

Abstract
The innate immune kinase TBK1 initiates inflammatory responses to combat infectious pathogens by driving production of type I interferons. TBK1 also controls metabolic processes and promotes oncogene‐induced cell proliferation and survival. Here, we demonstrate that TBK1 activates mTOR complex 1 (mTORC1) directly. In cultured cells, TBK1 associates with and activates mTORC1 through site‐specific mTOR phosphorylation (on S2159) in response to certain growth factor receptors (i.e., EGF‐receptor but not insulin receptor) and pathogen recognition receptors (PRRs) (i.e., TLR3; TLR4), revealing a stimulus‐selective role for TBK1 in mTORC1 regulation. By studying cultured macrophages and those isolated from genome edited mTOR S2159A knock‐in mice, we show that mTOR S2159 phosphorylation promotes mTORC1 signaling, IRF3 nuclear translocation, and IFN‐β production. These data demonstrate a direct mechanistic link between TBK1 and mTORC1 function as well as physiologic significance of the TBK1‐mTORC1 axis in control of innate immune function. These data unveil TBK1 as a direct mTORC1 activator and suggest unanticipated roles for mTORC1 downstream of TBK1 in control of innate immunity, tumorigenesis, and disorders linked to chronic inflammation.
Keywords: IFN‐β, mTOR, mTORC1, TBK1
Subject Categories: Cancer, Immunology, Signal Transduction
Introduction
TANK‐binding kinase 1 (TBK1; aka NAK or T2K) and IKKε (IkB kinase ε) (aka IKKi, for “inducible”) represent non‐canonical IKK‐related innate immune kinases that mediate production of interferons and interferon‐stimulated genes (ISGs) downstream of Toll‐like receptors (TLRs), which function as pathogen recognition receptors (PRRs; Hacker & Karin, 2006; Clement et al, 2008; Mogensen, 2009). Upon PRR activation by pathogen‐associated molecular patterns (PAMPs), TLR signaling initiates host defense responses that eliminate pathogenic microbes (Mogensen, 2009; O'Neill et al, 2013). TANK‐binding kinase 1 expression is ubiquitous while IKKε expression is tissue‐restricted but inducible (Hacker & Karin, 2006; Clement et al, 2008). While the canonical IKKs (i.e.,IKKα; IKKβ) promote NF‐κB‐mediated pro‐inflammatory gene expression, TBK1 and IKKε phosphorylate the transcription factors IRF3 and IRF7 and promote their dimerization, nuclear translocation, and transcriptional activation, resulting in transcriptional induction of type I interferons (e.g., IFN‐α; IFN‐β; Fitzgerald et al, 2003; Hacker & Karin, 2006; Karin, 2009; Mogensen, 2009). TLR3 and TLR4 signal through the adaptor TRIF to activate TBK1 by an incompletely defined mechanism that involves TBK1 dimerization, K63‐linked polyubiquitination, and activation loop phosphorylation by an unknown kinase and/or by auto‐phosphorylation (Hacker & Karin, 2006; Mogensen, 2009; Ma et al, 2012; Tu et al, 2013). In addition to well‐defined roles in innate immunity, emerging work suggests that TBK1 and IKKε contribute to various pathological conditions including autoimmune diseases, obesity‐associated metabolic disorders, and cancer (Chiang et al, 2009; Grivennikov et al, 2010; Shen & Hahn, 2011; Yu et al, 2012; Reilly et al, 2013). In oncogenic KRAS transformed cells, TBK1 is required for anchorage‐independent cell proliferation and survival, as well as the growth of tumor explants in vivo (Chien et al, 2006; Clement et al, 2008; Barbie et al, 2009; Ou et al, 2011; Xie et al, 2011). In addition, IKKε contributes to cell transformation and exhibits amplification in breast cancer cells (Boehm et al, 2007).
Mechanistic target of rapamycin (mTOR), an evolutionarily conserved serine/threonine protein kinase, integrates diverse environmental signals and translates these cues into appropriate cellular responses (Huang & Fingar, 2014; Dibble & Cantley, 2015; Saxton & Sabatini, 2017). Mechanistic target of rapamycin promotes cell growth, proliferation, and survival and modulates immune function and cell metabolism. Consequently, aberrant mTOR signaling has been linked to myriad pathologic states including cancer and obesity‐linked diabetes (Zoncu et al, 2011; Laplante & Sabatini, 2012; Cornu et al, 2013). Despite the clear physiologic and therapeutic importance of mTOR, the biochemical pathways and molecular mechanisms that regulate mTOR function in response to diverse cellular cues and physiologic contexts remain incompletely deciphered. mTOR forms the catalytic core of two functionally distinct complexes defined by distinct partner proteins and sensitivities to the allosteric mTOR inhibitor rapamycin. The mTOR interacting protein raptor defines mTOR complex 1 (mTORC1; sensitive to acute rapamycin) while rictor defines mTOR complex 2 (mTORC2; insensitive to acute rapamycin; Hara et al, 2002; Kim et al, 2002; Sarbassov et al, 2004; Thoreen & Sabatini, 2009; Laplante & Sabatini, 2012; Cornu et al, 2013). Well‐described substrates of mTORC1 include S6K1 (ribosomal protein S6 kinase 1) and the translational repressor 4EBP1 (eIF4E‐binding protein 1; Jacinto & Lorberg, 2008; Magnuson et al, 2012; Huang & Fingar, 2014). Anabolic cellular signals such as growth factors (i.e., insulin; EGF) and nutrients (i.e., amino acids, glucose) promote mTORC1 signaling to increase protein, lipid, and nucleotide synthesis (Ma & Blenis, 2009; Dibble & Manning, 2013; Howell et al, 2013; Ricoult & Manning, 2013). While the regulation of mTORC2 remains less defined, mTORC2 phosphorylates Akt to modulate cell metabolism and promote cell survival (Sarbassov et al, 2005; Alessi et al, 2009).
Here, we demonstrate that the innate immune kinase TBK1 associates with and promotes mTOR complex 1 (mTORC1) catalytic activity and downstream signaling directly through site‐specific mTOR phosphorylation, thus unveiling new crosstalk between these important signaling systems. Although not mechanistically defined here, our data also indicate that TBK1 promotes mTORC2 signaling, which supports published work (Ou et al, 2011; Xie et al, 2011). EGF‐ but not insulin‐receptor‐mediated mTORC1 signaling requires TBK1 and mTOR S2159 phosphorylation, indicating a stimulus‐selective role for TBK1 in mTORC1 regulation by growth factors. During innate immune signaling, TRL3‐ and TLR4‐induced mTORC1 signaling also requires TBK1 and mTOR S2159 phosphorylation. By studying IFN‐β production in cultured and primary macrophages, we demonstrate that the TBK1‐mTORC1 axis controls physiologically relevant innate immune function. mTORC1 inhibition with rapamycin suppresses TLR3‐induced IFN‐β production by blunting the translocation of IRF3 from the cytosol to nucleus. Moreover, primary macrophages isolated from genome edited mice bearing a germline mTOR S2159A knock‐in allele show impaired mTORC1 signaling, IRF3 translocation, and IFN‐β production. Taken together, these data unveil TBK1 as a direct activator of mTORC1 and suggest roles for mTORC1 downstream of TBK1, which may improve our understanding of innate immunity as well as disorders linked to chronic low‐grade inflammation such as cancer, diabetes, and autoimmune diseases.
Results
TBK1 interacts with and phosphorylates mTOR within mTORC1
In previous work, we demonstrated that dual mTOR phosphorylation on S2159 and T2164 promotes mTOR complex 1 (mTORC1) signaling and mTORC1‐mediated cell growth (Ekim et al, 2011). To identify upstream mTOR kinases for these sites, we performed in vitro kinome screens. Roughly 300 recombinant active kinases were tested for their ability to phosphorylate recombinant GST‐mTOR (32 amino acids; 2,144–2,175) in a site‐specific manner. Mechanistic target of rapamycin phosphorylation was measured by dot‐blot analysis with mTOR phospho‐specific antibodies (Ekim et al, 2011; Fig EV1A) and by incorporation of [32P] on wild‐type GST‐mTOR but not on phospho‐deficient Ala substitution mutant (Fig EV1B). TBK1 and IKKε provided the strongest site‐specific mTOR S2159 phosphorylation. Indeed, the mTOR amino acid sequence surrounding S2159 fits consensus motifs found in defined TBK1 substrates (i.e., IRF3; IRF7; optineurin; suppressor of IKKε [SIKE]; Wild et al, 2011; Marion et al, 2013) and bears similarity to an IKKε consensus phosphorylation motif (Fig 1A; Peters et al, 2000; Hutti et al, 2009). At this time, the upstream mTOR T2164 kinase remains unknown. We validated these screens with conventional in vitro kinase assays. Recombinant active TBK1 and IKKε each phosphorylated GST‐mTOR S2159 in vitro in a manner sensitive to the TBK1/IKKε pharmacologic inhibitors amlexanox, BX‐795 and MRT‐67307 (a derivative of BX‐795) (Clark et al, 2009, 2011; Reilly et al, 2013; Fig 1B). Recombinant active TBK1 and IKKε also phosphorylated full‐length wild‐type but not S2159A Myc‐mTOR directly in vitro (Fig 1C). When immunoprecipitated from HEK293 cells, transfected wild‐type (WT) but not kinase dead (KD) Flag‐TBK1 and Flag‐IKKε phosphorylated GST‐mTOR S2159 in vitro (Fig EV1C). These data confirm the site specificity of the P‐S2159 antibody (demonstrated by us previously; Ekim et al, 2011) and show that TBK1 and IKKε phosphorylate full‐length mTOR as well as a truncated mTOR fragment.
Figure EV1. An in vitro human kinome screen identified TBK1 and IKKε as mTOR S2159 kinases that interact with mTORC1 (related to Fig 1).
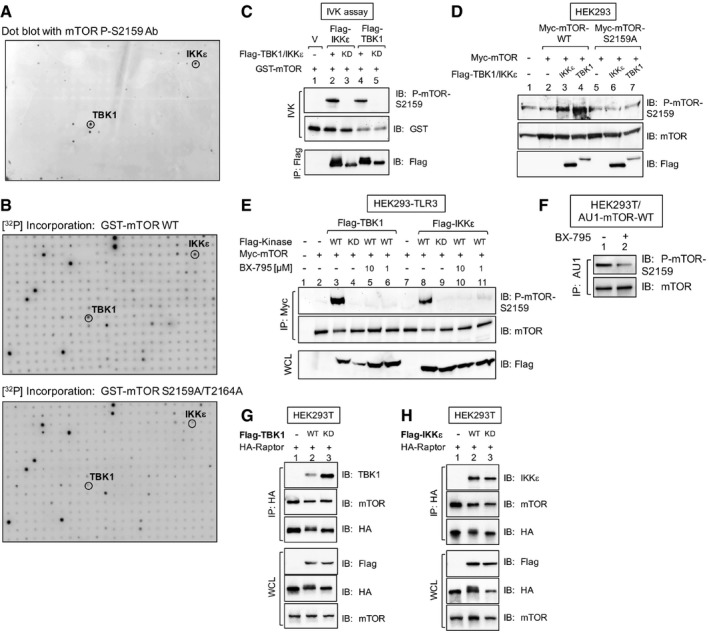
-
AIn vitro kinome screen with recombinant GST‐mTOR substrate and ˜300 recombinant active kinases. Substrate phosphorylation was detected with mTOR P‐S2159 antibodies.
-
BSimilar to (A), except that GST‐mTOR wild type (WT) or GST‐mTOR S2159A/T2164A (AA) was used as substrate, and [γ‐32P]‐ATP was included in the reactions. [32P] incorporation was detected by autoradiography.
-
CTBK1 and IKKε immune complex in vitro kinase (IVK) assays. Flag‐TBK1 or Flag‐IKKε WT (+) or kinase dead (KD) was immunoprecipitated from transfected HEK293 cells and incubated with GST‐mTOR substrate. IVK reactions were performed by incubating the Flag‐TBK1 or Flag‐IKKε immunoprecipitates (IP) with GST‐mTOR substrate [200 ng] for 30 min at 30°C. Immunoprecipitates (IPs) were immunoblotted (IB) as indicated.
-
DCellular overexpression of TBK1 and IKKε in cells increases mTOR P‐S2159. HEK293 cells were co‐transfected with Myc‐mTOR (WT or S2159A) together with Flag‐IKKε or Flag‐TBK1 or plasmids. Whole‐cell lysate (WCL) was immunoblotted as indicated.
-
EOverexpression of TBK1 and IKKε in cells increases mTOR P‐S2159 in a BX‐795‐sensitive manner. HEK293‐TLR3 cells were co‐transfected with Myc‐mTOR and Flag‐TBK1 or Flag‐IKKε wild type (+) or kinase dead (KD) and then treated with BX‐795 [10 μM or 1 μM] (2 h). Myc‐mTOR was immunoprecipitated, and IPs and WCL were immunoblotted as indicated.
-
FCellular BX‐795 treatment decreases mTOR S2159 phosphorylation. HEK293T cells stably expressing AU1‐mTOR were pre‐treated with BX‐795 [10 μM] (2 h). AU1‐mTOR was immunoprecipitated and immunoblotted as indicated.
-
G, HFlag‐TBK1 and Flag‐IKKε co‐immunoprecipitate with HA‐raptor and mTOR. HEK293T cells stably expressing AU1‐mTOR were transfected with Flag‐TBK1 (G) or Flag‐IKKε (H) wild‐type (+) or kinase‐dead (KD) plasmids together with HA‐raptor. HA‐raptor was immunoprecipitated and immunoblotted as indicated.
Source data are available online for this figure.
