ʹ to the 1aR and along the entire undamaged strand (Fig 2C).
Figure 2. AlkC encircles damaged DNA .
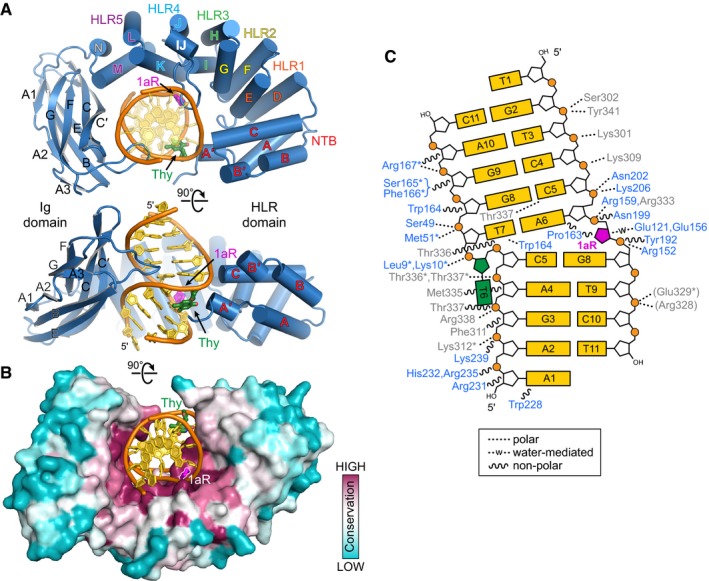
- Two orthogonal views of the PfAlkC/1aR‐DNA complex crystal structure. The protein is colored blue, DNA gold, 1′‐aza‐2′,4′‐dideoxyribose (1aR) magenta, and opposite thymine green. NTB, N‐terminal helical bundle; HLR, HEAT‐like repeat.
- AlkC sequence conservation (purple, high; cyan, low) superposed onto the protein surface.
- Schematic illustration of AlkC‐DNA interactions. Dashed and wavy lines denote polar and non‐polar interactions, respectively. Residues from HLR and Ig‐like domains are blue and gray, respectively. Contacts to the protein backbone are marked with an asterisk, and symmetry‐related contacts are in parentheses.
The AlkC Ig‐like domain is a unique DNA binding motif in bacteria
Ig‐like domains are prevalent in both bacteria and eukaryotes as a generic scaffold (Bork et al, 1994; Halaby & Mornon, 1998; Halaby et al, 1999; Bodelón et al, 2013) and are important for sequence‐specific DNA binding in some eukaryotic transcription factors (Cho et al, 1994; Cramer et al, 1997; Becker et al, 1998; Chen et al, 1998; Nagata et al, 1999; Bravo et al, 2001; Rudolph & Gergen, 2001; Tahirov et al, 2001; Lamoureux et al, 2002; Rohs et al, 2010). To our knowledge, no bacterial Ig‐like domains have been reported to bind DNA or to function in DNA repair. The Ig‐like domain of PfAlkC is composed of a nine‐strand (βA1‐βG) antiparallel β‐sandwich, in which βA1, βA2, βA3, βB, and βE form one β‐sheet packed against a second sheet formed by strands βC, βCʹ, βF, and βG (Fig 2A). The topology is consistent with the C2 subtype (Fig 3B) but with very low sequence similarity, a highly kinked βA strand (βA1‐βA2‐βA3), and a longer βCʹ strand than other C2‐type Ig‐like domains (Halaby et al, 1999; Bodelón et al, 2013). The closest structural homolog of AlkC's Ig‐like domain as judged by a Dali search (Holm & Laakso, 2016) is found in a bacterial β‐glucosidase and has been proposed to contribute to substrate binding and dimerization of that enzyme (McAndrew et al, 2013).
Figure 3. The Ig‐like domain is important for AlkCβ function.
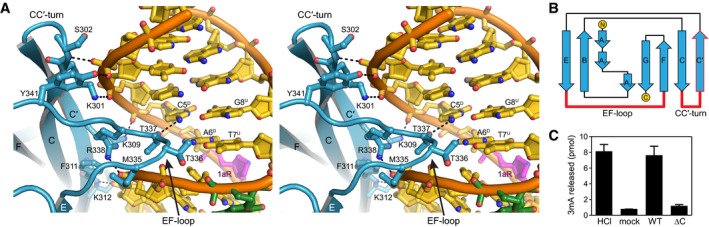
- Stereoimage of the Ig‐like domain (blue) interactions with the major grove of DNA (gold). DNA‐interacting side chains are shown as sticks, and hydrogen bonds are shown as dashed lines. Superscripts in nucleotide labels refer to damaged (D) and undamaged (U) strands.
- Topology of AlkCβ Ig‐like domain. Regions contacting the DNA are highlighted in red.
- Release of 3mA from methylated genomic DNA after a 5‐min incubation with either HCl, no enzyme (mock), PfAlkC (WT), or PfAlkC∆C (∆C). Values are mean ± SD (n = 3).
The Ig‐like domains of eukaryotic transcription factors mediate DNA binding through the loops between strands βA‐βB and βE‐βF (AB‐ and EF‐loops) and the C‐terminal tail regions, all of which emanate from the β‐sandwich core (Fig EV1) (Cho et al, 1994; Cramer et al, 1997; Becker et al, 1998; Chen et al, 1998; Nagata et al, 1999; Bravo et al, 2001; Rudolph & Gergen, 2001; Tahirov et al, 2001; Lamoureux et al, 2002; Rohs et al, 2010). Although we have no evidence for sequence‐specific binding by AlkC, AlkC's Ig‐like domain also mediates DNA binding by the EF‐loop, which in PfAlkC contains a conserved Met‐Thr‐Thr‐Arg motif (residues 336–338) that contacts nucleobases on both strands in the major groove (Fig 3A). Side‐ and main‐chain groups within this motif interact with the phosphoribose groups on the undamaged strand immediately 5ʹ to the orphaned thymidine T6, and Thr337 is engaged with cytosine C5 on the damaged strand. In contrast to the eukaryotic transcription factors, AlkC's AB‐loop does not contact DNA, despite its orientation toward the DNA (Fig EV1). Instead, PfAlkC makes unique DNA binding interactions using the βCʹ strand and the preceding CCʹ‐turn (Fig 3A and B). Side chains of Lys309, Phe311, and Lys312 on the βCʹ strand interact with the backbones of both strands, and Lys301 and Ser302 on the CCʹ‐turn form polar interactions with phosphates on the 5ʹ‐side of the lesion strand. As a consequence of these contacts to both DNA strands, the Ig‐like domain in AlkC plays an important role in stabilizing the kinked DNA conformation toward the major groove.
Figure EV1. Comparison of DNA binding Ig folds.
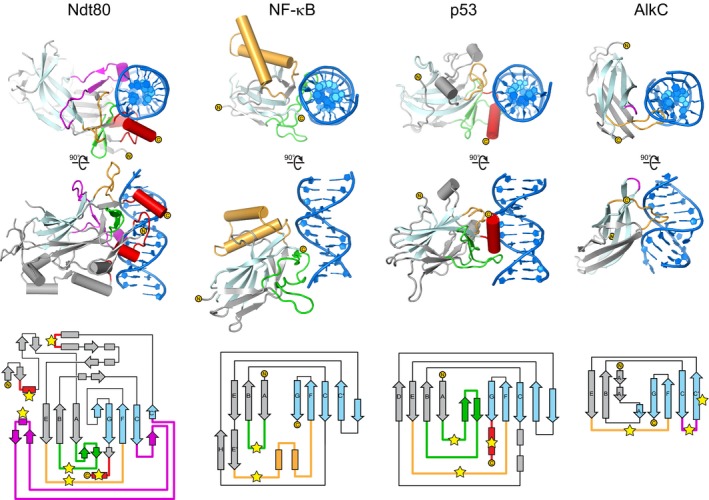
The top two rows show orthogonal views of DNA‐bound structures of Ig‐like domains from three eukaryotic transcription factors Ndt80 (PDB 1MNN), NF‐κB (PDB 1A3Q), and p53 (PDB 1TUP), compared to that of PfAlkC (this work). Topology diagrams of each structure are shown at the bottom and are colored the same as the crystal structures. The two β‐sheets within the Ig core are colored gray (A, B, D, E, Eʹ, and H) and pale cyan (C, Cʹ, F, and G). Regions contacting the DNA are colored green (AB‐loop), magenta (CCʹ‐loop), orange (EF‐loop), and red. Points of contact with the DNA in are marked with yellow stars in the topology diagram.