Figure 1. TBK1 interacts with and phosphorylates mTOR within mTORC1.

- The mTOR S2159 sequence fits TBK1 and IKKε consensus phosphorylation motifs.
- TBK1/IKKε phosphorylate mTOR S2159 directly in vitro. In vitro kinase (IVK) assays with recombinant (re) active TBK1 or IKKε [50 ng] (Invitrogen) and recombinant GST‐mTOR substrate [200 ng] for 30 min at 30°C. Reactions were pre‐incubated on ice 30 min with amlexanox [500, 250 or 50 μM], BX‐795 [10 μM] or MRT‐67307 [10 μM] and immunoblotted (IB) as indicated.
- TBK1/IKKε phosphorylate full‐length mTOR on S2159. Myc‐mTOR wild type (WT) and S2159A were immunoprecipitated (IP) from transfected HEK293 cells and incubated with re‐TBK1 or re‐IKKε. IVK assays were performed as above and immunoblotted (IB) as indicated.
- TBK1 overexpression increases mTOR P‐S2159, and poly(I:C) boosts this phosphorylation further. HEK293‐TLR3 cells were co‐transfected with Flag‐TBK1 and Myc‐mTOR. Cells were serum‐starved (20 h) and stimulated −/+ poly(I:C) [50 μg/ml] (2 h). Myc‐mTOR immunoprecipitates were immunoblotted (IB) as indicated.
- TBK1 and IKKε overexpression increases mTOR P‐S2159 within mTORC1. HEK293‐TLR3 cells were co‐transfected with Flag‐TBK1 or Flag‐IKKε, Myc‐mTOR, and HA‐raptor. HA‐raptor immunoprecipitates and whole‐cell lysates (WCL) were immunoblotted (IB) as indicated.
- Flag‐TBK1 and Flag‐IKKε co‐immunoprecipitate with endogenous mTORC1. HEK293T cells were transfected with Flag‐TBK1 or Flag‐IKKε wild type (+) or kinase dead (KD). Endogenous raptor immunoprecipitates and WCL were immunoblotted (IB) as indicated.
- mTOR is phosphorylated on S2159 in wild type but not TBK1 null MEFs. TBK1+/+ and TBK1−/− MEFs were serum‐starved (20 h) and stimulated ± EGF [25 ng/ml]. WCL was immunoblotted (IB) as indicated. The arrow indicates mTOR phosphorylated on S2159.
- The TBK1‐ and IKKε‐activating agonists poly(I:C) and LPS increase mTOR P‐S2159 in a BX‐795‐sensitive manner in cultured macrophages. RAW264.7 macrophages were pre‐treated with BX‐795 [10 μM] (2 h) and stimulated −/+ poly(I:C) [30 μg/ml] or LPS [100 ng/ml] (60 min).
- Poly(I:C) and LPS increase mTOR P‐S2159 in primary bone marrow‐derived macrophages (BMDMs). BMDMs were stimulated −/+ poly(I:C) [30 μg/ml] or LPS [100 ng/ml] (60 min).
- LPS increases mTOR P‐S2159 in vivo. Mice (C57BL6, 6 weeks old) were injected intraperitoneally with PBS or LPS [1 mg/kg BW] (2 h). mTOR was immunoprecipitated from spleen tissue, and IPs and WCL were immunoblotted as indicated. The graph depicts levels of mTOR P‐S2159 relative to total mTOR in spleen tissue −/+ LPS. n = 8 from 3 independent experiments ± SD. *P = 0.004 relative to PBS‐treated control mice by paired t‐test (two‐tailed).
Source data are available online for this figure.
We next investigated whether TBK1 and IKKε increase mTOR S2159 phosphorylation in intact cells. Overexpression of Flag‐TBK1 in HEK293 cells expressing TLR3 increased P‐S2159 on Myc‐mTOR, while treatment with poly(I:C) (polyinosinic: polycytidylic acid), a double‐stranded RNA mimetic that binds to and activates TLR3, increased mTOR P‐S2159 further (Fig 1D). Overexpression of wild‐type but not kinase‐dead Flag‐TBK1 and Flag‐IKKε in HEK293 cells increased mTOR S2159 phosphorylation on wild‐type but not S2159A Myc‐mTOR in a manner sensitive to BX‐795 (Fig EV1D and E). Treatment of HEK293 cells expressing endogenous TBK1 with BX‐795 reduced mTOR P‐S2159 (Fig EV1F). These data indicate that TBK1 promotes mTOR S2159 phosphorylation in intact cells. Flag‐TBK1 and Flag‐IKKε overexpression also increased mTOR S2159 phosphorylation on HA‐raptor‐associated Myc‐mTOR (Fig 1E), indicating that TBK1/IKKε increase mTOR phosphorylation within mTORC1. Flag‐TBK1 and Flag‐IKKε interacted with both endogenous raptor (Fig 1F) and exogenously expressed HA‐raptor (Fig EV1G and H) by co‐immunoprecipitation assay. Importantly, immortalized TBK1−/− MEFs exhibited reduced mTOR P‐S2159 relative to littermate‐matched TBK1+/+ MEFs (Fig 1G; see also Fig 2H). Taken together, these data suggest that these innate immune kinases interact with and phosphorylate mTOR and within mTORC1.
Figure 2. TBK1 promotes growth factor‐induced mTORC1 signaling in a stimulus‐selective manner.

- EGF increases mTORC1 signaling in a TBK1‐dependent manner. TBK1+/+ and TBK1−/− MEFs were serum‐starved (20 h), pre‐treated with Ku‐0063794 [1 μM] (30 min), and stimulated −/+ EGF [25 ng/ml] (0–60 min). Whole‐cell lysates (WCL) were immunoblotted (IB) as indicated.
- Ectopic expression of TBK1 rescues mTORC1 signaling in TBK1−/− MEFs. TBK1−/− MEFs were transiently transfected with vector control (V), wild type (WT), or kinase‐dead (KD) Flag‐TBK1, serum‐starved (20 h), and analyzed as above.
- EGF‐receptor signaling remains intact in TBK1−/− MEFs. TBK1+/+ and TBK1−/− MEFs were serum‐starved (20 h) and stimulated −/+ EGF [25 ng/ml] (0, 5, or 15 min).
- TBK1 is required for EGF‐stimulated mTORC1 and mTORC2 signaling. TBK1+/+ and TBK1−/− MEFs were stimulated with EGF as in (A).
- TBK1 is not required for insulin‐stimulated mTORC1 signaling. TBK1+/+ and TBK1−/− MEFs were stimulated with insulin as in (C).
- Side‐by‐side comparison of EGF‐ vs. insulin‐stimulated mTORC1 and mTORC2 signaling in TBK1+/+ vs. TBK1−/− MEFs. MEFs were treated as in (C).
- Pharmacologic TBK1 inhibition reduces EGF‐induced mTORC1 signaling. HEK293 cells were serum‐starved (20 h), pre‐treated with Ku‐0063794 [1 μM] (30 min) or amlexanox [50 μM] (2 h), and stimulated −/+ EGF [25 ng/ml] (30 min) or insulin (INS) [100 nM] (30 min).
- EGF but not insulin increases mTOR P‐S2159. TBK1+/+ vs. TBK1−/− MEFs: MEFs were treated as in (C). The arrow indicates mTOR phosphorylated on S2159.
Source data are available online for this figure.
We next analyzed TBK1/IKKε‐mediated mTOR phosphorylation in cultured and primary macrophages, cells in which TLR3 and TLR4 engagement with microbial PAMPs activates TBK1 and IKKε during an innate immune response. Activation of TBK1/IKKε in cultured RAW264.7 macrophages upon treatment with poly(I:C) and LPS (lysophosphatidic acid) (a bacterial cell wall component that binds to and activates TLR4) increased mTOR P‐S2159 in a BX‐795 sensitive manner (Fig 1H). Similarly, TBK1/IKKε activation in primary bone marrow‐derived macrophages (BMDMs) with poly(I:C) and LPS increased mTOR P‐S2159 (Fig 1I). Importantly, we confirmed activation of TBK1 and IKKε by measuring phosphorylation on their activation loop sites (S172) in cultured and primary macrophages. To extend these results to an in vivo setting, we injected mice acutely (2 h) with LPS. LPS increased mTOR S2159 phosphorylation in spleen tissue, a source of abundant monocytes (Fig 1J). As expected, LPS activated TBK1 in spleen, as determined by increased phosphorylation of TBK1 P‐S172. Moreover, LPS increased mTORC1 signaling (as measured by the phosphorylation of the mTORC1 substrate S6K1 on T389; Fig 1J), consistent with an earlier finding that LPS administered to mice in vivo increased mTORC1 signaling in several tissues in a rapamycin‐sensitive manner (e.g., liver; lung; kidney; Lee et al, 2010). We also found that LPS administered in vivo increased mTORC2 signaling (as measured by the phosphorylation of the mTORC2 substrate Akt (on S473). These data demonstrate that TLR4 signaling in vivo activates TBK1 and promotes mTOR S2159 phosphorylation, events that correlate with increased mTORC1 and mTORC2 signaling. Taken together, the data demonstrate that TBK1/IKKε associate with and phosphorylate mTOR within mTORC1. Whether TBK1/IKKε phosphorylates other sites on mTOR or mTORC1 components in addition to S2159 remains an open question.
TBK1 promotes growth factor‐induced mTORC1 signaling in a stimulus‐selective manner
We next focused on the role of TBK1 in control of growth factor‐induced mTORC1 signaling by studying TBK1+/+ and TBK1−/− MEFs. An EGF time course (1–60 min) revealed substantially stronger EGF‐stimulated mTORC1 signaling in TBK1+/+ wild‐type MEFs relative to TBK1−/− null MEFs at all time points (Fig 2A; see also Fig 2D). These data indicate that TBK1 contributes to EGF‐receptor signaling to mTORC1. As immortalized MEFs can possess clonal differences due to divergence in culture, we confirmed that re‐introduction of wild‐type Flag‐TBK1 into TBK1−/− MEFs rescued EGF‐induced S6K1 T389 phosphorylation (Fig 2B). Re‐introduction of kinase‐dead Flag‐TBK1 rescued mTORC1 signaling modestly, suggesting that while TBK1 kinase activity is important for mTORC1 signaling, TBK1 may also provide kinase‐independent scaffolding function (Fig 2B). Importantly, we also confirmed that EGF‐receptor signaling remains intact in these immortalized TBK1−/− MEFs. EGF‐stimulated phosphorylation of EGF‐receptor (Y1173, an auto‐phosphorylation site), P‐STAT3 (Y705), and MAPK (T202/Y204) remained intact in TBK1 null MEFs relative to wild‐type MEFs (Fig 2C). We noted that TBK1−/− MEFs exhibit increased basal levels of P‐STAT3‐Y705 relative to TBK1+/+ MEFs. While we do not know the reason for this phenomenon at this time, we speculate that it may result from reduced TBK1‐ or mTORC1‐mediated negative feedback in TBK1−/− MEFs, leading to elevated P‐STAT3. Consistent with a role for TBK1 in EGF‐stimulated mTORC1 signaling, pre‐treatment of TBK1+/+ MEFs with the TBK1/IKKε inhibitor amlexanox reduced EGF‐stimulated S6K1 phosphorylation (Fig 2D); as expected, the mTOR catalytic inhibitor Ku‐0063794 ablated S6K1 phosphorylation. Thus, genetic or pharmacologic inactivation of TBK1 reduces EGF‐stimulated mTORC1 signaling. It is important to note that we avoided use of the better known TBK1/IKKε inhibitors BX‐795 and MRT‐67307 (Clark et al, 2009, 2011) for analysis of mTORC1 signaling due to documented TBK1 independent inhibitory effects on regulatory mTORC1‐S6K1 pathway components. BX‐795 was originally developed as an inhibitor of PDK1, the kinase that phosphorylates S6K1 on its activation loop site (T229). As PDK1‐mediated phosphorylation of T229 was reported to promote mTORC1‐mediated phosphorylation of S6K1 T389 (Keshwani et al, 2011), inhibition of PDK1 with BX‐795 (or its derivative MRT‐67037) could reduce mTORC1‐mediated S6K1 P‐T389 through a TBK1 independent mechanism. To avoid this complication, we employed the more recently identified TBK1/IKKε inhibitor amlexanox (Reilly et al, 2013), as no inhibitory effects on mTORC1‐S6K1 pathway components have been reported.
Unlike EGF‐induced mTORC1 signaling, insulin‐induced mTORC1 signaling was similar in TBK1+/+ and TBK1−/− MEFs, and amlexanox had no inhibitory effect on S6K1 T389 phosphorylation (Fig 2E). These data indicate that TBK1 is not required for insulin‐receptor signaling to mTORC1. We also compared insulin‐ and EGF‐stimulated mTORC1 signaling side by side and included analysis of 4EBP1, another well‐studied mTORC1 substrate. Relative to TBK1+/+ MEFs, TBK1−/− MEFs exhibited reduced mTORC1 dependent S6K1 (T389) and 4EBP1 (T70; S65; T46) phosphorylation in response to EGF but not insulin (Fig 2F). Similar to MEFs, amlexanox reduced EGF‐ but not insulin‐stimulated mTORC1 signaling in HEK293 cells (Fig 2G). To begin to understand why the EGF but not the insulin pathway requires TBK1 to promote mTORC1 signaling, we examined mTOR S2159 phosphorylation in response to EGF vs. insulin. We found that EGF increases mTOR P‐S2159 to a greater extent than insulin (Fig 2H), suggesting that EGF but not insulin signaling either activates TBK1 or alternately primes mTOR for TBK1‐mediated phosphorylation. Previous work demonstrated that cellular EGF treatment increased the kinase activity of immunoprecipitated TBK1 toward His‐Akt1 substrate in vitro (Ou et al, 2011), suggesting that EGF increases TBK1 intrinsic catalytic activity. Unexpectedly, EGF stimulation of MEFs and HEK293‐TLR3 cells failed to increase TBK1 phosphorylation on the activation loop site (S172) while increasing mTORC1 signaling (Fig EV2A and B). As expected, innate immune agonists (i.e., LPS; poly(I:C)) increased P‐TBK1‐S172 and mTORC1 signaling concomitantly (Fig EV2A and B). These data require further investigation but suggest that either TBK1 activation loop site phosphorylation does not represent an accurate readout of TBK1 kinase activity in response to EGF or that EGF‐receptor signaling does not activate TBK1. Finally, we asked if TBK1 plays a role in amino acid activated mTORC1 signaling. We found that acute amino acid stimulation of amino acid‐deprived cells increases P‐S6K1 T389 similarly in TBK1+/+ and TBK1−/− MEFs, indicating that TBK1 is not required for activation of mTORC1 by amino acids (Fig EV2C). As a final cautionary note, we noted that overexpression of Flag‐TBK1 suppressed mTORC1 signaling (Fig EV2D), likely due to inappropriate interaction of overexpressed TBK1 with mTORC1 or regulatory factors. Taken together, the data indicate that TBK1 contributes in a stimulus‐selective manner to activation of mTORC1 signaling, with TBK1 playing a dominant role in the EGF‐ but not insulin‐ or amino acid‐mediated activation of mTORC1.
Figure EV2. LPS but not EGF increases TBK1 activation loop site phosphorylation in MEFs; TBK1 is not required for amino acid stimulated mTORC1 signaling; ectopic TBK1 overexpression suppresses mTORC1 signaling (related to Fig 2).
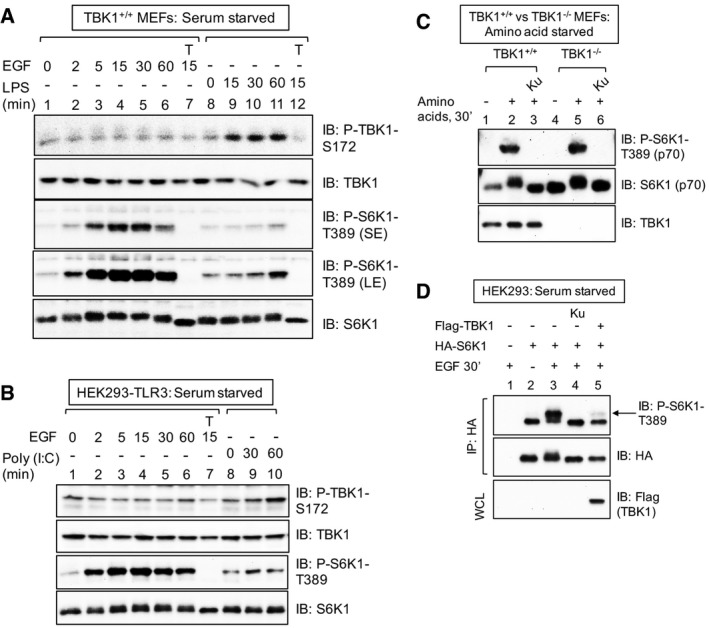
- EGF fails to increase TBK1 S172 phosphorylation in MEFs. Cells were serum deprived (20 h) and stimulated −/+ EGF [25 ng/ml] or LPS [100 ng/ml] for the times indicated. Whole‐cell lysate (WCL) was immunoblotted as indicated. SE: short exposure; LE: long exposure.
- EGF fails to increase TBK1 S172 phosphorylation in HEK293/TLR3 cells. Cells were serum deprived (20 h) and stimulated −/+ EGF [25 ng/ml] or poly(I:C) [50 ng/ml] for the times indicated.
- TBK1 is not required for amino acid‐stimulated mTORC1 signaling. TBK1+/+ and TBK1−/− MEFs were deprived of amino acids by incubation in D‐PBS + glucose + dialyzed FBS [10%] (60 min). Amino acids were added back by incubating the cells in DMEM/FBS [10%] (30 min).
- Cellular overexpression of TBK1 inhibits mTORC1 signaling. HEK293 cells were co‐transfected with HA‐S6K1 together with Flag‐TBK1, serum‐starved (20 h), pre‐treated with Ku‐0063794 [1 μM] (30 min), and stimulated −/+ EGF [25 ng/ml] (30 min). HA‐S6K1 was immunoprecipitated, and IPs and WCL were immunoblotted (IB) as indicated.
We noted that phosphorylation of Akt S473, a site phosphorylated directly by mTORC2, was reduced in TBK1−/− MEFs relative to TBK1+/+ MEFs in response to EGF (Fig 2A, D and F), consistent with published work (Ou et al, 2011; Xie et al, 2011). As expected, the mTOR inhibitor Ku‐0063794 suppressed Akt S473 phosphorylation strongly (Fig 2A, D and F; Sarbassov et al, 2005). Collectively, the data indicate that the TBK1‐mTOR axis promotes both mTORC1 and mTORC2 signaling, with TBK1 positioned as a critical effector of EGF‐receptor signaling. As Akt positively regulates mTORC1 through inhibitory phosphorylation of Tsc2 (Inoki et al, 2002; Manning et al, 2002) and PRAS40 (van der Haar et al, 2007; Sancak et al, 2007), we sought to exclude the possibility that reduced mTORC1 signaling in TBK1−/− MEFs results indirectly from reduced Akt S473 phosphorylation and activity. PDK1‐mediated phosphorylation of Akt on its activation loop site (T308) is essential for its catalytic activity (Pearce et al, 2010) whereas mTORC2‐mediated phosphorylation of Akt on its hydrophobic motif site (S473) boosts catalytic activity and directs Akt substrate preference toward certain substrates (i.e., Foxo3) but not others (i.e., PRAS40; GSK3; Tsc2; Guertin et al, 2006; Jacinto et al, 2006; Pearce et al, 2010). Published work shows that cells lacking rictor, an mTOR partner critical for mTORC2 function, maintain mTORC1 signaling despite extremely low Akt S473 phosphorylation (Sarbassov et al, 2005; Guertin et al, 2006; Jacinto et al, 2006). By studying rictor−/− MEFs reconstituted with either vector control or HA‐rictor, we confirmed that EGF‐stimulated S6K1 T389 phosphorylation remains intact in cells with reduced Akt S473 phosphorylation (Fig 3A). Despite modestly reduced Akt T308 phosphorylation, often observed in cells with reduced Akt S473 phosphorylation (Sarbassov et al, 2005), phosphorylation of the Akt substrates PRAS40, GSK3, and Tsc2 remained intact (Fig 3A). Taken together, these data indicate that Akt retains significant catalytic activity in cells with an impaired mTORC2‐Akt P‐S473 axis (Fig 3A). Moreover, knockdown of TBK1 using shRNA reduced EGF‐stimulated S6K1 T389 phosphorylation in rictor−/− MEFs (Fig 3B), verifying an important role of TBK1 in mTORC1 activation in cells that lack mTORC2 function. Collectively, the data support a model in which TBK1 promotes mTORC1 signaling by a direct mechanism rather than an indirect mechanism involving modulation of Akt S473 phosphorylation.
Figure 3. TBK1 promotes mTORC1 signaling independently of mTORC2‐dependent Akt S473 phosphorylation.
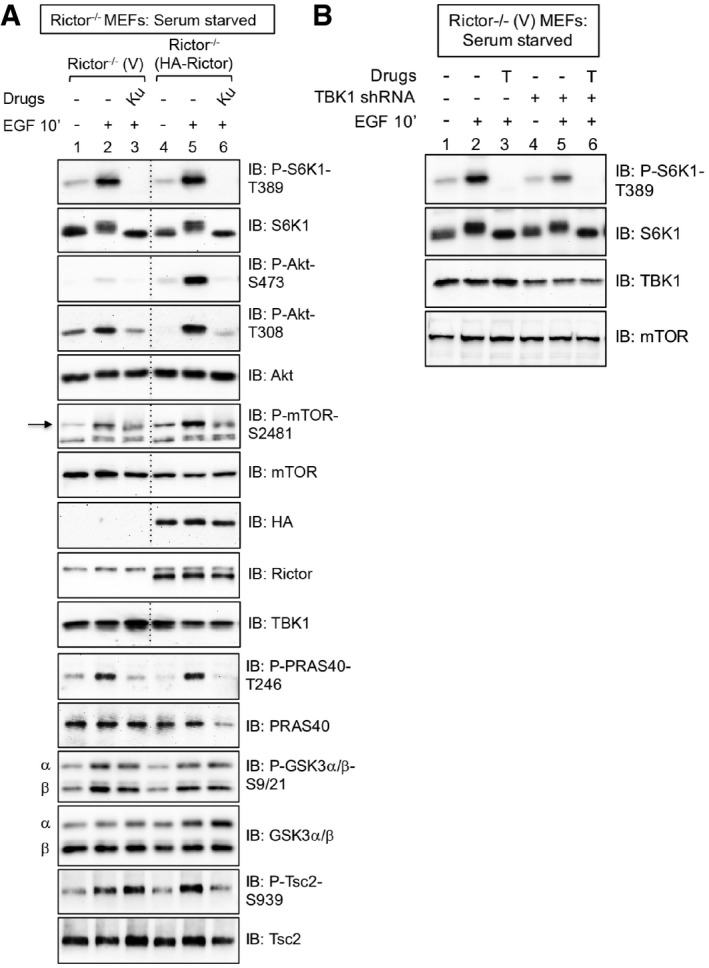
- mTORC1 signaling remains intact in MEFs lacking mTORC2 function. Rictor−/− MEFs stably expressing vector control (V) or rescued stably with HA‐rictor were serum‐starved (20 h), pre‐treated with Ku‐0063794 [1 μM] (30 min), and stimulated −/+ epidermal growth factor (EGF) [25 ng/ml] (30 min). Whole‐cell lysates (WCL) were immunoblotted as indicated. The arrow indicates mTOR auto‐phosphorylated on S2481.
- TBK1 is required for mTORC1 signaling in MEFs with reduced Akt S473 phosphorylation. TBK1 expression was reduced using lentivirally delivered TBK1 shRNA in Rictor−/− MEFs stably expressing vector control (V). Cells were EGF stimulated as above, except Torin1 [100 nM] (T) was used to inhibit mTOR.
In response to EGF‐receptor activation, TBK1 promotes mTORC1 signaling and catalytic activity in a manner that depends on mTOR S2159 phosphorylation
To determine whether EGF‐stimulated mTORC1 signaling requires phosphorylation of mTOR on the TBK1 site (S2159), we performed a chemical mTOR knockout‐rescue experiment using a rapamycin‐resistant (RR) allele of mTOR (S2035I) that cannot bind rapamycin and its obligate partner FKBP12 (Stan et al, 1994; Chen et al, 1995). Expression of RR‐mTOR enables the signaling capacity of mTORC1 containing exogenously expressed mutant mTOR alleles to be studied in the absence of endogenous mTORC1 function upon chemical knockout with rapamycin (Brown et al, 1995; Hara et al, 1997). As expected, rapamycin abrogated HA‐S6K1 phosphorylation in HEK293 cells expressing wild‐type (WT) AU1‐mTOR, and expression of rapamycin‐resistant (RR) AU1‐mTOR rescued HA‐S6K1 T389 phosphorylation during rapamycin treatment (Fig 4A). In response to EGF, HA‐S6K1 phosphorylation was reduced in cells expressing RR‐mTOR with a S2159A substitution relative to RR‐mTOR with a wild‐type backbone (Fig 4A). Moreover, substitution of a phospho‐mimetic Asp (D) residue at S2159 within RR‐mTOR (S2159D) rescued HA‐S6K1 phosphorylation relative to RR‐mTOR S2159A. These data indicate that mTOR S2159 phosphorylation is required for EGF‐stimulated mTORC1 signaling, at least in part.
Figure 4. TBK1 promotes EGF‐stimulated mTORC1 signaling and catalytic activity in a manner dependent on site‐specific mTOR phosphorylation.
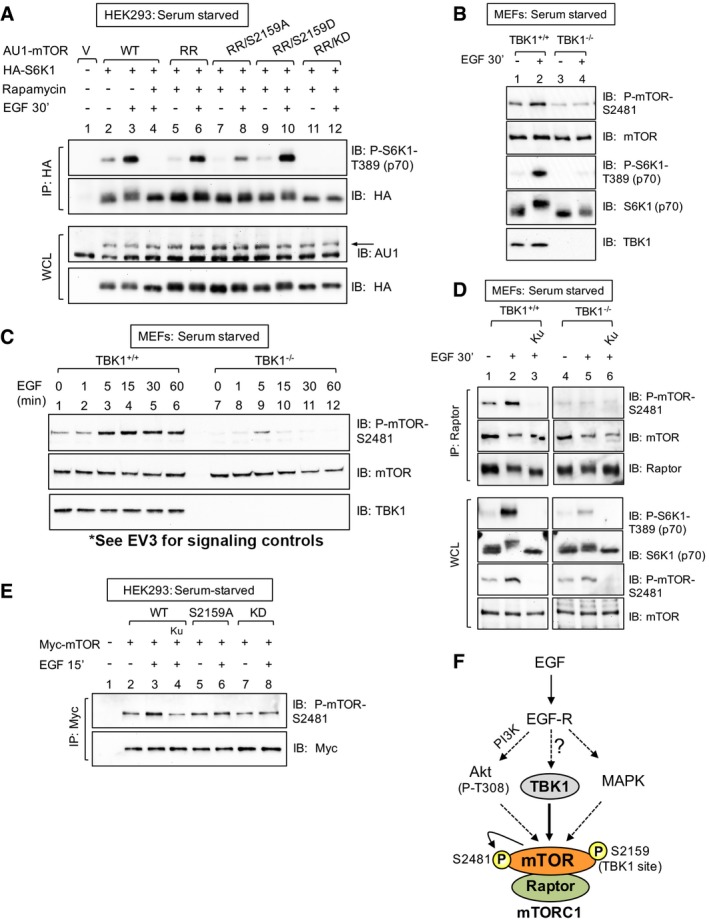
- mTOR S2159 phosphorylation is required for EGF‐stimulated mTORC1 signaling. HEK293 cells were co‐transfected with vector control, wild‐type, or rapamycin‐resistant (RR) AU1‐mTOR alleles (RR or RR/S2159A) together with HA‐S6K1. Cells were serum‐starved (20 h), treated −/+ rapamycin (30 min) to ablate endogenous mTORC1 function, and stimulated −/+ EGF [25 ng/ml] (30 min). HA‐S6K1 was immunoprecipitated, and immunoprecipitates (IP) and whole‐cell lysates (WCL) were immunoblotted (IB) as indicated. The arrow indicates AU1‐mTOR.