Consistent with a functional importance of the Ig‐like domain, deleting it from PfAlkC (PfAlkC∆C, Fig EV2A and B) abrogated base excision activity (Fig 3C), likely owing to a severe decrease in DNA binding affinity relative to the wild‐type protein (Fig EV2C). The dependence of PfAlkC activity on the Ig‐like domain is interesting given the absence of this domain in the fully functional AlkCα proteins, which suggests that the AlkCα HLR domain contains a structural feature that compensates for DNA binding in the absence of the Ig‐like domain. Perhaps the most distinguishing feature of the AlkCα HLR primary structure is an 8–10 residue insertion within helix αE of HLR1 (Appendix Fig S1). Whereas HLR1 does not contact the DNA in the PfAlkC structure, a homology model of BcAlkC shows that the extra residues likely endow AlkCα proteins with a novel DNA binding interaction to the damaged strand (Fig EV3). Together with potential additional interactions between the N‐terminal helical bundle and the undamaged strand, these extra HLR1‐DNA contacts would help stabilize the DNA kink. Although the precise manner in which AlkCα proteins compensate for the lack of the Ig‐like domain has yet to be determined, it is clear that DNA binding by the Ig motif is essential for AlkCβ function.
Figure EV2. Characterization of PfAlkC proteins used in this study.
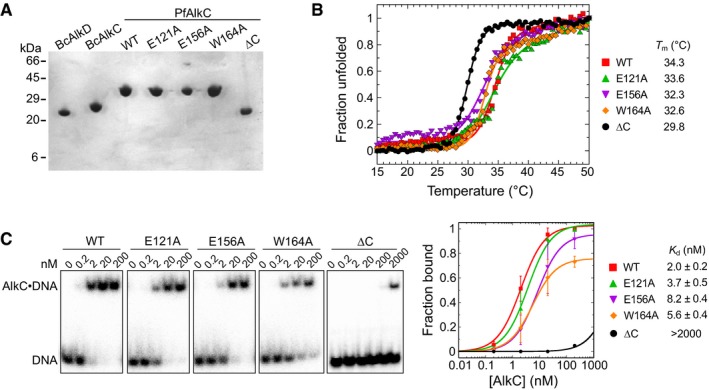
- SDS–PAGE of purified proteins, stained with Coomassie Blue.
- Thermal denaturation of wild‐type and PfAlkC mutants monitored by circular dichroism. The fraction of unfolded protein was expressed as the normalized molar ellipticity at 222 nm. Melting temperatures (T m) were derived by fitting the data to the equation (fraction unfolded) = 1/(1 + e(Tm−T)/k), where k denotes the cooperativity of the transition. Although PfAlkC∆C has a lower T m than the full‐length proteins, it is properly folded at 21°C at which biochemical assays were performed.
- Electrophoretic mobility shift assay for PfAlkC mutants binding to 1 nM THF‐DNA of the sequence 32P‐d(GACCACTACACT(THF)ATTCCTAACAAC)/d(GTTGTTAGGAAT(T)AGTGTAGTGGTC) in 25 mM HEPES (pH 7.5), 50 mM KCl, 5 mM DTT, 5% glycerol, and 0.05 mg/ml BSA at 21°C for 30 min. Concentrations of PfAlkC are shown at the top of the representative gels. Electrophoretic separation was carried out on a Novex™ TBE gel (ThermoFisher Scientific). Quantitation is plotted on the right, in which each value is the mean ± SD (n = 3). Equilibrium dissociation constants (K d) were extracted by fitting the data to a two‐state binding model.
Figure EV3. AlkCα proteins contain an insertion that may stabilize DNA bend in the absence of Ig‐like domain.
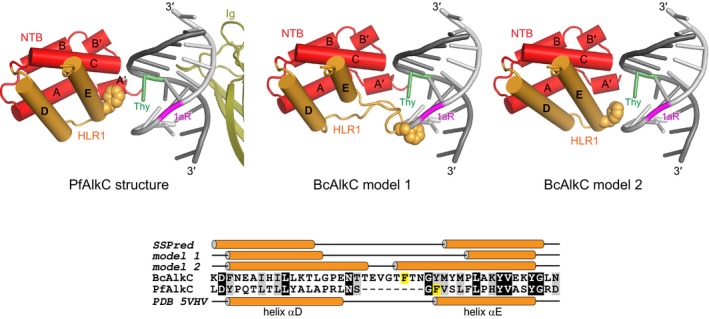
Two homology models for BcAlkC (center and right) are shown superimposed against the 1aR‐DNA from the PfAlkC/1aR‐DNA crystal structure (left). Only the N‐terminal helical bundle (NTB, red), HEAT‐like repeat 1 (HLR1, orange), and Ig‐like domain (olive) are shown for clarity. A phenylalanine side chain at the N‐terminus of helix αE in PfAlkC and in the 8‐residue insertion of BcAlkC is shown in spheres and highlighted yellow in the sequence alignment at the bottom. The secondary structural elements from the three models and from a secondary structure prediction of BcAlkC are shown against the sequences for each protein. The homology models were generated in SWISS‐MODEL (https://swissmodel.expasy.org/) using either the sequence alignment shown in Appendix Fig S1 (model 1) or generated by SWISS‐MODEL (model 2). In both models, the insertion makes contacts to the 1aR strand, either as a loop (model 1) or as an N‐terminal extension to helix αE (model 2).
AlkC inserts its active site into the DNA duplex in lieu of base flipping
Contacts between the HLR domain and the lesion help to explain AlkC's specificity for 3mA and its mechanism of base excision. At the DNA bend, both the 1aR lesion and its opposing thymidine on the undamaged strand (T6U) are displaced into the widened minor groove (Fig 4A). The 1aR is slightly rotated toward the protein to make electrostatic interactions and water‐bridging hydrogen bonds to the carboxylate side chains of Glu121 and Glu156 (Fig 4B). Contrary to base‐flipping glycosylases, there are no protein residues filling the void left by the missing nucleobase on the damaged strand. On the undamaged strand, the opposing thymidine T6U is displaced into the minor groove as a single nucleotide bulge as a consequence of the sharp kink in the DNA. The thymine base is no longer stacked with the flanking nucleotides and is tethered to the damaged strand by a hydrogen bond to guanine G8D and via Na+ coordination to T9D and C10D nucleobases (Figs 4A and EV4A). The minor groove is held in this opened conformation by the AlkC‐specific αIJ‐loop, at the tip of which Pro163 and Trp164 form van der Waals and hydrogen bonding contacts to the edges of the A6D•T7U base pair just 5′ to the lesion (Fig 4A). Residues on the αIJ‐loop and at the N‐terminal end of helix αG form a cavity at the hinge point in the DNA that is appropriately shaped for a 3mA nucleobase (Fig 4C). In the crystal structure, this cleft is filled by a pentaerythritol propoxylate solvent molecule from the crystallization buffer (Fig EV4B), but presumably would be occupied by the modified nucleobase prior to and immediately following nucleobase excision (Fig 1A).
Figure 4. AlkC inserts its active site into the DNA .
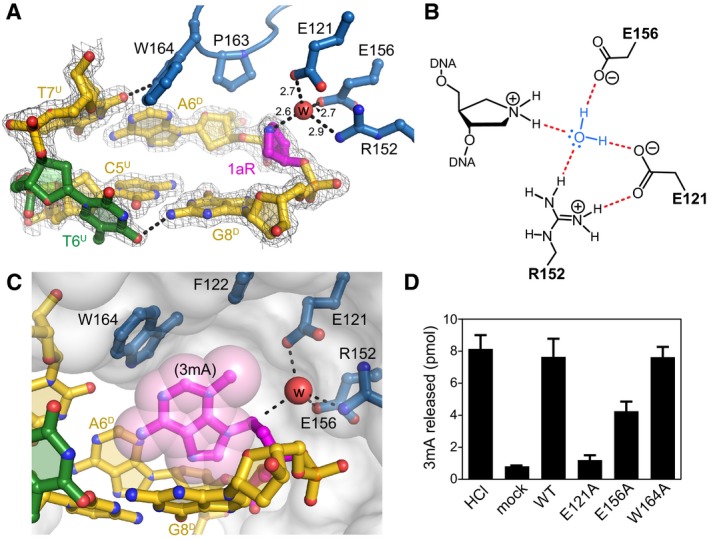
- Close‐up view of the AlkC active site (blue) bound to 1aR‐DNA (gold). The 1aR and opposite thymine are magenta and green, respectively. Water is shown as a red sphere, and hydrogen bonds are depicted as dashed lines. Composite omit electron density contoured to 1σ is superimposed against only the DNA for clarity. Superscripts in nucleotide labels refer to the 1aR‐containing, damaged (D) strand or the opposite, undamaged (U) strand. The pentaerythritol propoxylate molecule that occupies the active site has been omitted for clarity (see Fig EV4B).
- Schematic of the alignment of a catalytic water molecule (blue) against the 1aR oxocarbenium mimetic by AlkC active site residues. Hydrogen bonds are shown in red.