- TBK1 is required for EGF‐stimulated mTOR auto‐phosphorylation. TBK1+/+ and TBK1−/− MEFs were serum‐starved, EGF stimulated, and analyzed as in (A).
- EGF time course analysis of mTOR auto‐phosphorylation. TBK1+/+ and TBK1−/− MEFs were serum‐starved, EGF stimulated for 0‐60 min, and analyzed as in (A).
- TBK1 is required for EGF‐stimulated mTOR auto‐phosphorylation within mTORC1. TBK1+/+ and TBK1−/− MEFs were serum‐starved, pre‐treated with Ku‐0063794 [1 μM], and EGF stimulated as in (A). Raptor was immunoprecipitated, and IPs and WCL were analyzed.
- mTOR S2159 phosphorylation is required for EGF‐stimulated mTOR auto‐phosphorylation. HEK293 cells were transfected with Myc‐mTOR wild type (WT), S2159A, and kinase dead (KD). Cells were then serum‐starved, pre‐treated with Ku‐0063794 (30 min), and stimulated −/+ EGF as in (A).
- Model. EGF‐receptor signaling increases mTORC1 signaling through at least three pathways in MEFs: the PI3K/Akt, MAPK, and TBK1 pathways.
We next investigated the molecular mechanism by which TBK1 promotes EGF‐induced mTORC1 signaling. EGF‐stimulated mTOR S2481 auto‐phosphorylation was reduced in TBK1−/− MEFs compared to TBK1+/+ MEFs (Fig 4B; also, see Fig 2F). It is important to note that mTOR S2481 auto‐phosphorylation correlates with active mTORC1 and mTORC2 signaling and thus represents a simple method to monitor overall mTOR and complex specific catalytic activity in intact cells (Soliman et al, 2010). Time course analysis revealed that EGF increased mTOR S2481 auto‐phosphorylation from 5 to 60 min more strongly in TBK1+/+ than TBK1−/− MEFs (Fig 4C). As before, S6K1 T389 and Akt S473 phosphorylation were significantly reduced in TBK1−/− MEFs relative to TBK1+/+ MEFs (Fig EV3A). To determine whether TBK1 promotes mTORC1‐specific catalytic activity, we measured raptor‐associated mTOR S2481 auto‐phosphorylation. EGF increased mTORC1 catalytic activity in TBK1+/+ but not TBK1−/− MEFs in an mTOR‐dependent manner (Fig 4D). Moreover, EGF‐stimulated mTOR S2481 auto‐phosphorylation was reduced on Myc‐mTOR S2159A and kinase‐dead alleles relative to wild type (Fig 4E), indicating that mTOR S2159 phosphorylation contributes to mTOR catalytic activity. These data indicate that TBK1 increases mTORC1 catalytic activity to promote mTORC1 downstream signaling. By pharmacologically inhibiting class I PI3Kα (with BYL‐719), Akt (with MK‐2206), and MAPK (with CI‐1040; Fig EV3B and C), we demonstrated that the EGF‐receptor signals through at least three parallel pathways that converge on mTORC1‐ PI3Kα/Akt, MAPK, and here TBK1 (Fig 4F).
Figure EV3. PI3K, Akt, MAPK, and TBK1 activity is required for EGF‐stimulated mTORC1 signaling (related to Fig 4).
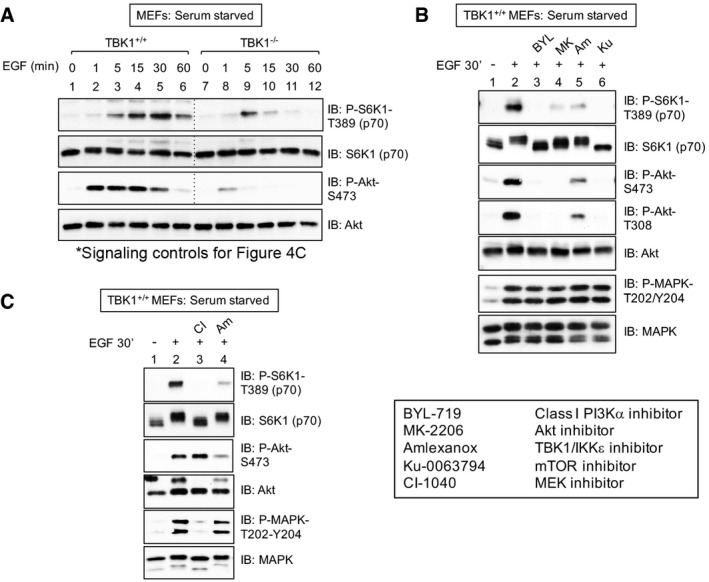
- mTORC1 signaling controls for Fig 4C. TBK1+/+ and TBK1−/− MEFs were serum‐starved (20 h) and stimulated without (−) or with (+) EGF [25 ng/ml] for 0–60 min. WCL was immunoblotted as indicated.
- PI3Kα, Akt, and TBK1 activity is required for EGF‐stimulated mTORC1 signaling. TBK1+/+ MEFs were serum‐starved (20 h), pre‐treated with the PI3Kα class I inhibitor BYL‐719 [10 μM] (30 min), the Akt inhibitor MK‐2206 [10 μM] (30 min), amlexanox [100 μM] (2 h), or Ku‐0063794 [1 μM] (30 min), and stimulated −/+ epidermal growth factor (EGF) [50 ng/ml] (30 min).
- MAPK and TBK1 activity is required for EGF‐stimulated mTORC1 signaling: TBK1+/+ MEFs were treated as above except they were pre‐treated with the MEK inhibitor CI‐1040 [10 μM] or amlexanox [100 μM] (30 min).
In response to TLR3 and TLR4 activation, TBK1 promotes mTORC1 signaling and catalytic activity in a manner dependent on mTOR S2159 phosphorylation
To examine mTORC1 regulation by TBK1 within innate immune signaling networks, we stimulated RAW264.7 macrophages with the TLR3 agonist poly(I:C) and the TLR4 agonist LPS. Both TLR agonists increased phosphorylation of the mTORC1 targets S6K1 (T389) and 4EBP1 (S65; T46; Fig 5A), consistent with an earlier report (Schmitz et al, 2008), in a manner sensitive to the TBK1 inhibitor amlexanox and the mTOR inhibitor Ku‐0063794. Amlexanox also blunted poly(I:C)‐ and LPS‐induced Akt S473 phosphorylation, indicating suppression of mTORC2 signaling. As expected, poly(I:C) and LPS activated TBK1, as monitored by increased phosphorylation of TBK1 on its activation loop site (S172) and amlexanox‐sensitive phosphorylation of the TBK1 substrate IRF3 (S396), the transcription factor that drives production of type I interferons (Fitzgerald et al, 2003) (Fig 5A). To confirm that amlexanox reduces mTORC1 signaling through inhibition of TBK1 and not due to an off‐target effect, we employed RNAi approaches to knockdown TBK1 expression in RAW264.7 macrophages (note that we found it difficult to knockdown IKKε). Knockdown of TBK1 with lentivirally delivered shRNA reduced both poly(I:C)‐ and LPS‐induced S6K1 T389 phosphorylation (Fig 5B) as did TBK1 knockdown with siRNA (Fig EV4A). Similar to RAW264.7 macrophages, amlexanox reduced mTORC1 signaling in primary bone marrow‐derived macrophages (BMDMs) in response to poly(I:C) and LPS (Fig 5C). In HEK293‐TLR3 cells stimulated with poly(I:C), amlexanox reduced both mTORC1 and mTORC2 signaling (Fig 5D). Lastly, LPS increased mTORC1 signaling in TBK1+/+ but not TBK1−/− MEFs in an amlexanox‐sensitive manner (Fig 5E). Taken together, these data indicate that TLR3‐ and TLR4‐mediated activation of TBK1 promotes mTORC1 and mTORC2 signaling in several cell types.
Figure 5. TLR3 and TLR4 promote mTORC1 signaling in a TBK1‐dependent manner in RAW264.7 macrophages and primary bone marrow‐derived macrophages (BMDMs).
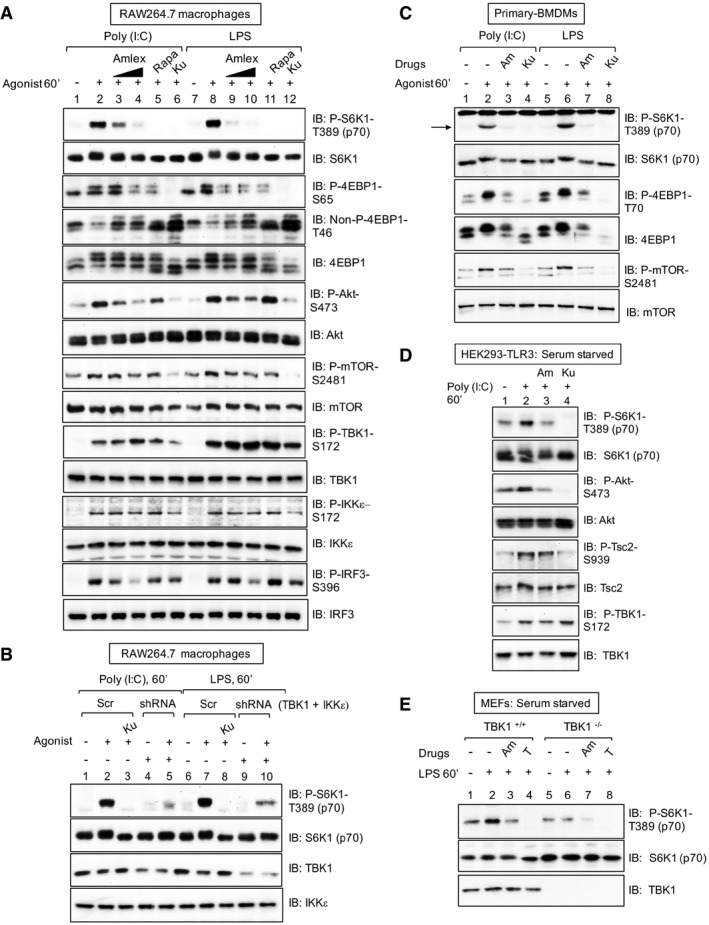
- Pharmacologic TBK1 inhibition reduces mTORC1 signaling upon activation of TLR3 and TLR4 in cultured macrophages. RAW264.7 cells cultured in full serum were pre‐treated with amlexanox [50 μM or 100 μM] (2 h), rapamycin [20 ng/ml] (30 min), or Ku‐0063794 [1 μM] (30 min) and stimulated −/+ poly(I:C) [30 μg/ml] or LPS [100 ng/ml] (60 min). Whole‐cell lysate (WCL) was immunoblotted as indicated.
- Knockdown of TBK1 with shRNA reduces TLR3‐ and TLR4‐stimulated mTORC1 signaling. RAW264.7 macrophages were co‐infected with shRNA‐containing lentiviruses targeting TBK1 and IKKε shRNA or infected with scrambled control (Scr), selected in puromycin, and treated as in (A).
- Pharmacologic TBK1 inhibition reduces mTORC1 signaling upon activation of TLR3 and TLR4 in primary macrophages. BMDMs were pre‐treated with amlexanox [100 μM] (2 h) or Ku‐0063794 [1 μM] (30 min) and stimulated −/+ poly(I:C) or LPS as in (A). The arrow indicates S6K1 phosphorylated on T389.
- Pharmacologic TBK1 inhibition reduces mTORC1 signaling upon activation of TLR3 in HEK293‐TLR3 cells. Cells were serum‐starved (20 h), pre‐treated with amlexanox or Ku‐0063794 as in (C) above, and stimulated −/+ poly(I:C) [50 μg/ml] (60 min).
- TLR4/LPS‐stimulated mTORC1 signaling requires TBK1. TBK1+/+ and TBK1−/− MEFs were serum‐starved (20 h), pre‐treated with amlexanox [50 μM] (2 h) or Torin1 [100 nM] (30 min), and stimulated −/+ LPS as in (A).
Figure EV4. TBK1 promotes mTORC1 signaling in RAW264.7 macrophages upon TLR3 and TLR4 activation (related to Figs 5 and 6).