- A hypothetical model for AlkC bound to a 3mA‐DNA substrate was generated by superimposing the 3mA deoxyribose ring onto that of 1aR in the crystal structure, followed by rotating about the 3mA χ (N‐glycosidic bond) torsion angle to maximize van der Waals interactions. The solvent accessible surface of AlkC is shown as a transparent white envelope.
- Release of 3mA from methylated genomic DNA after a 5‐min incubation with either HCl, no enzyme (mock), PfAlkC (WT), E121A, E156A, or W164A. Values are mean ± SD (n = 3).
Figure EV4. Crystallographic features of the active site.
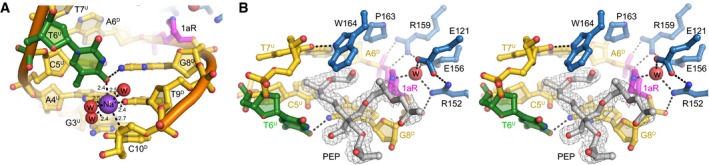
- Na+ ion coordination in the 1aR‐DNA structure. Na+ and water oxygens are shown as purple and red spheres, respectively. 1aR is magenta, and the opposite thymidine T6 is green. Superscripts in nucleotide labels refer to the 1aR‐containing, damaged (D) strand or the opposite, undamaged (U) strand. Hydrogen bonds are shown as dashed lines with interatomic distances in Ångstroms.
- Stereo‐views of the PfAlkC/1aR‐DNA active site. Active site residues are blue, and DNA is gold/magenta/green. Pentaerythritol propoxylate (PEP) sequestered from the solvent is in silver and superimposed against composite omit electron density contoured at 1σ. The putative catalytic water is shown as a red sphere. One arm of the PEP projects into the DNA kink between the 1aR and the flanking base pairs and thus may limit rotation of the 1aR ring back toward the DNA. The other two polymeric PEP arms project outward to solvent. The fourth PEP arm is not present; its terminal hydroxyl group forms a hydrogen bond to the displaced T6U thymine base. The position of the thymine is thus affected by the presence of the PEP in addition to its coordination by the Na+ ion. However, even in the absence of these stabilizing contacts, the T6U thymidine would be displaced into the minor groove as a result of the kink in the DNA. The space occupied by the PEP molecule would be occupied by the excised base both before and immediately after cleavage.
To gain a better idea of how the protein selects for 3mA, we modeled the nucleotide in the active site using 1aR as a guide (Fig 4C). Holding the deoxyribose ring in a fixed position, the 3mA nucleobase could adopt only a limited range of positions that varied within a 15° torsional rotation about the N‐glycosidic bond, constrained by π‐stacking against either its 3ʹ‐neighbor guanine G8D on one side or the protein surface on the other. Against the protein, the N1‐C2 edge of the 3mA nucleobase abuts the indole side chain of Trp164 to form a stabilizing cation‐π interaction. In this conformation, the N3‐methyl group resides in a pocket formed by Phe122 and Glu121 (Fig 4B). These contacts to the damaged nucleobase explain AlkC's inability to excise bulky minor groove YTMA lesions. Moreover, in this model, the Hoogsteen face of the purine ring is snugly nestled between the flanking nucleobases at the DNA kink, suggesting that purine N7‐adducts would be sterically disfavored from this collapsed major groove, providing a rationale for AlkC's low activity toward 7mG.
The position of the 1aR deoxyribose in the crystal structure provides a model for catalysis of base excision by AlkC. The N1 nitrogen of 1aR, which mimics the anomeric C1′ carbon in the oxocarbenium intermediate, is positioned perfectly for nucleophilic attack by the water molecule held in place by Glu121, Arg152, and Glu156 (Fig 4A and B). These putative catalytic protein–DNA contacts are made without flipping the modified nucleotide into an active site pocket. In fact, the nucleotide is sterically constrained from further rotation around the DNA backbone. A network of alternating charges between Glu121‐Arg152‐Glu156‐Arg159‐Asp203 spans the backbone in a 3′–5′ direction across the damaged nucleotide. Arg152 and Arg159 electrostatically stabilize the two phosphates flanking the 1aR and hold the carboxylate side chains of Glu121 and Glu156 in close proximity to the 1aR sugar ring (Fig EV4B). Thus, Glu121 and Glu156 are positioned to electrostatically stabilize the positive charge that develops on the deoxyribose during base excision and to orient and deprotonate the catalytic water nucleophile for attack of the anomeric C1′ carbon, without the need to flip the damaged nucleotide out of the DNA.
To validate this region of the protein as the active site, we tested the contribution of Glu121, Glu156, and Trp164 to catalysis by substituting individual residues with alanine and comparing the ability of the mutants to excise 3mA from methylated genomic DNA. We verified that the mutations did not compromise protein integrity or DNA binding (Fig EV2). The Glu121Ala substitution abrogated 3mA excision activity and Glu156Ala significantly decreased 3mA excision relative to wild‐type (Fig 4D), consistent with a catalytic role for these residues. In contrast, removal of the indole side chain from Trp164 had no effect on the total amount of 3mA excised after 5 min, suggesting that this residue either does not participate in catalysis or has a less detectable role in this assay. These structural and biochemical data clearly designate the region surrounding the 1aR as the active site and demonstrate that like AlkD, the AlkC protein uses a non‐base‐flipping mechanism to access its substrate.
DNA glycosylase product complexes are often remarkably similar in conformation to their substrate or intermediate structures, and consequently, these enzymes are typically inhibited by their abasic site products, which we recently showed to be true for the non‐base‐flipping glycosylase AlkD (Brooks et al, 2013; Mullins et al, 2015b, 2017). In an effort to trap an AlkC product complex, we crystallized PfAlkC with DNA containing a tetrahydrofuran (THF) abasic site mimetic. The PfAlkC/THF‐DNA crystal structure was refined to a crystallographic residual of 16.8% (R free = 22.5%) against X‐ray data extending to 2.4 Å (Appendix Table S1, Fig EV5). The asu of the THF structure contained two protein–DNA complexes with HLR and Ig‐like domains wrapping around highly bent DNA duplexes as in the 1aR structure. However, unlike the virtually identical protein–DNA complexes in the 1aR structure, each protein–DNA complex in the THF structure differed significantly in the relative position of their Ig‐like and HLR domains as well as in the conformation of the DNA, with a 60° bend angle in one and an 85° bend in the other (Fig EV5G). The two DNA duplexes in the asu were stacked on one another at the overhanging A1 nucleotides on one end, but did not make lattice contacts on the other, and thus, the different conformations likely indicate the flexible nature of DNA near the lesion. Most notably, there was a substantial difference in the positions of THF and 1aR bound to AlkC. The THF moieties were slipped out of the DNA helix as single nucleotide bulges, each adopting a distinct conformation and a set of contacts to AlkC outside of the active site (Fig EV5D–F). As a consequence, adenine A6D, immediately 5′ to the THF on the damaged (D) strand, formed an opportunistic base pair with thymine T6U opposite THF on the undamaged (U) strand, leaving T7U (instead of T6U) unpaired. The mismatched A6D•T6U base pair was observed in both Watson–Crick and Hoogsteen orientations in the two complexes in the asu (Fig EV5F). Because neither THF contacted the active site residues, similar to that observed in a non‐catalytic complex of THF‐DNA bound to AlkD (Rubinson et al, 2010), we conclude that the PfAlkC/THF‐DNA structure is not representative of the true PfAlkC product complex. Rather, the structural heterogeneity observed in this structure highlights a key aspect of the non‐base‐flipping mechanisms used by AlkC and AlkD. These enzymes lack the intercalating side chain used by base‐flipping enzymes to plug the gap left in the DNA as a result of the everted or excised base, and thus, AlkC and AlkD do not directly stabilize the conformation of the abasic DNA on their own. Rather, we previously showed that the excised nucleobase stabilizes intermediate and product complexes by remaining stacked in the DNA duplex from its position in the substrate (Mullins et al, 2015a). Attempts to trap ternary PfAlkC/1aR‐DNA/3mA and PfAlkC/THF‐DNA/3mA complexes containing free 3mA nucleobase were unsuccessful, likely owing to the lack of base stacking at the sharp kink in the DNA, and suggesting that the nucleobase readily dissociates following N‐glycosidic bond cleavage.
Figure EV5. Structure of the PfAlkC/THF‐DNA complex and its comparison to PfAlkC/1aR‐DNA .
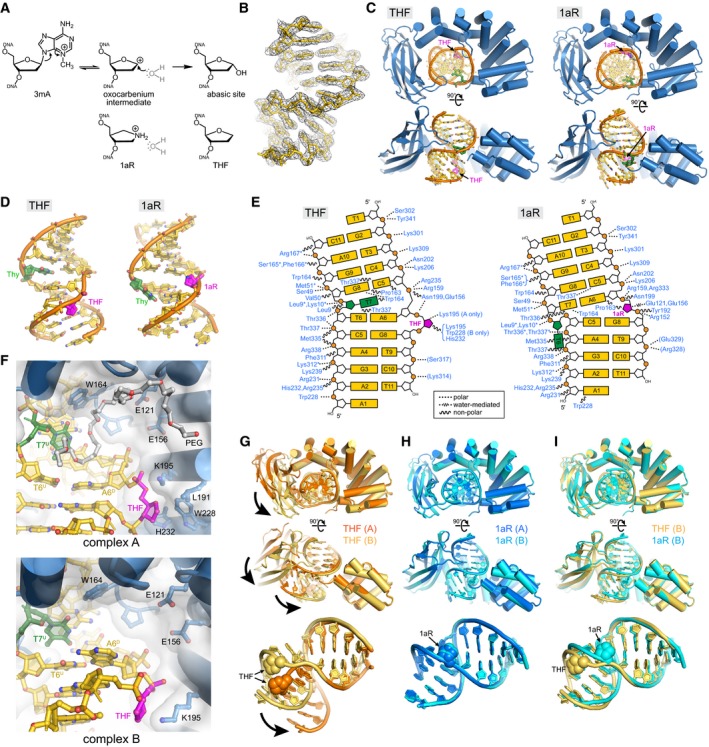
- DNA glycosylase‐catalyzed reaction together with chemical structures of intermediate and product mimetics used in this study.
- Annealed composite omit electron density contoured to 1σ is superposed onto the THF‐DNA crystallographic model.
- Two views of the crystal structures, with protein in blue, DNA in gold, THF/1aR in magenta, and the thymidine opposite THF/1aR in green.
- DNA structures extracted from the complexes.
- Schematic of protein–DNA contacts. Contacts to the protein backbone are marked with an asterisk, and symmetry‐related contacts are in parentheses. PfAlkC maintains similar contacts with the DNA in THF and 1aR complexes relative to the position of the DNA bend and not to the position of the 1aR/THF.
- Details of THF‐DNA bound outside of the PfAlkC active site. Both complexes in the crystallographic asymmetric unit are shown. A PEG 4,000 molecule (white carbons) fills the void in the catalytic pocket in complex A.
- Superposition of the two protomers in the THF complex. Bold arrows highlight the difference in positions of the Ig‐like domain relative to the HLR domain and in the two DNA conformations.
- Superposition of the two 1aR complexes.
- Superposition of one protomer from each of the THF (gold) and 1aR (cyan) complexes.
Structural differences between AlkC and AlkD
With crystal structures for both AlkC and AlkD in hand, we are able to understand the basis for their differences in locating and discriminating DNA alkylation damage. Despite the low sequence conservation between the HLR domains of BcAlkD and PfAlkC (16.3% identity and 27.1% similarity), they adopt the same general C‐shaped architecture that complements the DNA helix (Fig 5A–C). Neither protein flips the target nucleobase out of the helix, but they use different mechanisms to interrogate the minor groove for lesion recognition. The modified nucleobase in the BcAlkD complex does not contact the protein, but remains stacked within a DNA duplex that is only modestly bent (Appendix Fig S3), which allows the enzyme to tolerate different positions and sizes of alkyl substituents. The bulky YTMA lesion in particular resides in a cleft between the enzyme and the widened minor groove (Mullins et al, 2017). In contrast, PfAlkC imposes a much sharper bend in the DNA duplex, which disrupts base stacking and exposes the modified nucleobase to protein contacts from the minor groove (Appendix Fig S3). A defining feature of AlkC that distinguishes it from AlkD is the highly conserved αIJ‐loop insertion that projects into the kink and traps the lesion between the surface of the protein and the kinked DNA (Fig 5A–C). This loop occupies the space in the minor groove that would be occupied by YTMA and explains why AlkC is unable to excise bulky minor groove lesions. Because of its direct contact to the modified nucleobases, the presence of the αIJ‐loop also restricts the size of the lesion that can be accommodated (Fig 4C).
Figure 5. Comparison of AlkC and AlkD.
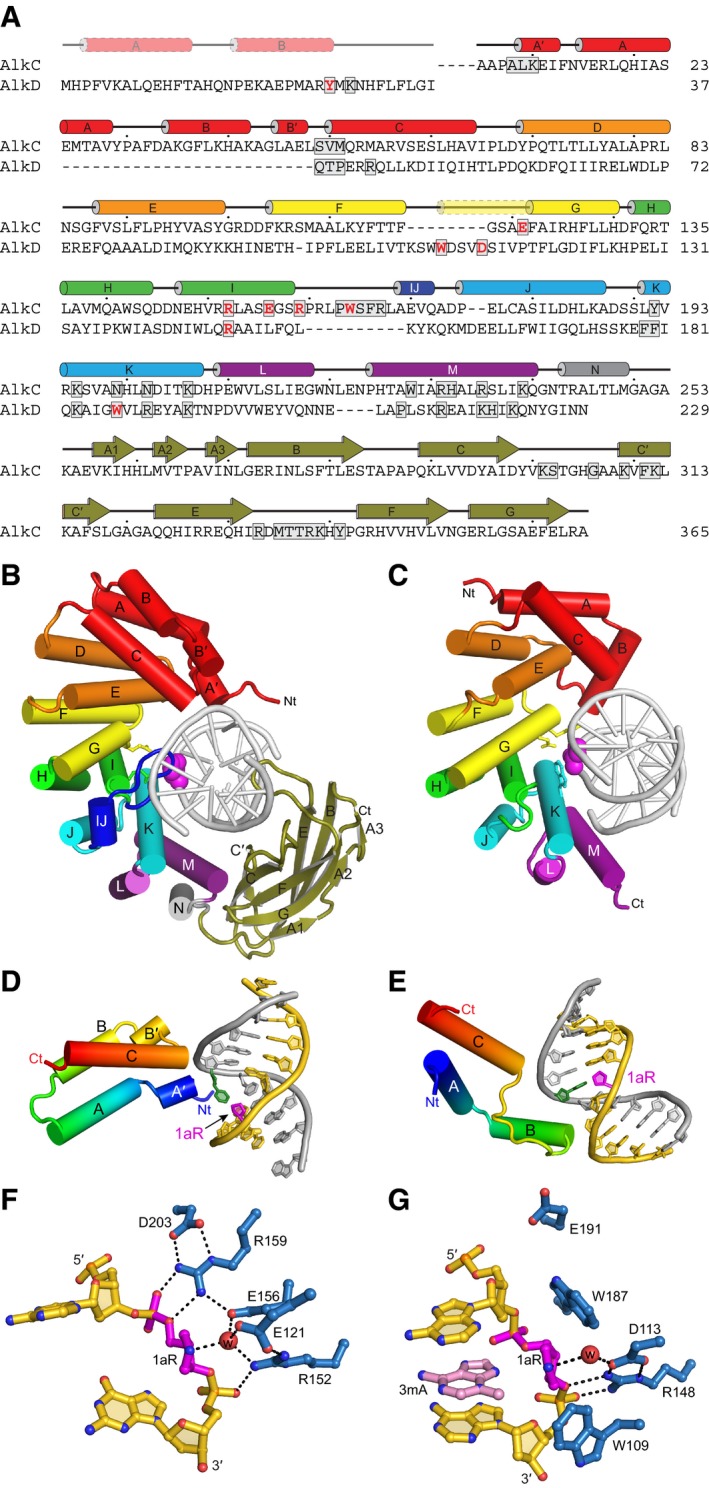
-
AStructure‐based sequence alignment of PfAlkC and BcAlkD. Active site residues are red and DNA‐interacting residues are boxed. Secondary structures derived from the crystal structures are shown above the sequences. The transparent N‐terminal segment of helix αG (yellow) is unique to AlkD and helix αIJ (dark blue) is unique to AlkC.
-
B, CCrystal structures of 1aR‐DNA complexes of PfAlkC (B) and BcAlkD (C, PDB ID 5CLD). The N‐terminal helical bundles are red; HEAT‐like repeats are orange, yellow, green, cyan, and purple; and the Ig‐like domain of AlkC is olive. Side chains of active site residues are shown as sticks, and the 1aR moieties are shown as magenta spheres.
-
D, EInteractions between the N‐terminal helical bundles of PfAlkC (D) and BcAlkD (E) and DNA. Protein is colored rainbow from N‐ (blue) to C‐terminus (red). DNA strands are colored gold and silver, and 1aR and opposite thymine are magenta and green, respectively.
-
F, GActive sites of PfAlkC (F) and BcAlkD (G). Protein residues are colored blue, DNA gold, 1aR magenta, and 3mA nucleobase pink. The catalytic water is shown as a red sphere, and hydrogen bonds are depicted as dashed lines.