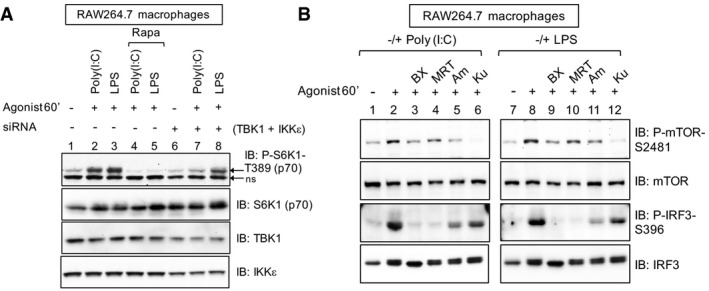
- siRNA‐mediated knockdown of TBK1/IKKε reduces TLR3/4‐stimulated mTORC1 signaling. RAW264.7 macrophages were transfected with siRNAs targeting TBK1 and IKKε. After 5 days, cells were pre‐treated with rapamycin [20 ng/ml] (30 min) and stimulated −/+ poly(I:C) [30 μg/ml] or LPS [100 ng/ml] (60 min). ns = non‐specific band.
- Pharmacologic inhibition of TBK1 reduces mTOR catalytic activity as measured by mTOR S2481 auto‐phosphorylation. RAW264.7 macrophages were pre‐treated with the TBK1 inhibitors BX‐795 [10 μM] (30 min), MRT‐67307 [10 μM] (30 min), amlexanox [100 μM] (2 h) or with the mTOR inhibitor Ku‐0063794 [1 μM] (30 min) and stimulated −/+ poly(I:C) [30 μg/ml] or LPS [100 ng/ml] (60 min). WCL was immunoblotted as indicated.
To test a requirement for site‐specific mTOR S2159 phosphorylation in TLR3‐ and TLR4‐ induced mTORC1 signaling, we again performed mTOR chemical knockout‐rescue experiments utilizing rapamycin‐resistant (RR) mTOR alleles. Phosphorylation of HA‐S6K1 in RAW264.7 macrophages expressing RR‐mTOR‐S2159A was reduced relative to those expressing RR‐mTOR in response to poly(I:C) (Fig 6A) and LPS (Fig 6B). These data demonstrate that mTOR S2159 phosphorylation promotes TLR3‐ and TLR4‐stimulated mTORC1 signaling, at least in part. We next asked whether TLR3 and TLR4 signaling increases mTORC1 catalytic activity in a TBK1‐dependent manner. Amlexanox reduced raptor‐associated mTOR S2481 auto‐phosphorylation in response to poly(I:C) and LPS (Fig 6C). Amlexanox also reduced S2481 auto‐phosphorylation on total mTOR in response to poly(I:C) and LPS in RAW264.7 macrophages (see Fig 5A) and primary BMDMs (see Fig 5C), as did BX‐795 and MRT‐67037 (Fig EV4B). These data demonstrate that TBK1 is required for mTORC1 catalytic activity in response to TLR3 and TLR4 signaling. By pharmacologically inhibiting class I PI3Kα (with BYL‐719), Akt (with MK‐2206), and TBK1 (with amlexanox), we found that TLR3 and TLR4 signaling require PI3Kα and TBK1 but not Akt for mTORC1 activation (Fig 6D).
Figure 6. TBK1 and mTOR S2159 phosphorylation are required for TLR3‐ and TLR4‐stimulated mTORC1 catalytic activity and signaling.
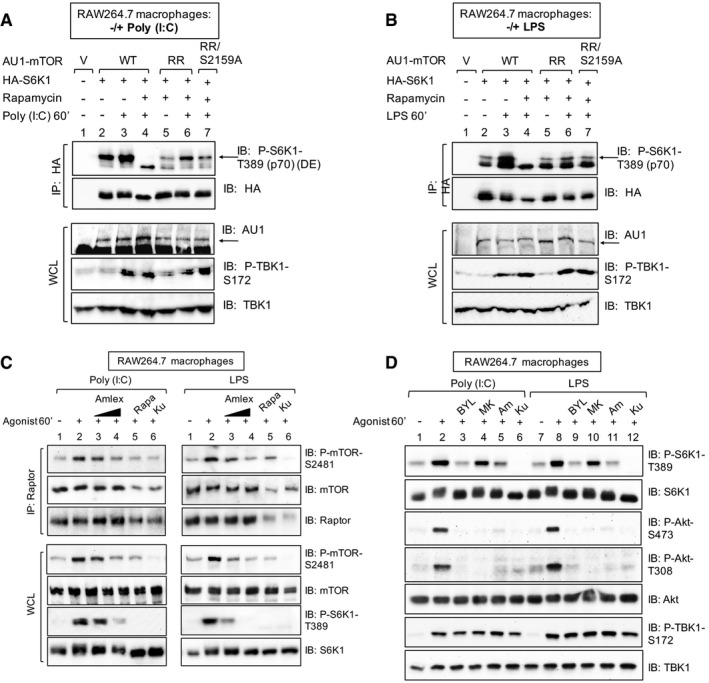
-
A, BmTOR S2159 phosphorylation is required for TLR3‐ and TLR4‐stimulated mTORC1 signaling. RAW264.7 macrophages were co‐transfected with vector control, wild‐type, or rapamycin‐resistant (RR) AU1‐mTOR alleles (RR or RR/S2159A) together with HA‐S6K1. Cells were treated with rapamycin (+) to ablate endogenous mTORC1 function and stimulated with poly(I:C) (A) or LPS (B) as in (C). HA‐S6K1 was immunoprecipitated, and IPs and WCL was immunoblotted as indicated. The arrows indicate S6K1 phosphorylated on T389.
-
CTBK1 activity is required for TLR3‐ and TLR4‐stimulated mTORC1 catalytic activity. RAW264.7 macrophages were pre‐treated with amlexanox [50 μM] or [100 μM] (2 h), rapamycin [20 ng/ml] (30 min), or Ku‐0063794 [1 μM] (30 min) and treated −/+ poly(I:C) [30 μg/ml] or LPS [100 ng/ml] (60 min). Raptor was immunoprecipitated (IP), and IPs and whole‐cell lysates (WCL) were immunoblotted (IB) as indicated.
-
DPharmacologic inhibition of PI3Kα but not Akt reduces TLR3‐ and TLR4‐stimulated mTORC1 signaling. RAW264.7 macrophages were pre‐treated with the PI3Kα class I inhibitor BYL‐719 [10 μM], the Akt inhibitor MK‐2206 [10 μM], amlexanox [100 μM], or Ku‐0063794 [1 μM] (30 min) and stimulated −/+ poly(I:C) or LPS as in (C).
The TBK1‐mTORC1 axis induces IFN‐β production by promoting IRF3 nuclear translocation through mTOR S2159 phosphorylation
TLR3‐ and TLR4‐mediated activation of TBK1 and IKKε induce the production of type I interferons to initiate innate immune responses against invading microbes (Fitzgerald et al, 2003; Hacker & Karin, 2006; Yu et al, 2012; Schneider et al, 2014). We therefore investigated a role for mTORC1 in production of IFN‐β upon poly(I:C) and LPS treatment of RAW264.7 macrophages and primary BMDMs. As expected, inhibition of TBK1/IKKε with amlexanox suppressed IFN‐β production in response to poly(I:C) (Fig 7A) and LPS (Fig EV5A) (6 h), as measured by ELISA. The mTORC1‐specific inhibitor rapamycin also suppressed IFN‐β production in response to both agonists, as did the mTOR inhibitor Ku‐0063794 (Figs 7A and EV5A). Importantly, these pharmacologic agents maintained inhibition of mTORC1 signaling at 6 h of agonist stimulation, the time point used to induce IFN‐β (Fig EV5B). To determine whether mTORC1 inhibition suppresses IFN‐β production transcriptionally or post‐transcriptionally, we employed qRT–PCR to measure IFN‐β gene expression. As expected, amlexanox reduced IFN‐β mRNA levels strongly in response to poly(I:C) (Fig 7B) and LPS (Fig EV5C) (3 h). Rapamycin and Ku‐0063794 also reduced levels of IFN‐β mRNA (Figs 7B and EV5C), although not to the same extent as amlexanox, suggesting that mTORC1 inhibition suppresses IFN‐β production by both transcriptional and post‐transcriptional mechanisms. Importantly, rapamycin suppressed IFN‐β protein production in primary BMDMs in response to poly(I:C) (Fig 7C) and LPS (Fig EV5D) (6 h), indicating that mTORC1 promotes IFN‐β production in both cultured and primary macrophages.
Figure 7. mTORC1 function and mTOR S2159 phosphorylation are required for TLR3‐stimulated IFN‐β production in cultured and primary macrophages by promoting IRF3 nuclear translocation.
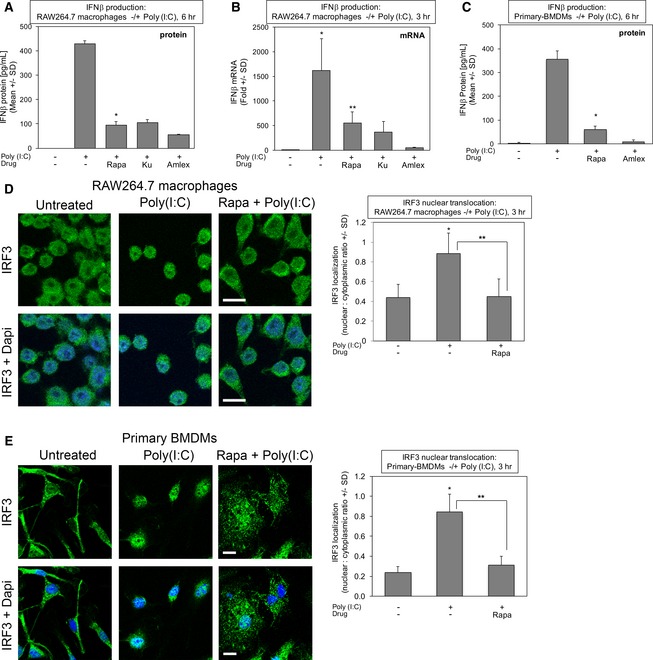
- Rapamycin suppresses TLR3‐stimulated IFN‐β protein production in cultured macrophages. RAW264.7 macrophages were pre‐treated with rapamycin [20 ng/ml] (30 min), Ku‐0063794 [1 μM] (30 min), or amlexanox [50 μM] (2 h) and stimulated −/+ poly(I:C) [30 μg/ml] for 6 h. The secretion of IFN‐β was measured by ELISA. Results represent the mean ± SD of quadruplicate samples from one experiment. *P = 0.0009 relative to +poly(I:C) by paired t‐test (two‐tailed).
- Rapamycin suppresses TLR3‐stimulated IFN‐β mRNA production in cultured macrophages. RAW264.7 macrophages were treated as above but stimulated −/+ poly(I:C) [30 μg/ml] for 3 h. IFN‐β gene expression was measured by qRT–PCR. Results represent the mean ± SD of triplicate samples from one experiment. *P = 0.03 relative to no poly(I:C) by paired t‐test (one‐tailed); **P = 0.04 relative to +poly(I:C) by paired t‐test (one‐tailed).
- Rapamycin suppresses TLR3‐stimulated IFN‐β protein production in primary macrophages. BMDMs were pre‐treated with rapamycin or amlexanox and analyzed as in (A). Results represent the mean ± SD of triplicate samples from one experiment. *P = 0.002 relative to +poly(I:C) by paired t‐test (two‐tailed).
- Rapamycin suppresses TLR3‐stimulated IRF3 nuclear translocation in RAW264.7 macrophages. Cells were pre‐treated with rapamycin and stimulated −/+ poly(I:C) for 3 h as in (A), fixed, and processed for confocal immunofluorescence microscopy using an anti‐IRF3‐Alexa 488 antibody and DAPI staining. The graph represents the mean ± SD of at least 400 cells total from three independent experiments. *P = 0.002 relative to no poly(I:C) by paired t‐test (two‐tailed); **P = 0.003 relative to +poly(I:C) by paired t‐test (two‐tailed). Scale bar = 10 μm.
- Rapamycin suppresses TLR3‐stimulated IRF3 nuclear translocation in primary BMDMs. Cells were pre‐treated with rapamycin and stimulated −/+ poly(I:C) for 3 h as in (A), fixed, and processed for confocal immunofluorescence microscopy using an anti‐IRF3‐Alexa 488 antibody and DAPI stain. The graph represents the mean ± SD of at least 380 cells total from three independent experiments. *P = 0.003 relative to no poly(I:C) by paired t‐test (two‐tailed); **P = 0.005 relative to +poly(I:C) by paired t‐test (two‐tailed). Scale bar = 10 μm. Note: the untreated control image is the same as the untreated control image in Fig EV5E.
Figure EV5. mTORC1 function is required for IFN‐β production and IRF3 nuclear translocation in RAW264.7 macrophages upon TLR4 activation (related to Fig 7).