The highly kinked DNA important for lesion recognition in AlkC is stabilized by both the Ig‐like domain and the N‐terminal helical bundle, the conformation and topology of which is distinct from AlkD and other HLR proteins AlkD2 and AlkF/AlkG (Fig 5D and E) (Rubinson et al, 2008; Backe et al, 2013). In AlkD's three‐helical bundle (αA‐αC), helix αB interacts with the damaged DNA strand and is essential for binding affinity (Mullins et al, 2015a). In contrast, PfAlkC's N‐terminal helical bundle does not contact the damaged strand. PfAlkC's helix αA and αB, which are broken into 2‐helix segments to give αA′, αA, αB, and αB′, run antiparallel to one another and with opposite polarity with respect to BcAlkD so that αB does not contact the DNA. Instead, the N‐terminal end of helix αA′ points toward the undamaged strand DNA, similar to the helix αC in both PfAlkC and BcAlkD (Fig 5D and E). As noted above, this region of AlkC may work together with the Ig‐like domain to stabilize the bent DNA conformation.
The active sites of AlkC and AlkD are surprisingly different. AlkC contains a network of alternating charged residues along the damaged DNA backbone, whereas AlkD uses a Trp‐Asp‐Trp motif to cradle the backbone around the damaged nucleotide (Fig 5F and G). Each of the charged AlkC residues that span the damaged DNA backbone is invariant except for Glu121 (Appendix Fig S1). Glu121 is positioned closest to the anomeric C1′ carbon and is spatially aligned with AlkD's catalytic Asp113 on helix αG (Fig 5A), consistent with its essential role in PfAlkC activity. Interestingly, among AlkC orthologs, the position of Glu121 is almost always either a glutamate or a tryptophan. We recently established that the two tryptophan side chains that flank the catalytic Asp113 in AlkD contribute to catalysis, presumably by further stabilizing the development of positive charge on the deoxyribose as the N‐glycosidic bond is broken (Mullins et al, 2015b; Parsons et al, 2016). In contrast, invariant Glu156 is positioned closer to the O4′ of the deoxyribose and is sandwiched between two arginine side chains from the ionic network, significantly lowering its predicted pK a (2.9) relative to that of Glu121 (4.3). We therefore speculate that Glu121 deprotonates the water nucleophile and that Glu156 plays more of a role in stabilizing positive charge that develops on the deoxyribose during base excision.
AlkC catalyzes base excision of 3mC and 1mA from duplex DNA
During our phylogenetic analysis, we noticed that most AlkC‐containing bacteria lacked an AlkB ortholog, implying that these bacteria would be unable to repair 1‐methyladenine (1mA) and 3‐methylcytosine (3mC) lesions by oxidative demethylation. Out of 834 completely sequenced and annotated bacterial genomes that contain either an AlkB or AlkC ortholog, only 6% contained both proteins (Fig 6A). Given this distribution and the apparent specificity of AlkC for cationic methylbases, we tested the ability of purified BcAlkC (α) and PfAlkC (β) proteins to excise 1mA and 3mC from a double‐stranded oligodeoxynucleotide substrate under single‐turnover conditions. Compared to a no‐enzyme control, BcAlkC and PfAlkC exhibited robust activity for 3mC and modest activity for 1mA, while AlkD had no activity toward either 3mC or 1mA (Fig 6B and C, and Appendix Fig S4A and B). AlkC excision of 1mA was comparable to its weak activity for 7mG (Appendix Table S2), suggesting that 3mC is a more biologically relevant substrate for AlkC than is 1mA. Consistent with this, we were able to easily model 3mC, but not 1mA, into our crystal structure. Positioning 3mC in the active site places the N3‐methyl group in van der Waals contact with Trp164 and the O2 oxygen in the Phe122/Glu121 pocket, whereas the N1‐methyl group on 1mA sterically clashed with Trp164. 3mC and 1mA excisions were abrogated by Glu121Ala and Glu156Ala mutants and impaired by Trp164Ala substitution (Appendix Fig S4C and D), consistent with a catalytic role for the glutamates and a possible role for Trp164 in discrimination of different alkylated substrates. AlkC repair of these unusual lesions and the low percentage of bacteria that contain both AlkB and AlkC raises the interesting possibility that AlkC repairs 3mC and 1mA in bacteria that do not contain the AlkB oxidative demethylase (Fig 6D). Similarly, AlkA orthologs from the archaeon Archaeoglobus fulgidus and the archaea‐related bacterium Deinococcus radiodurans, neither of which contain an AlkB homolog, are capable of 1mA and 3mC excision (Leiros et al, 2007; Moe et al, 2012). We did not observe 3mC or 1mA excision activity from ssDNA, nor did we observe excision of N1‐methylguanine (1mG) or N3‐methylthymine (3mT) (Appendix Fig S4E), all of which are substrates for AlkB (Falnes et al, 2002, 2004; Trewick et al, 2002; Delaney & Essigmann, 2004; Koivisto et al, 2004), indicating that AlkB and AlkC are not strict functional orthologs.
Figure 6. AlkC excises 3‐methylcytosine (3mC) and 1‐methyladenine (1mA) from DNA .
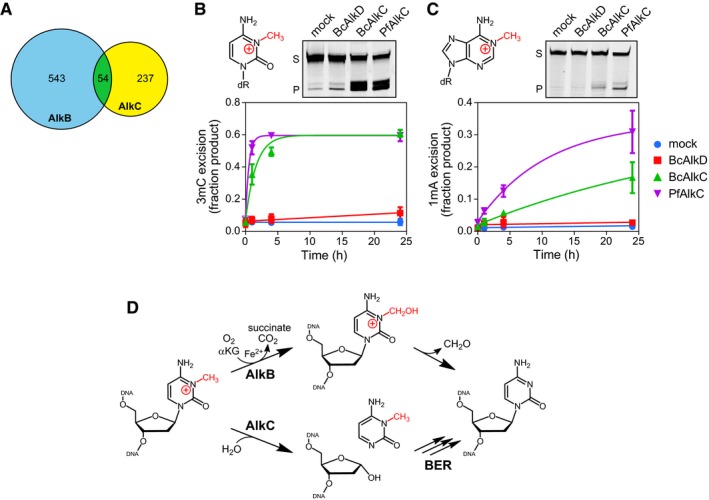
-
AVenn diagram of the numbers of bacterial species containing either AlkB (blue), AlkC (yellow), or both (green).
-
B, CChemical structures and in vitro base excision of 3mC (B) and 1mA (C) from 25‐mer double‐stranded oligodeoxyribonucleotides. Representative denaturing electrophoresis gels show substrate (S) and product (P) after a 24‐h incubation with either no enzyme (mock), BcAlkD, BcAlkC (AlkCα), or PfAlkC (AlkCβ). Plots show quantified time courses from three experiments (values are mean ± SD).
-
D3mC may be repaired in bacteria by either AlkB‐catalyzed oxidative demethylation or AlkC‐catalyzed base excision. αKG, α‐ketoglutarate.
Conclusion
Previous work defined the HLR domain as a new enzymatic scaffold for duplex DNA containing bulky lesions (Rubinson et al, 2008, 2010; Rubinson & Eichman, 2012; Mullins et al, 2015b). We now show how decoration of this DNA repair fold with additional motifs adapts it for specific, discreet lesions. The majority of AlkC orthologs extend the DNA binding surface beyond the HLR domain using an Ig‐like domain, which works together with the HLR N‐terminal helical bundle to severely distort the DNA duplex and expose the nucleobase lesion. Projection of the AlkC‐unique αIJ‐loop into this kinked DNA helix limits the size of alkylbases that can be excised, while two glutamate side chains enable AlkC to excise 3mC and 1mA, providing an alternative means of removing these modified nucleobases by BER, in addition to oxidative demethylation. In contrast, AlkD utilizes a Trp‐Asp‐Trp triad to recognize DNA damage without contacting the nucleobase or disrupting the DNA base stack, enabling AlkD to excise both major and minor groove lesions of various sizes. Thus, although AlkC and AlkD are related evolutionarily through their HLR architectures, and neither enzyme flips its target substrate into an active site pocket, they have developed different strategies to recognize and excise specific sets of alkylated lesions.
Material and Methods
Phylogenetic analysis
AlkC and AlkD orthologs were identified by the PSI‐BLAST search against the NCBI non‐redundant protein sequence database (last accessed on March 14, 2016) using PfAlkC (NCBI reference WP_012723400.1) and BcAlkD (NCBI reference CAJ31885.1) as queries with a cutoff e‐value of 10−5. If multiple protein sequences were included for a single species, a simple neighbor‐joining tree and the corresponding HLR domain were used to remove the potentially redundant and paralogous sequences. All AlkC and AlkD orthologous sequences were aligned using Clustal Omega (Sievers et al, 2011), and the phylogenetic tree constructed using FASTTREE (Price et al, 2010) initiated with 100 random starting trees using a Whelan and Goldman + GAMMA amino acid model of substitution. The distribution of AlkB and AlkC sequences in bacteria was culled from the complete genomes using RefSeq representative genomes of NCBI TBLASTN (last accessed on March 14, 2016) with a cutoff e‐value of 10−5. The corresponding HLR and AlkB conserved domain were used to manually remove species that have potential paralogous sequences.
Protein purification
Cloning and purification of B. cereus (ATCC 14579) AlkC and AlkD were described previously (Rubinson et al, 2008; Mullins et al, 2013). AlkC genes from Bacillus anthracis (NCBI WP_000521720.1), Bacillus mycoides (WP_003190106.1), Bacillus weihenstephanensis (WP_012261200.1), Sphingobacterium sp. 21 (WP_013668283.1), P. fluorescens SBW25 (WP_012723400.1), and Streptomyces albus (WP_003946460.1) were synthesized into expression vector pJ434 (DNA2.0). Constructs used in the genomic DNA base excision assay were overexpressed in E. coli C41(DE3) cells at 16°C overnight upon induction with 500 μM IPTG. For all other biochemical assays and X‐ray crystallography, P. fluorescens alkC was subcloned into pBG100 (Vanderbilt Center for Structural Biology) encoding a cleavable N‐terminal hexahistidine tag and overexpressed in E. coli HMS174(DE3) at 16°C overnight with 500 μM IPTG. Cells were lysed by sonication at 4°C in buffer A (50 mM Tris–HCl pH 8.5, 500 mM NaCl, and 10% (v/v) glycerol). AlkC orthologs were purified from soluble lysate by Ni‐NTA (Qiagen) affinity chromatography by sequentially washing and eluting in buffer A containing 20 and 500 mM imidazole, respectively. Pooled fractions were digested with Rhinovirus 3C (PreScission) Protease overnight at 4°C to remove the N‐terminal hexahistidine tag. Cleaved protein was diluted 10‐fold in buffer B (50 mM Tris–HCl pH 8.5, 10% (v/v) glycerol, 2 mM DTT, and 0.1 mM EDTA) and purified over heparin sepharose (GE Healthcare) using a 50–1,000 mM NaCl linear gradient. The protein was further purified using gel filtration chromatography (Superdex 200, GE Healthcare) in buffer C (20 mM Tris–HCl pH 8.5, 150 mM NaCl, 10% (v/v) glycerol, 2 mM DTT, and 0.1 mM EDTA). Purified proteins were concentrated, flash‐frozen, and stored at −80°C in buffer C.
PfAlkC mutants were constructed using the Q5 Site‐Directed Mutagenesis Kit (New England BioLabs, Inc.). PfAlkC∆C (residues 1–249) was generated by mutating the codon corresponding to residue 250 to a stop codon. Purification of PfAlkC mutants was conducted in the same manner as wild‐type PfAlkC except that PfAlkC Glu121Ala was overexpressed in E. coli ArcticExpress(DE3) cells. Selenomethionine (SeMet)‐incorporated PfAlkC was overexpressed and purified the same as wild‐type PfAlkC, except overexpression was carried out in M9 minimal media supplemented with 0.4% (w/v) dextrose; 1 mM MgSO4; 0.1 mM CaCl2; 1 mg/l thiamine; 60 mg/l selenomethionine; 50 mg/l each of leucine, isoleucine, and valine; and 100 mg/l each of phenylalanine, lysine, and threonine. Integrity of mutant proteins was verified by thermal denaturation, monitored by circular dichroism molar ellipticity at 222 nm on a Jasco J‐810 spectropolarimeter using 7.5 μM protein in 50 mM HEPES pH 8.5, 100 mM KCl, and 10% (v/v) glycerol.
Base excision assays
Base excision using genomic DNA and oligonucleotide substrates was carried out as previously described (Mullins et al, 2013). Briefly, to measure release of methylbases from genomic DNA by AlkC orthologs, 5 μM protein was incubated with 10 μg of MNU‐treated calf thymus DNA at 37°C for 1 h in 50 mM HEPES pH 7.5, 100 mM KCl, 5% (v/v) glycerol, 10 mM DTT, 2 mM EDTA and 0.1 mg/ml BSA in a 50 μl reaction and products quantified by HPLC‐MS/MS. The activity of PfAlkC and mutants were tested in the same manner except that the assay was carried out at 21°C for 5 min in a 30 μl reaction consisting of 10 μM protein, 50 mM HEPES pH 8.5, 100 mM KCl, 5% (v/v) glycerol, and 6 μg of MNU‐treated calf thymus DNA from a different batch. For oligonucleotide‐based assays, 7mG, 1mA, 3mC, 1mG, or 3mT were incorporated into the sequence d(GACCACTACACCXATTCCTTACAAC) at the underlined position either enzymatically (7mG) (Mullins et al, 2013) or by solid‐phase synthesis by Midland Certified Reagent Company (1mA, 1mG, and 3mT) or ChemGenes Corporation (3mC) and annealed to the complementary strand in annealing buffer (10 mM MES pH 6.5 and 40 mM NaCl). The YTMA lesion was generated in the sequence [d(CGGGCGGCGGCA(YTMA)AGGGCGCGGGCC)/d(GGCCCGCGCCCTTTGCCGCCGCCCG)] as described previously (Mullins et al, 2015b). Glycosylase reactions contained 100 nM 6‐carboxyfluorescein (FAM) labeled‐DNA and 10 μM protein and were carried out either at 21°C in 50 mM HEPES pH 8.5, 100 mM KCl and 10% (v/v) glycerol (PfAlkC) or at 35°C in 25 mM HEPES pH 7.5, 50 mM KCl and 5% (v/v) glycerol (BcAlkC and BcAlkD).
X‐ray crystallography
Crystallization of the seven purified AlkC orthologs was first screened against a library of double‐stranded oligonucleotides (Integrated DNA Technologies) ranging in length from 8 to 15 nucleotides and containing a centralized THF across from thymine. Diffracting crystals were obtained from the PfAlkC protein grown in the presence of an 11‐mer THF‐DNA [d(TGTCCA(THF)GTCT)/d(AAGACTTGGAC)]. The PfAlkC/THF‐DNA structure was determined using single‐wavelength dispersion (SAD) phases from SeMet‐incorporated protein and refined to 2.4‐Å resolution (Appendix Table S1, Fig EV5). Crystals of the 1aR‐DNA complex were obtained by replacing THF with 1aR in the same 11‐mer sequence, which was synthesized as previously described (Chu et al, 2011). The PfAlkC/1aR‐DNA structure was determined by molecular replacement using the refined protein coordinates from the PfAlkC/THF‐DNA complex as a search model.
Protein–DNA complexes were assembled by incubating 0.12 mM PfAlkC with 0.15 mM DNA on ice for 30 min. Crystals were grown using the hanging‐drop vapor diffusion method at 21°C. For the THF complex, 1 μl of SeMet‐PfAlkC/THF‐DNA solution was mixed with 1 μl of reservoir solution (100 mM Tris–HCl pH 8.5, 18% PEG 4,000, and 5% (v/v) glycerol). For the 1aR complex, 1 μl of wild‐type PfAlkC/1aR‐DNA solution was mixed with 1 μl of reservoir solution (18% pentaerythritol propoxylate and 100 mM MES pH 6.1). Drops were equilibrated against 500 μl of reservoir solution. Crystals were flash‐frozen in liquid N2 in either mother liquor supplemented with 30% (v/v) glycerol (THF complex) or in mother liquor alone (1aR complex).
X‐ray diffraction data were collected at Advanced Photon Source beamline 21‐ID‐F (THF complex) and Advanced Light Source SIBYLS beamline (1aR complex). All data were processed with HKL2000 (Otwinowski & Minor, 1997). Phases for the PfAlkC/THF‐DNA complex were determined by Se‐SAD from positions of 18 Se atoms using AutoSHARP (Vonrhein et al, 2007). PfAlkC/1aR‐DNA phases were determined by molecular replacement using the refined coordinates of the SeMet‐PfAlkC protein as a search model in the program Phaser (McCoy et al, 2007). Atomic models were built in Coot (Emsley & Cowtan, 2004), and atomic positions, individual B‐factors, TLS parameters, and occupancies were refined using Phenix (Adams et al, 2010). The final models were validated with MolProbity (Davis et al, 2007) and contained no residues (THF complex) or one residue (1aR complex) in the disallowed regions of the Ramachandran plot (Appendix Table S1). The identity of the sodium ion was verified by ligand distances, coordination, and geometry using the CheckMyMetal web server (Zheng et al, 2014).
Structural biology software was curated by SBGrid (Morin et al, 2013). Structure figures were prepared in PyMOL (Schrödinger, 2016). Conservation from 527 AlkCβ sequences was mapped onto the structure of PfAlkC using the ConSurf server (http://bental.tau.ac.il/new_ConSurfDB/). DNA geometric parameters were measured using Web 3DNA (Zheng et al, 2009). Structure‐based pK a calculations were carried out in Rosetta (Kilambi & Gray, 2012; Lyskov et al, 2013).