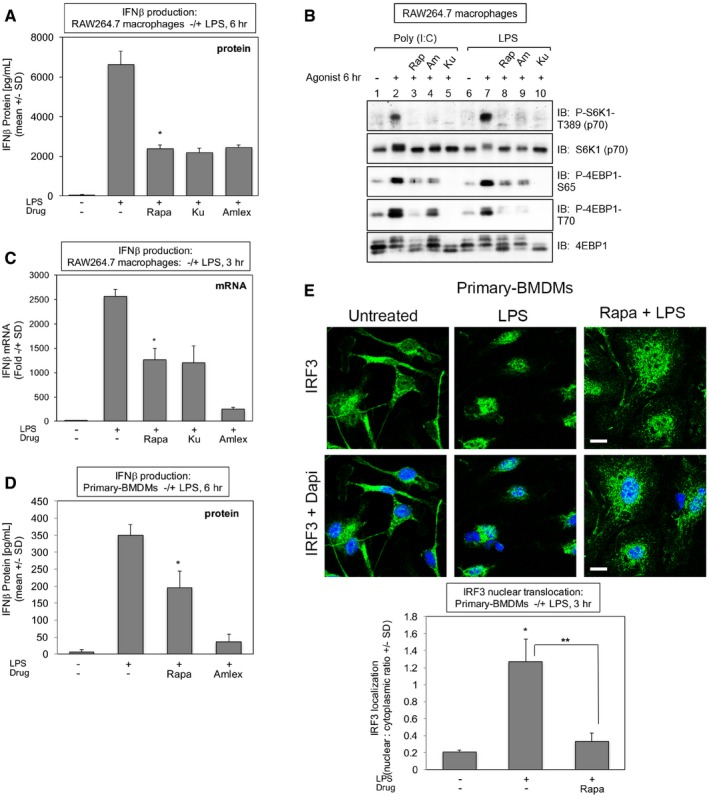
- mTORC1 inhibition with rapamycin suppresses TLR4‐induced IFN‐β protein production in RAW264.7 macrophages. Cells were pre‐treated with rapamycin [20 ng/ml] (30 min), Ku‐0063794 [1 μM] (30 min), or amlexanox [50 μM] (2 h) and stimulated −/+ LPS [100 ng/ml] (6 h). The secretion of IFN‐β into the culture media was measured by ELISA. Results represent the mean ± SD of quadruplicate samples from one experiment. *P = 0.007 relative to +LPS by paired t‐test (two‐tailed).
- Rapamycin maintains suppression of TLR3/4‐induced mTORC1 signaling at 6 h of treatment in RAW264.7 macrophages. Cells were treated as in (A). WCL was immunoblotted as indicated.
- Rapamycin suppresses TLR4‐induced IFN‐β mRNA production in RAW264.7 macrophages. Cells were treated as in (A) but LPS treatment lasted 3 h. IFN‐β gene expression was measured by qRT–PCR. Results represent the mean ± SD of triplicate samples from one experiment. *P = 0.002 relative to +LPS by paired t‐test (two‐tailed).
- Rapamycin suppresses TLR4‐stimulated IFN‐β protein production in primary macrophages. BMDMs were pre‐treated with rapamycin or amlexanox and analyzed as in (A). Results represent the mean ± SD of triplicate samples from one experiment. *P = 0.02 relative to +LPS no rapamycin by paired t‐test (two‐tailed).
- Rapamycin suppresses TLR4‐stimulated IRF3 nuclear translocation in primary macrophages. BMDMs were treated with LPS for 3 h as in (A), fixed, and processed for confocal immunofluorescence microscopy using an anti‐IRF3‐Alexa488 antibody and DAPI staining. The graph represents the mean ± SD of at least 95 cells from one experiment. *P = 0.002 relative to no LPS by paired t‐test (two‐tailed); **P = 0.005 relative to +LPS by paired t‐test (two‐tailed). Scale bar = 10 μm. Note: the control image is the same as the control image shown in Fig 7E.
We next sought to understand the mechanism by which mTORC1 promotes IFN‐β production. Upon TLR3 and TLR4 activation by innate immune agonists, TBK1 phosphorylates IRF3, the transcription factor responsible for induction of type I interferons, and promotes IRF3 dimerization, translocation from the cytosol to nucleus, and transcriptional activity (Hiscott, 2007; McWhirter et al, 2004; Mori et al, 2004; TenOever et al, 2004; Ikushima et al, 2013). By employing confocal immunofluorescence microscopy to investigate the subcellular localization of IRF3, we found that rapamycin blunted the nuclear translocation of IRF3 in response to poly(I:C) in RAW264.7 macrophages (Fig 7D) and BMDMs (Fig 7E). Rapamycin also blunted LPS‐induced IRF3 translocation in primary BMDMs (Fig EV5E). Our finding that mTORC1 promotes IRF3 nuclear translocation provides a mechanistic basis for how rapamycin reduces IFN‐β gene expression.
To investigate a direct mechanistic link between TBK1 and mTOR with regards to mTORC1 signaling and cellular innate immune function, we studied primary BMDMs isolated from genome edited mice bearing an alanine knock‐in substitution at S2159 in the mTOR gene using CRISPR/Cas9 technology (Fig EV6A and B). In response to TLR3 (Fig 8A) and TLR4 (Fig 8B) activation with poly(I:C) and LPS, respectively, mTORC1 signaling was impaired in homozygous mutant mTORA/A primary BMDMs compared to wild‐type mTOR+/+ macrophages. We next investigated a role for mTOR S2159 phosphorylation in IFN‐β production and IRF3 nuclear translocation. In primary BMDMs cultured in full serum‐containing media, we noted differences in IFN‐β production by mTOR+/+ vs. mTORA/A macrophages in some experiments but not others. To reduce this variability, we serum‐starved the macrophages gently (6 h) prior to stimulation with poly(I:C) to reduce the potential effects of growth factor action on mTORC1. mTORA/A macrophages produced less IFN‐β in response to poly(I:C) than mTOR+/+ macrophages (Fig 8C). Moreover, IRF3 nuclear translocation was reduced in mTORA/A macrophages relative to mTOR+/+ macrophages (Fig 8D). Collectively, these data support a model whereby TBK1‐mediated mTOR phosphorylation on S2159 increases mTORC1 catalytic activity and signaling upon activation of TLR3 or TLR4. mTORC1 then induces the translocation of IRF3 from the cytoplasm to the nucleus and thus cooperates with TBK1 to promote IFN‐β production (Fig 8E).
Figure EV6. Generation and genotyping mTORA/A genome‐edited mice (related to Fig 8).
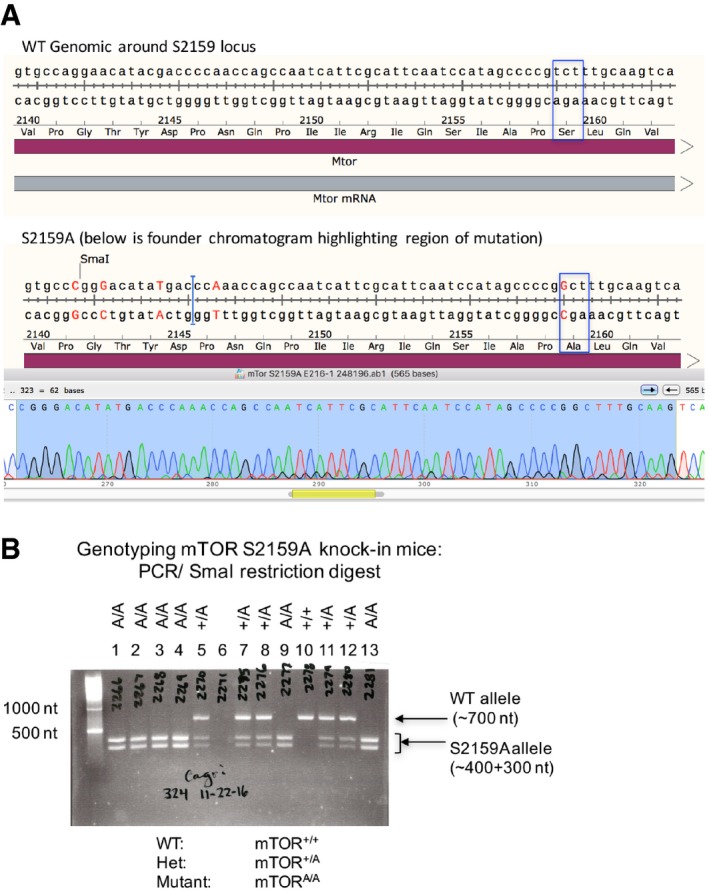
- TOPO cloning and DNA sequencing of founder mTORA/A mice. Sequence analysis confirmed the presence of an edited mTOR gene containing an Ala substitution at Ser2159. The blue box highlights the S2159A point mutation. Sequencing also confirmed the introduction of a new SmaI restriction site to facilitate genotyping and several silent mutations to prevent re‐targeting of edited genomic DNA.
- Representative DNA agarose gel depicting genotyping of wild‐type (mTOR+/+), heterozygous (mTOR+/A), and homozygous mutant S2159A knock‐in mice (mTORA/A). A ˜700 nt fragment of genomic DNA surrounding the mTOR S2159 locus was PCR amplified and digested with SmaI. Restriction products of ˜400 nt and ˜300 nt indicate the presence of the S2159A knock‐in allele.
Figure 8. mTOR S2159 phosphorylation promotes mTORC1 signaling, IRF3 nuclear translocation, and IFN‐β production upon TLR3 activation in primary macrophages.
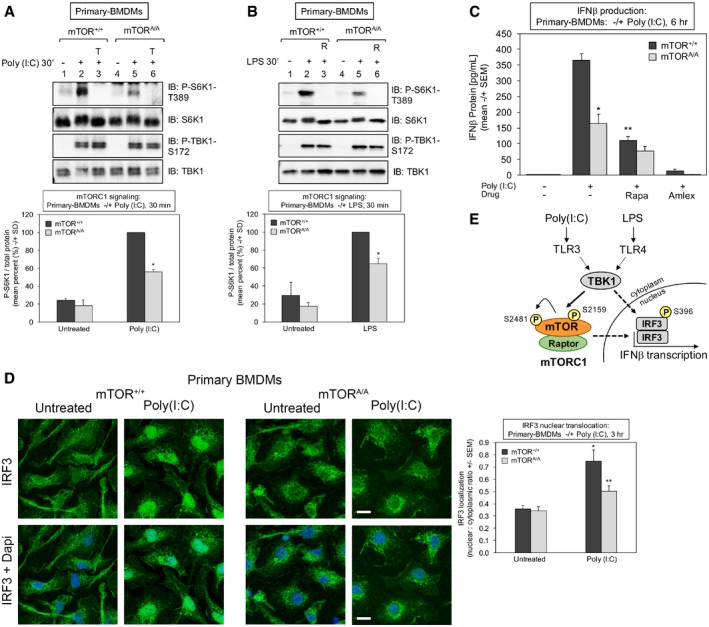
- Reduced TLR3‐induced mTORC1 signaling in mTOR S2159A knock‐in primary macrophages (mTORA/A) relative to wild type (mTOR+/+): BMDMs were pre‐treated with Torin 1 [100 nM] (30 min) and stimulated −/+ poly(I:C) [30 μg/ml] for 30 min. Whole‐cell lysate (WCL) was immunoblotted as indicated. The graph quantitates three independent experiments each with n = 1 (n = 3 total). The level of P‐S6K1‐T389 normalized to total protein in mTOR+/+ macrophages stimulated with poly(I:C) was set at 100%. The other bars represent mean ± SD as a relative percent. *The confidence interval at 95% (52.7–59.1) indicates statistical significance between mTOR+/+ vs. mTORA/A BMDMs.
- Reduced TLR4‐induced mTORC1 signaling in mTORA/A primary macrophages. BMDMs were treated as in (A) except stimulated −/+ LPS [100 ng/ml] (30 min). The graph quantitates three independent experiments each with n = 1 (n = 3 total). The level of P‐S6K1‐T389 normalized to total protein in mTOR+/+ macrophages stimulated with LPS was set at 100%. The other bars represent mean ± SD as a relative percent. *The confidence interval at 95% (58.6–71.2%) indicates statistical significance between mTOR+/+ vs. mTORA/A BMDMs.
- Reduced TLR3‐stimulated IFN‐β protein production in mTORA/A primary macrophages. BMDMs were serum‐starved for 6 h and stimulated −/+ poly(I:C) [30 μg/ml] for 6 h. The secretion of IFN‐β was measured by ELISA. Results represent the mean ± SEM from three independent experiments, n = 8 samples total. *P = 0.0002 relative to mTOR+/+ + poly(I:C) by unpaired t‐test (equal variance; two‐tailed). **P = 0.0001 relative to mTOR+/+ + poly(I:C) by paired t‐test (two‐tailed).
- Reduced TLR3‐stimulated IRF3 nuclear translocation in mTORA/A primary macrophages. BMDMs were treated −/+ poly(I:C) for 3 h as in (A), fixed, and processed for confocal immunofluorescence microscopy using an anti‐IRF3‐Alexa 488 antibody and DAPI stain. The graph represents the mean ± SEM of at least 270 cells total from four independent experiments. *P = 0.001 relative to mTOR+/+ no poly(I:C) by unpaired t‐test (equal variance; two‐tailed). **P = 0.02 relative to mTOR+/+ + poly(I:C) by unpaired t‐test (equal variance; two‐tailed). Scale bar = 10 μm.
- Model. TLR3 and TLR4 signaling increases TBK1‐mediated mTOR S2159 phosphorylation, resulting in increased mTORC1 catalytic activity, mTORC1 downstream signaling, IRF3 nuclear translocation, and IFN‐β production.