Data availability
Structure factors and coordinates were deposited in the Protein Data Bank under accession codes 5VHV (1aR complex) and 5VI0 (THF complex), and the corresponding X‐ray diffraction images deposited in the SBGrid Data Bank (Meyer et al, 2016).
Author contributions
BFE conceived the project; RS, EAM, and BFE designed experiments; RS performed biochemical and structural experiments; EAM purified and analyzed activity of AlkC orthologs and generated preliminary PfAlkC‐DNA crystals; KTL, PKY, and SSD synthesized 1aR‐oligonucleotides; XS, RS, and AR performed phylogenetic analysis; RS, EAM, XS, AR, and BFE analyzed data; RS, EAM, and BFE wrote the manuscript.
Conflicts of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
This work was funded by the National Science Foundation (MCB‐1517695 to B.F.E., CHE‐1610721 to S.S.D.) and the National Institutes of Health (R01 ES019625). Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy Office of Science by Argonne National Laboratory, was supported by the U.S. Department of Energy (DE‐AC02‐06CH11357). Use of LS‐CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri‐Corridor (085P1000817). The Advanced Light Source operated by Lawrence Berkeley National Laboratory on behalf of the Department of Energy, Office of Basic Energy Sciences, through the Integrated Diffraction Analysis Technologies (IDAT) program, was supported by DOE Office of Biological and Environmental Research. E.A.M. was supported by the Vanderbilt Training Program in Environmental Toxicology (T32ES07028). K.T.L. was supported by National Institutes of Health training program in chemical biology (T32 GM113770).
The EMBO Journal (2018) 37: 63–74
References
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L‐W, Kapral GJ, Grosse‐Kunstleve RW (2010) PHENIX: a comprehensive Python‐based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66: 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alseth I, Rognes T, Lindback T, Solberg I, Robertsen K, Kristiansen KI, Mainieri D, Lillehagen L, Kolsto AB, Bjoras M (2006) A new protein superfamily includes two novel 3‐methyladenine DNA glycosylases from Bacillus cereus, AlkC and AlkD. Mol Microbiol 59: 1602–1609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backe PH, Simm R, Laerdahl JK, Dalhus B, Fagerlund A, Okstad OA, Rognes T, Alseth I, Kolsto AB, Bjoras M (2013) A new family of proteins related to the HEAT‐like repeat DNA glycosylases with affinity for branched DNA structures. J Struct Biol 183: 66–75 [DOI] [PubMed] [Google Scholar]
- Becker S, Groner B, Muller CW (1998) Three‐dimensional structure of the Stat3β homodimer bound to DNA. Nature 394: 145 [DOI] [PubMed] [Google Scholar]
- Birkeland NK, Anensen H, Knaevelsrud I, Kristoffersen W, Bjoras M, Robb FT, Klungland A, Bjelland S (2002) Methylpurine DNA glycosylase of the hyperthermophilic archaeon Archaeoglobus fulgidus . Biochemistry 41: 12697–12705 [DOI] [PubMed] [Google Scholar]
- Bjelland S, Bjoras M, Seeberg E (1993) Excision of 3‐methylguanine from alkylated DNA by 3‐methyladenine DNA glycosylase I of Escherichia coli . Nucleic Acids Res 21: 2045–2049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjelland S, Birkeland NK, Benneche T, Volden G, Seeberg E (1994) DNA glycosylase activities for thymine residues oxidized in the methyl group are functions of the AlkA enzyme in Escherichia coli . J Biol Chem 269: 30489–30495 [PubMed] [Google Scholar]
- Bodelón G, Palomino C, Fernández LÁ (2013) Immunoglobulin domains in Escherichia coli and other enterobacteria: from pathogenesis to applications in antibody technologies. FEMS Microbiol Rev 37: 204–250 [DOI] [PubMed] [Google Scholar]
- Bork P, Holm L, Sander C (1994) The immunoglobulin fold: structural classification, sequence patterns and common core. J Mol Biol 242: 309–320 [DOI] [PubMed] [Google Scholar]
- Bravo J, Li Z, Speck NA, Warren AJ (2001) The leukemia‐associated AML1 (Runx1)–CBFβ complex functions as a DNA‐induced molecular clamp. Nat Struct Biol 8: 371–378 [DOI] [PubMed] [Google Scholar]
- Brooks SC, Adhikary S, Rubinson EH, Eichman BF (2013) Recent advances in the structural mechanisms of DNA glycosylases. Biochim Biophys Acta 1834: 247–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Derfler B, Maskati A, Samson L (1989) Cloning a eukaryotic DNA glycosylase repair gene by the suppression of a DNA repair defect in Escherichia coli . Proc Natl Acad Sci USA 86: 7961–7965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Glover JN, Hogan PG, Rao A, Harrison SC (1998) Structure of the DNA‐binding domains from NFAT, Fos and Jun bound specifically to DNA. Nature 392: 42–48 [DOI] [PubMed] [Google Scholar]
- Cho Y, Gorina S, Jeffrey PD, Pavletich NP (1994) Crystal structure of a p53 tumor suppressor‐DNA complex: understanding tumorigenic mutations. Science 265: 346–355 [DOI] [PubMed] [Google Scholar]
- Chu AM, Fettinger JC, David SS (2011) Profiling base excision repair glycosylases with synthesized transition state analogs. Bioorg Med Chem Lett 21: 4969–4972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer P, Larson CJ, Verdine GL, Muller CW (1997) Structure of the human NF‐κB p52 homodimer‐DNA complex at 2.1 Å resolution. EMBO J 16: 7078–7090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalhus B, Helle IH, Backe PH, Alseth I, Rognes T, Bjoras M, Laerdahl JK (2007) Structural insight into repair of alkylated DNA by a new superfamily of DNA glycosylases comprising HEAT‐like repeats. Nucleic Acids Res 35: 2451–2459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis IW, Leaver‐Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, Snoeyink J, Richardson JS (2007) MolProbity: all‐atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res 35(suppl 2): W375–W383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaney JC, Essigmann JM (2004) Mutagenesis, genotoxicity, and repair of 1‐methyladenine, 3‐alkylcytosines, 1‐methylguanine, and 3‐methylthymine in alkB Escherichia coli . Proc Natl Acad Sci USA 101: 14051–14056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drohat AC, Maiti A (2014) Mechanisms for enzymatic cleavage of the N‐glycosidic bond in DNA. Org Biomol Chem 12: 8367–8378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K (2004) Coot: model‐building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60: 2126–2132 [DOI] [PubMed] [Google Scholar]
- Falnes PO, Johansen RF, Seeberg E (2002) AlkB‐mediated oxidative demethylation reverses DNA damage in Escherichia coli . Nature 419: 178–182 [DOI] [PubMed] [Google Scholar]
- Falnes P, Bjørås M, Aas PA, Sundheim O, Seeberg E (2004) Substrate specificities of bacterial and human AlkB proteins. Nucleic Acids Res 32: 3456–3461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedberg EC, Aguilera A, Gellert M, Hanawalt PC, Hays JB, Lehmann AR, Lindahl T, Lowndes N, Sarasin A, Wood RD (2006) DNA repair: from molecular mechanism to human disease. DNA Repair 5: 986–996 [DOI] [PubMed] [Google Scholar]
- Halaby DM, Mornon JP (1998) The immunoglobulin superfamily: an insight on its tissular, species, and functional diversity. J Mol Evol 46: 389–400 [DOI] [PubMed] [Google Scholar]
- Halaby DM, Poupon A, Mornon J (1999) The immunoglobulin fold family: sequence analysis and 3D structure comparisons. Protein Eng 12: 563–571 [DOI] [PubMed] [Google Scholar]
- Hollis T, Ichikawa Y, Ellenberger T (2000) DNA bending and a flip‐out mechanism for base excision by the helix‐hairpin‐helix DNA glycosylase, Escherichia coli AlkA. EMBO J 19: 758–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L, Laakso LM (2016) Dali server update. Nucleic Acids Res 44: W351–W355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SP, Bartek J (2009) The DNA‐damage response in human biology and disease. Nature 461: 1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilambi KP, Gray JJ (2012) Rapid calculation of protein pK a values using Rosetta. Biophys J 103: 587–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koivisto P, Robins P, Lindahl T, Sedgwick B (2004) Demethylation of 3‐methylthymine in DNA by bacterial and human DNA dioxygenases. J Biol Chem 279: 40470–40474 [DOI] [PubMed] [Google Scholar]
- Krokan HE, Bjørås M (2013) Base excision repair. Cold Spring Harb Perspect Biol 5: a012583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamoureux JS, Stuart D, Tsang R, Wu C, Glover JM (2002) Structure of the sporulation‐specific transcription factor Ndt80 bound to DNA. EMBO J 21: 5721–5732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawley PD (1976) Carcinogenesis by alkylating agents. Chem Carcinog 1: 303–484 [Google Scholar]
- Leiros I, Nabong MP, Grosvik K, Ringvoll J, Haugland GT, Uldal L, Reite K, Olsbu IK, Knaevelsrud I, Moe E, Andersen OA, Birkeland NK, Ruoff P, Klungland A, Bjelland S (2007) Structural basis for enzymatic excision of N1‐methyladenine and N3‐methylcytosine from DNA. EMBO J 26: 2206–2217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I, Bork P (2011) Interactive Tree Of Life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res 39(suppl_2): W475–W478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyskov S, Chou F‐C, Conchúir SÓ, Der BS, Drew K, Kuroda D, Xu J, Weitzner BD, Renfrew PD, Sripakdeevong P (2013) Serverification of molecular modeling applications: the Rosetta Online Server that Includes Everyone (ROSIE). PLoS One 8: e63906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAndrew RP, Park JI, Heins RA, Reindl W, Friedland GD, D'haeseleer P, Northen T, Sale KL, Simmons BA, Adams PD (2013) From soil to structure, a novel dimeric β‐glucosidase belonging to glycoside hydrolase family 3 isolated from compost using metagenomic analysis. J Biol Chem 288: 14985–14992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy TV, Karran P, Lindahl T (1984) Inducible repair of O‐alkylated DNA pyrimidines in Escherichia coli . EMBO J 3: 545–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ (2007) Phaser crystallographic software. J Appl Crystallogr 40: 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer PA, Socias S, Key J, Ransey E, Tjon EC, Buschiazzo A, Lei M, Botka C, Withrow J, Neau D (2016) Data publication with the structural biology data grid supports live analysis. Nat Commun 7: 10882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishina Y, He C (2006) Oxidative dealkylation DNA repair mediated by the mononuclear non‐heme iron AlkB proteins. J Inorg Biochem 100: 670–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moe E, Hall DR, Leiros I, Monsen VT, Timmins J, McSweeney S (2012) Structure‐function studies of an unusual 3‐methyladenine DNA glycosylase II (AlkA) from Deinococcus radiodurans . Acta Crystallogr D Biol Crystallogr 68: 703–712 [DOI] [PubMed] [Google Scholar]
- Morin A, Eisenbraun B, Key J, Sanschagrin PC, Timony MA, Ottaviano M, Sliz P (2013) Collaboration gets the most out of software. Elife 2: e01456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins EA, Rubinson EH, Pereira KN, Calcutt MW, Christov PP, Eichman BF (2013) An HPLC‐tandem mass spectrometry method for simultaneous detection of alkylated base excision repair products. Methods 64: 59–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins EA, Shi R, Kotsch LA, Eichman BF (2015a) A new family of HEAT‐like repeat proteins lacking a critical substrate recognition motif present in related DNA glycosylases. PLoS One 10: e0127733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins EA, Shi R, Parsons ZD, Yuen PK, David SS, Igarashi Y, Eichman BF (2015b) The DNA glycosylase AlkD uses a non‐base‐flipping mechanism to excise bulky lesions. Nature 527: 254–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins EA, Shi R, Eichman BF (2017) Toxicity and repair of DNA adducts produced by the natural product yatakemycin. Nat Chem Biol 13: 1002–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata T, Gupta V, Sorce D, Kim W‐Y, Sali A, Chait BT, Shigesada K, Ito Y, Werner MH (1999) Immunoglobulin motif DNA recognition and heterodimerization of the PEBP2/CBF Runt domain. Nat Struct Mol Biol 6: 615–619 [DOI] [PubMed] [Google Scholar]
- O'Brien PJ, Ellenberger T (2004) The Escherichia coli 3‐methyladenine DNA glycosylase AlkA has a remarkably versatile active site. J Biol Chem 279: 26876–26884 [DOI] [PubMed] [Google Scholar]
- O'Connor TR, Laval J (1991) Human cDNA expressing a functional DNA glycosylase excising 3‐methyladenine and 7‐methylguanine. Biochem Biophys Res Commun 176: 1170–1177 1645538 [Google Scholar]
- Otwinowski Z, Minor W (1997) Processing of X‐ray diffraction data collected in oscillation mode. Methods Enzymol 276: 307–326 [DOI] [PubMed] [Google Scholar]
- Parsons ZD, Bland JM, Mullins EA, Eichman BF (2016) A catalytic role for C‐H/π interactions in base excision repair by Bacillus cereus DNA glycosylase AlkD. J Am Chem Soc 138: 11485–11488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MN, Dehal PS, Arkin AP (2010) FastTree 2‐approximately maximum‐likelihood trees for large alignments. PLoS One 5: e9490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon JT, Sancar A (2005) Nucleotide excision repair. Prog Nucleic Acid Res Mol Biol 79: 183–235 [DOI] [PubMed] [Google Scholar]
- Riazuddin S, Lindahl T (1978) Properties of 3‐methyladenine‐DNA glycosylase from Escherichia coli . Biochemistry 17: 2110–2118 [DOI] [PubMed] [Google Scholar]
- Rohs R, Jin X, West SM, Joshi R, Honig B, Mann RS (2010) Origins of specificity in protein‐DNA recognition. Annu Rev Biochem 79: 233–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinson EH, Metz AH, O'Quin J, Eichman BF (2008) A new protein architecture for processing alkylation damaged DNA: the crystal structure of DNA glycosylase AlkD. J Mol Biol 381: 13–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinson EH, Gowda AS, Spratt TE, Gold B, Eichman BF (2010) An unprecedented nucleic acid capture mechanism for excision of DNA damage. Nature 468: 406–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinson EH, Eichman BF (2012) Nucleic acid recognition by tandem helical repeats. Curr Opin Struct Biol 22: 101–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph MJ, Gergen JP (2001) DNA‐binding by Ig‐fold proteins. Nat Struct Biol 8: 384–386 [DOI] [PubMed] [Google Scholar]
- Saparbaev M, Kleibl K, Laval J (1995) Escherichia coli, Saccharomyces cerevisiae, rat and human 3‐methyladenine DNA glycosylases repair 1, N6‐ethenoadenine when present in DNA. Nucleic Acids Res 23: 3750–3755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramm VL (2011) Enzymatic transition states, transition‐state analogs, dynamics, thermodynamics, and lifetimes. Annu Rev Biochem 80: 703–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrödinger L (2016) The PyMOL molecular graphics system, Version 1.7.4 Schrödinger, LLC.
- Sedgwick B (2004) Repairing DNA‐methylation damage. Nat Rev Mol Cell Biol 5: 148–157 [DOI] [PubMed] [Google Scholar]
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J (2011) Fast, scalable generation of high‐quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7: 539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahirov TH, Inoue‐Bungo T, Morii H, Fujikawa A, Sasaki M, Kimura K, Shiina M, Sato K, Kumasaka T, Yamamoto M (2001) Structural analyses of DNA recognition by the AML1/Runx‐1 Runt domain and its allosteric control by CBFβ. Cell 104: 755–767 [DOI] [PubMed] [Google Scholar]
- Thomas L, Yang CH, Goldthwait DA (1982) Two DNA glycosylases in Escherichia coli which release primarily 3‐methyladenine. Biochemistry 21: 1162–1169 [DOI] [PubMed] [Google Scholar]
- Trewick SC, Henshaw TF, Hausinger RP, Lindahl T, Sedgwick B (2002) Oxidative demethylation by Escherichia coli AlkB directly reverts DNA base damage. Nature 419: 174–178 [DOI] [PubMed] [Google Scholar]
- Vonrhein C, Blanc E, Roversi P, Bricogne G (2007) Automated structure solution with autoSHARP. Methods Mol Biol 364: 215–230 [DOI] [PubMed] [Google Scholar]
- Wyatt MD, Pittman DL (2006) Methylating agents and DNA repair responses: methylated bases and sources of strand breaks. Chem Res Toxicol 19: 1580–1594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Huang W, He QL, Zhao ZX, Zhang F, Wang RX, Kang JW, Tang GL (2012) Self‐resistance to an antitumor antibiotic: a DNA glycosylase triggers the base‐excision repair system in yatakemycin biosynthesis. Angew Chem 51: 10532–10536 [DOI] [PubMed] [Google Scholar]
- Zheng G, Lu X‐J, Olson WK (2009) Web 3DNA‐a web server for the analysis, reconstruction, and visualization of three‐dimensional nucleic‐acid structures. Nucleic Acids Res 37(suppl_2): W240–W246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Chordia MD, Cooper DR, Chruszcz M, Müller P, Sheldrick GM, Minor W (2014) Validating metal binding sites in macromolecule structures using the CheckMyMetal web server. Nat Protoc 9: 156 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File
Data Availability Statement
Structure factors and coordinates were deposited in the Protein Data Bank under accession codes 5VHV (1aR complex) and 5VI0 (THF complex), and the corresponding X‐ray diffraction images deposited in the SBGrid Data Bank (Meyer et al, 2016).