Discussion
In addition to classical roles in innate immunity and inflammation to combat infectious pathogens, TBK1 and IKKε have been linked to tumorigenesis during oncogenic stress and metabolic control during obesity (Chiang et al, 2009; Shen & Hahn, 2011; Helgason et al, 2013; Reilly et al, 2013). The role of TBK1/IKKε in diverse cellular processes suggests that these kinases likely possess multiple substrates that control cell physiology in cell context‐dependent manners. Here we demonstrate that TBK1 phosphorylates mTOR directly to increase mTORC1 catalytic activity and signaling, thus identifying mTORC1 as a new TBK1 substrate and TBK1 as a new mTORC1 activator. While challenging to prove definitively that a kinase phosphorylates a substrate directly in intact cells, our collective data support the notion that mTORC1 represents a bona fide TBK1 substrate. For example, recombinant TBK1 as well as Flag‐TBK1 immunoprecipitated from intact cells phosphorylates mTOR in vitro, Flag‐TBK1 expressed ectopically in cells increases mTOR phosphorylation, treatment of cells with EGF increases mTOR phosphorylation in TBK1+/+ but not TBK1−/− MEFs, and TBK1 interacts with mTORC1 in intact cells. Multiple approaches including genetic knockout and knock‐in, pharmacological inhibition, and RNAi demonstrate that mTORC1 signaling requires TBK1 downstream of EGF‐receptor and TLR3/4. By studying cultured cells expressing an ectopic mTOR S2159 Ala substitution mutant or primary macrophages derived from genetically modified mice bearing germline mTOR S2159A knock‐in mutation (mTORA/A), we show that phosphorylation of mTOR on a TBK1 site (S2159) promotes mTORC1 signaling in response to EGF and innate immune agonists. These data demonstrate a direct mechanistic link between TBK1 and mTORC1 and cross talk between these important signaling systems. To gain greater insight into the physiologic relevance of the TBK1‐mTORC1 axis in control of innate immune function, we measured IFN‐β production, a major cellular function controlled by TBK1. Pharmacological inhibition of mTORC1 with rapamycin in cultured macrophages (RAW264.7) and primary macrophages (BMDMs) suppressed IFN‐β production upon TLR3 or TLR4 activation through a mechanism involving reduced translocation of the transcription factor IRF3 from the cytosol into the nucleus. Analysis of primary macrophages derived from our mTORA/A knock‐in mice revealed that TBK1 promotes IRF3 translocation and IFN‐β production through site‐specific mTOR phosphorylation.
Consistent with our work, other studies reported that TBK1 interacts with mTOR (Kim et al, 2013; Hasan et al, 2017). Paradoxically, however, these studies concluded that TBK1 inhibits mTORC1. While we do not fully understand this discrepancy, the cellular contexts of these studies were quite different from ours. Kim et al (2013) studied prostate cancer cells and found that overexpression of TBK1 reduced S6K1 T389 phosphorylation. Similarly, we find that TBK1 overexpression inhibits mTORC1 signaling (see Fig EV2B). We have thus avoided TBK1 overexpression as an approach when studying mTORC1 signaling. It is important to note that raptor, a scaffolding protein and mTOR partner essential for mTORC1 signaling, also suppresses S6K1 phosphorylation when overexpressed. Hasan et al (2017) studied MEFs from mice lacking Trex, a clinical model of autoimmune/autoinflammatory disease. Knockout of the Trex exonuclease in MEFs activates the cytosolic DNA sensing cGAS‐STING‐TBK1 pathway; these MEFs also exhibit reduced mTORC1 signaling relative to Trex+/+ MEFs (Hasan et al, 2017). As both TBK1 and mTORC1 engage in negative feedback, it is possible that chronic loss of Trex re‐wires cell signaling that impacts the mTORC1 pathway negatively.
Our work demonstrates a dominant role for TBK1 in EGF‐ but not insulin‐stimulated mTORC1 signaling, revealing a stimulus‐selective role for TBK1 in mTORC1 regulation by growth factors. Interestingly, data shown but not discussed in an earlier report agrees with this finding (Ou et al, 2011). Consistently, cells reliant on an mTOR S2159A allele show impaired mTORC1 signaling in response to EGF but not insulin [see Fig 4A and Ekim et al (2011)]. How TBK1 contributes selectively to mTORC1 activation by growth factors remains an important question. EGF but not insulin may activate TBK1. Indeed, an earlier report found that cellular EGF stimulation increases the kinase activity of immunoprecipitated TBK1 toward substrate in vitro (Ou et al, 2011), suggesting that EGF‐receptor signaling increases TBK1 intrinsic catalytic activity. Consistently, we found that EGF but not insulin increases mTOR S2159 phosphorylation in intact cells (see Fig 2H). As TBK1 interacts with mutually exclusive scaffolding partners, TBK1 activation may be governed by recruitment of TBK1 to specific signaling platforms (Ma et al, 2012; Helgason et al, 2013). These platforms may contain a TBK1 activating kinase or promote local clustering of TBK1, enabling kinase domain interaction and activation loop swapping, leading to trans auto‐phosphorylation and auto‐activation (Ma et al, 2012; Helgason et al, 2013). Alternately, the EGF but not insulin pathway may drive co‐localization of TBK1 with substrate (i.e., mTOR) (Helgason et al, 2013). It is important to note that while LPS or poly(I:C) increased TBK1 S172 phosphorylation and mTORC1 signaling in MEFs and HEK293/TLR3 cells, respectively, EGF failed to increase P‐TBK1‐S172 (see Fig EV2A and B) while it increased mTORC1 signaling. What do these unexpected observations mean? While we do not know at the moment, these data may suggest that EGF‐receptor signaling does not increase TBK1 intrinsic catalytic activity, which can be monitored by phosphorylation on the TBK1 activation loop site (S172). In this case, basal TBK1 kinase activity would “prime” mTOR for EGF‐stimulated activation of mTORC1. Alternately, the data may suggest that the mechanism by which innate immune agonists vs. EGF activate TBK1 differs; in this scenario, immunoblotting with P‐TBK1‐S172 antibodies does not represent a reliable readout for TBK1 activation in response to EGF.
Downstream of EGF‐receptor, KRAS signaling activates TBK1 through Ral‐GEF, the guanine nucleotide exchange factor for the RalA and RalB GTPases (Zhu et al, 2014; Kitajima et al, 2016). Active GTP‐loaded RalB and its effector Sec5 recruit and activate TBK1 by an unclear mechanism (Chien et al, 2006). Interestingly, RalB and Sec5 were reported to promote mTORC1 signaling through an unknown mechanism (Martin et al, 2014). Our work suggests that TBK1 may represent the missing link between RalB/Sec5 and mTORC1. Our data also demonstrate that EGF and insulin increase Akt S473 phosphorylation in a TBK1‐ and mTOR‐dependent manner (see Fig 2F). While not defined mechanistically at this time, these data indicate that TBK1 contributes to mTORC2 activation in response to both EGF and insulin. It is important to note that our observed dependency of Akt S473 phosphorylation on TBK1 agrees with published work (Joung et al, 2011; Ou et al, 2011; Xie et al, 2011). Two of these studies concluded, however, that TBK1 phosphorylates Akt S473 directly independently of mTORC2 (Ou et al, 2011; Xie et al, 2011). In the cells studied here, however (i.e., MEFs; HEK293; RAW264.7), Akt S473 phosphorylation stimulated by growth factors or innate immune agonists depended strongly on mTOR. Perhaps TBK1 phosphorylates Akt directly in certain physiologic or pathological contexts.
Our results provide greater mechanistic insight into how mTORC1 modulates innate immune function, identifying mTORC1 as a direct effector of TBK1 that promotes IRF3 nuclear translocation and IFN‐β production. While it is well established that TBK1 induces IFN‐β production downstream of TLR3 and TLR4 by promoting the dimerization, nuclear translocation, and transcriptional activation of IRF3 in cooperation with the co‐activator CBP/p300 (McWhirter et al, 2004; Mori et al, 2004; TenOever et al, 2004; Hiscott, 2007), many unresolved issues remain regarding the kinases that phosphorylate IRF3 on its many sites and the functional consequences of these phosphorylation events. It has been suggested that TBK1‐mediated phosphorylation of a cluster of C‐terminal sites (S396; S398; S402; T404; S405) alleviates structural auto‐inhibition and increases transcriptional activity, enabling IRF3 phosphorylation on nearby sites (S385; S386), which induces dimerization and nuclear translocation (McWhirter et al, 2004; Mori et al, 2004; Panne et al, 2007). As mTORC1 function is required for IRF3 nuclear translocation, it is tempting to speculate that mTORC1 itself or a downstream kinase (i.e., S6K1) may phosphorylate IRF3 to promote dimerization and/or nuclear translocation, thus cooperating with TBK1 to drive IFN‐β production.
It is important to note that a limited number of reports in the literature have noted connections between the mTORC1 pathway and TLR‐mediated innate immune responses. Rapamycin suppressed IFN‐β production upon TLR3 activation in human oral keratinocytes (Zhao et al, 2010), and rapamycin administered in vivo delayed the mortality of mice injected with a lethal dose of LPS (aka endotoxin) by blunting production of several cytokines including IFN‐γ (Lee et al, 2010). In response to vesicular stomatitis virus (VSV), which activates TBK1 (TenOever et al, 2007), mice and MEFs lacking S6K1 and 2 were more susceptible to infection than wild‐type controls due to impaired production of type I interferons (Alain et al, 2010). In addition, MEFs lacking other mTORC1 substrates‐ 4EBP1 and 4EBP2, repressors of eIF4E controlled cap‐dependent translation‐ produced higher levels of type I IFNs in response to TLR3 activation (Colina et al, 2008; Erickson & Gale, 2008). Moreover, these 4EBP1/2 knockout mice exhibited resistance to VSV infection (Colina et al, 2008; Erickson & Gale, 2008). Increased translational efficiency of IRF7, which drives IFN‐β expression to high levels after an initial wave of IRF3‐mediated transcriptional induction of IRF7 (through a positive feedback loop), was found to underlie these effects in 4EBP1/2 null MEFs (Colina et al, 2008). Thus, mTORC1 promotes IFN‐β production through parallel effector pathways involving S6Ks and 4EBPs. As cap‐dependent translation represents a major cellular function controlled by mTORC1 (Ma & Blenis, 2009), it will be interesting to investigate whether mTORC1 plays a more global role in innate immunity via translational control. More recently, the cytosolic DNA sensing cGAS‐STING‐TBK1 pathway was shown to activate IRF3 in a manner that required the mTORC1 substrate S6K1 (but not its kinase activity; Wang et al, 2016). Other work reveals roles for mTORC1 in control of interferon production downstream of TLR7 and TLR9 (Cao et al, 2008; Schmitz et al, 2008; Boor et al, 2013). Beyond its role in innate immunity, TBK1 promotes tumorigenic processes and modulates metabolism. TBK1 is required for anchorage‐independent proliferation and survival of non‐small cell lung cancer (NSCLC) cells and cultured cells transformed with oncogenic KRAS (Chien et al, 2006; Barbie et al, 2009; Ou et al, 2011; Xie et al, 2011). With regard to metabolic control, tissue from obese mice experiencing chronic low‐grade inflammation (i.e., liver; adipocytes; adipose tissue macrophages) exhibits elevated expression of TBK1 and IKKε downstream of NF‐κB; in addition, TBK1/IKKε have been linked to increased glucose uptake in adipocytes, increased whole‐body energy storage during obesity, and suppression of catecholamine‐induced lipolysis (Chiang et al, 2009; Mowers et al, 2013; Reilly et al, 2013; Uhm et al, 2017). Curiously, certain phenotypes resulting from tissue‐specific knockout of raptor (mTORC1) or rictor (mTORC2) from metabolic tissues (i.e., adipose; liver; skeletal muscle) in mice align with these metabolic functions of TBK1 (Kumar et al, 2008, 2010; Polak et al, 2008; Hagiwara et al, 2012; Lee et al, 2016; Kleinert et al, 2017).
Our identification of TBK1 as a direct upstream activator of mTORC1‐ and possibly mTORC2‐ suggests new roles for mTORCs as downstream TBK1 effectors that control innate immunity and contribute to disorders such as tumorigenesis, metabolic diseases, and autoimmune diseases (Shen & Hahn, 2011; Yu et al, 2012; Reilly et al, 2013). Our mTORA/A mouse model represents an important tool for future investigation into roles for the TBK1‐mTOR axis in vivo in control of normal physiology and pathophysiology. Additional important questions remain. The upstream signaling intermediates controlling the activation state of the TBK1‐mTORC1 axis during growth factor and innate immune signaling remain incompletely defined as does the relative contribution of IKKε to mTORC regulation. Moreover, the molecular mechanisms by which TBK1 promotes mTORC2 signaling and by which mTORC1 promotes IRF3 nuclear translocation remain unresolved. Collectively our work reveals new crosstalk between two important signaling systems that coordinate cellular responses to growth factors and innate immune agonists.
Materials and Methods
Materials
All chemicals were from either Fisher Chemicals or Sigma. Protein A‐ and G‐Sepharose Fast Flow and glutathione‐Sepharose beads were from GE Healthcare; 3‐[(3‐cholamidopropyl)‐dimethylammonio]‐1‐propanesulfonate (CHAPS) was from Pierce; Immobilon‐P polyvinylidene difluoride (PVDF) membrane (0.45 μM) was from Millipore, and reagents for enhanced chemiluminescence (ECL) were from Millipore (Immobilon Western chemiluminescent horseradish peroxidase [HRP] substrate) or Advansta (WesternBright Sirius HRP substrate). Recombinant TBK1 (#PV3504) and recombinant IKKε (#PV4875) proteins were from Invitrogen/Life Technologies.
Antibodies
Myc‐9E10 (#MMS‐150P) and HA.11 (#MMS‐101P) monoclonal antibodies for immunoprecipitation and immunoblotting were from Covance, now Biolegend. Flag‐M2 monoclonal antibody was from Sigma (#F3165). AU1 monoclonal antibody was from Biolegend (#903101). The following commercial antibodies were from Cell Signaling Technology: mTOR (#2972); P‐S6K1‐T389 (rabbit monoclonal 108D2; #9234); P‐4EBP1‐T37/46 (#9459); P‐4EBP1‐T70 (#9455); P‐4EBP1‐S65 (#9451); non‐P‐4EBP1‐T46 (#4923); 4EBP1 (#9452); GST (#2625); P‐Akt‐S473 (#4060); Akt (#9272); P‐TBK1‐S172 (#5483); TBK1 (#3504); P‐IKKε‐S172 (#8766); IKKε (#3416); P‐IRF3‐S396 (#4947); IRF3 (#4302). P‐mTOR‐S2481 was from Millipore (#09‐343). Commercial polyclonal antibodies to raptor were from Millipore (#09‐217). Several polyclonal antibodies to the following proteins were generated in‐house using a Covance custom antibody service, as described in (Acosta‐Jaquez et al, 2009): Raptor (amino acids 1–17 or 885–901; human); mTOR (amino acids 221–237; rat); rictor (amino acids 6–20; human); S6K1 (amino acids 485–502 of the 70 kDa isoform; rat). mTOR P‐S2159 antibodies (amino acids 2154–2163; rat) were generated in collaboration with Millipore (#ABS79), as described (Ekim et al, 2011) (note that the mTOR P‐S2159 possesses weak phospho reactivity). Donkey anti‐rabbit‐HRP secondary antibody was from Jackson (#711‐095‐152), and sheep anti‐mouse‐HRP was from GE Healthcare (#NA931V).
Plasmids
pRK5/Myc‐mTOR and pRK5/HA‐raptor plasmids were obtained from D. Sabatini via Addgene (#1861 and 8513, respectively); pcDNA3/AU1‐mTOR (wild‐type and rapamycin‐resistant (S2035I) alleles) were from R. Abraham (Burnham Institute of Medical Research, La Jolla, CA); pRK7/HA‐S6K1 was from J. Blenis (Weill Cornell Medical College, New York, NY); pcDNA3/Flag‐TBK1, pcDNA3/Flag‐TBK1‐kinase‐dead (K38A), pcDNA3/Flag‐IKKε, and pcDNA3/Flag‐IKKε‐kinase‐dead (K38A) plasmids were from A. Saltiel (University of Michigan, Ann Arbor, MI). mTOR S2159A and S2159D mutants in the rapamycin‐resistant (S2035I) backbone of AU1‐mTOR were generated as described previously using site‐directed mutagenesis (QuikChange II XL; Stratagene; Ekim et al, 2011).
Cell culture, transfection, and drug treatments
HEK293‐TLR3 cells were obtained from K. Fitzgerald (University of Massachusetts Medical School, Worcester, MA). TBK1+/+ and TBK1−/− MEFs were from K.L. Guan (University of California San Diego, La Jolla, CA). RAW264.7 murine macrophages were from A. Saltiel (University of Michigan, Life Sciences Institute, Ann Arbor, MI). All cell lines were cultured in DMEM that contained high glucose [4.5 g/l], glutamine [584 mg/l], and sodium pyruvate [110 mg/l] (Life Technologies/Invitrogen) supplemented with 10% fetal bovine serum (FBS) (Gibco/Invitrogen; except that heat‐inactivated FBS was used for RAW264.7 murine macrophages) and incubated at 37°C in a humidified atmosphere containing 5% CO2. HEK293 cells were transfected according to manufacturer's directions using TransIT‐LT1 (Mirus). TBK1−/− MEFs and RAW264.7 macrophages were transfected using JetPRIME transfection reagent (Polyplus Transfection). Cells were lysed ~24–48 h post‐transfection. TBK1+/+ and TBK1−/− MEFs that had been serum‐starved for ~20 h (in DMEM containing 20 mM Hepes pH7.2) were stimulated with insulin [100 nM] (Invitrogen; #12585) or epidermal growth factor (EGF) [100 μg/ml] (Sigma; #E4127) for 30 min. Serum‐starved HEK293‐TLR3 cells were stimulated with poly(I:C) (Sigma; #P1530) [50 μg/ml] for 2 h RAW264.7 macrophages cultured under steady‐state conditions (DMEM/FBS) were stimulated with poly I:C [30 μg/ml] or ultrapure LPS [100 ng/ml] (InVivo Gen #tlrl‐3pelps) for times indicated in the figure legend. The Invitrogen Flp‐In system was used to generate HEK293T cell lines that express stably AU1‐mTOR, as described (Ekim et al, 2011). The following drugs were employed: Amlexanox [100 μM] (Tocris #485710), BX‐795 [10 μM] (Millipore/CalBiochem #204011), MRT‐67307 [10 μM] (Millipore/CalBiochem #506306), rapamycin [20 ng/ml] (Calbiochem #553210); Ku‐0063794 [100 nM] (Tocris #3725); BYL‐719 [10 μM] (Selleck #S1020); MK‐2206 [10 μM] (Selleck #S1078); CI‐1040 [10 μM] (Selleck #S2814).
Cell lysis, immunoprecipitation, and immunoblotting
Unless indicated otherwise, cells were washed twice with ice‐cold PBS and lysed in ice‐cold buffer A containing NP‐40 [0.5%] and Brij35 [0.1%], as described (Acosta‐Jaquez et al, 2009). To maintain the detergent sensitive mTOR‐raptor interaction, cells were lysed in ice‐cold buffer A containing CHAPS [0.3%]. Lysates were spun at 16,100 rcf for 5 min at 4°C, and the post‐nuclear supernatants were collected. Bradford assay was used to normalize protein levels for immunoprecipitation and immunoblot analysis. For immunoprecipitation, whole‐cell lysates were incubated with antibodies for 2 h at 4°C, followed by incubation with Protein G‐ or A‐Sepharose beads for 1 h. Sepharose beads were washed three times in lysis buffer and resuspended in 1× sample buffer. Samples were resolved on SDS–PAGE and transferred to PVDF membranes by using Towbin transfer buffer. Immunoblotting was performed by blocking PVDF membranes in Tris‐buffered saline (TBS) pH 7.5 with 0.1% Tween 20 (TBST) containing 3% non‐fat milk and incubating the membranes in TBST with 2% bovine serum albumin (BSA) containing primary antibodies or secondary HRP‐conjugated antibodies. Blots were developed by ECL and detected digitally with a Chemi‐Doc‐It System (UVP).
In vitro kinase assays
Generation of recombinant GST‐mTOR for in vitro kinase assays
A fragment of mTOR encoding amino acids 2,144–2,175 (wild type and a S2159A/T2164A mutant) was subcloned via PCR into vector pGEX‐20T for production of GST fusion proteins in the bacterial strain BL21(DE3)LysS. The following primers were used to PCR amplify the mTOR fragment: Primer 1, 5′‐gactggatcctatgaccccaaccagccaatc‐3′; primer 2, 5′‐gactgaattcgcccatcagggtcagcttccg‐3′. GST‐mTOR was affinity purified on glutathione‐sepharose beads via a standard protocol and dialyzed against 10 mM Tris pH 7.4, 100 mM NaCl, 1 mM EDTA, 154 mg/L DTT, and 5% glycerol.
In vitro kinome screen
The in vitro kinome screen was performed in collaboration with Invitrogen/Life Technologies. ~300 recombinant human kinases arrayed on a 384‐well plate were incubated with GST‐mTOR substrate [0.125 mg/ml] substrate in reactions containing 25 nM recombinant kinases and ATP [1 mM, 50 mM HEPES pH 7.5, 10 mM MgCl2, 1 mM EGTA, and 0.01% Brij‐35. Reactions were incubated at room temperature for 1 h. Dot blots of the kinase reactions were imaged after incubation with P‐mTOR‐S2159 primary antibody and Alexa Fluor 488 anti‐rabbit secondary antibody. Incorporation of [32P]‐ATP was performed similarly, except that reactions contained 20 nM recombinant kinase, 0.11 mg/ml GST‐mTOR (WT or AA), cold ATP [0.1 mM], and trace [γ32P]‐ATP. After 1 h incubation at room temperature, reactions were spotted on nitrocellulose, washed with phosphoric acid and water, and then imaged.
Conventional in vitro kinase (IVK) assays
In vitro kinase assays were performed by incubating recombinant GST‐mTOR ~ [200 ng] or immunoprecipitated Myc‐mTOR substrate with ATP [250 μM] and recombinant [~50 ng/reaction] or immunoprecipitated TBK1/IKKε in kinase buffer containing 50 mM Tris pH 7.5, 12 mM MgCl2, and 1 mM β‐glycerophosphate. Reactions were incubated at 30°C for 30 min and stopped by addition of sample buffer followed by incubation at 95°C for 5 min. Samples were resolved on SDS–PAGE, transferred to PVDF membrane, and immunoblotted with P‐mTOR S2159 antibodies. For drug pre‐treatments, recombinant kinases were pre‐incubated with amlexanox [100 μM], BX‐795 [10 μM], MRT‐67307 [10 μM], or in kinase buffer on ice for 30 min.
In vivo LPS treatment
Mice (C57BL6) were housed in a specific pathogen‐free facility with a 12‐h light/12‐h dark cycle and given free access to food and water. All animal use was in compliance with the Institute of Laboratory Animal Research Guide for the Care and Use of Laboratory Animals and approved by the University Committee on Use and Care of Animals at the University of Michigan. To determine the response upon LPS, mice at 6‐week‐old were administered by an intraperitoneal injection of PBS, LPS [1 mg/kg BW] for 2 h. Spleens were dissected and homogenized for western blot analysis.
Generation of rictor−/− MEFs stably expressing HA‐Rictor by lentiviral transduction
A HA‐tagged rictor cDNA was subcloned into a modified lentiviral vector, pHAGE‐Puro‐MCS (pPPM) (modified by Amy Hudson; Medical College of WI). Lentivirus particles were packaged in HEK293T cells by co‐transfecting empty pPPM vector or pPPM/HA‐Rictor together with pRC/Tat, pRC/Rev, pRC/gag‐pol and pMD/VSV‐G using Mirus TransIT‐LT1 transfection reagent. Supernatants containing viral particles were collected 48 h post‐transfection and filtered through a 0.45‐μm filter. Rictor−/− MEFs were infected with fresh supernatants with 8 μg/ml polybrene. 24 h post‐infection, cells were selected in DMEM/10% FBS supplemented with 3 μg/ml puromycin.
shRNA interference
RAW264.7 macrophages were co‐infected with lentiviral shRNAs targeting TBK1 and IKKε (Sigma; mouse TBK1 # TRCN0000323444; mouse IKBKE (IKKε) # TRCN 0000026722; non‐targeting # SHC016V) and then selected in puromycin [8 μg/ml] for 4 days.
siRNA interference
RAW264.7 macrophages were transfected with On‐TARGETplus siRNA SMARTpool reagents [100 nM] (Dharmacon‐GE Healthcare) targeting TBK1 and IKKε using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's instructions (mouse TBK1 #L‐063162; mouse IKBKE (IKKε) #L‐040798; non‐targeting #D‐001810). Cells were re‐fed 2 h prior to lysis 5 days post‐transfection.
IFN‐β ELISA
RAW264.7 macrophages or primary BMDMs (from 8‐ to 16‐week‐old mice) were treated with poly(I:C) [30 μg/ml] or LPS [100 ng/ml] (6 h). Medium was collected to quantify IFN‐β secretion using the mouse IFN‐β ELISA Kit (R&D Systems; 42400‐1) according to manufacturer's directions. Protein assays were used to normalized cell number per well to compare IFN‐β production in wild‐type vs. S2159 primary BMDMs.
qRT–PCR
Total RNA was extracted from RAW264.7 cells using an RNeasy Plus Mini Kit (Qiagen) according to manufacturer's instruction, and 250 ng total RNA was reverse transcribed into cDNA using High Capacity RNA to cDNA Kit (Applied Biosystems). Quantitative polymerase chain reaction (qPCR) was performed using TaqMan Fast Advanced Master Mix on a StepOne Plus Real‐Time PCR System (Applied Biosystems). cDNA was diluted ten times prior to qRT–PCR. Relative quantification was performed by methodology (Livak and Schmittgen, 2001). TaqMan primer/probe sets for mouse IFN‐β1 (Mm00439552‐s1) and GAPDH (Mm99999915‐g1) genes were purchased from Applied Biosystems. Amplification specificity was confirmed with agarose gel electrophoresis of the reaction products. cDNA was diluted ten times for qRT–PCR using TaqMan Fast Advanced Master Mix. StepOne Plus Real‐Time PCR System (Applied Biosystems). GAPDH was used as an endogenous control for normalization.
Isolation of primary bone marrow‐derived macrophages–monocytes
Bone marrow was harvested by flushing femora, tibiae and humeri from 8‐week‐old WT mice with 1 ml of ice‐cold PBS with a 30G needle (BD) under sterile conditions. Bone marrow cells were suspended in MEM with l‐glutamine supplemented with 10% HI‐FBS, 50 U/ml penicillin, 50 μg/ml streptomycin, and 20 ng/ml M‐CSF (R&D Systems) and were plated into 6‐well tissue culture plates with a density of 3 × 106/well. Cells were incubated at 7.5% CO2, at 37°C, and medium was replaced every 2 days until the 7th day when the experiments were done at around 50% confluency.
IRF3 translocation by confocal microscopy
Cells were seeded in 6‐well plates containing glass coverslips in appropriate culture medium. Following various treatments, cells were washed with PBS and fixed with 3.7% formaldehyde for 10 min. Upon quenching the fixative 5 min with 50 mM NH4Cl, cells were permeabilized 5 min in 0.2% TX‐100 and blocked 1 h in 0.2% fish skin gelatin. Coverslips were inverted on Alexa 488‐conjugated anti‐IRF3 antibody (1:100) (Abcam # ab204647) for 1 h, followed by mounting in Prolong Gold with DAPI (Invitrogen). Slides were visualized using a Nikon A1 confocal microscope. To quantify IRF3 nuclear to cytosolic subcellular distribution, DAPI‐stained nuclear regions of individual cells and the whole field of view containing all cells were each selected using a drawing tool in Image J. Background FITC signal was calculated from cell‐free regions and subtracted from calculations. Cytoplasmic IRF3 was determined by subtracting the total nuclear FITC intensity from the total FITC intensity of the whole area. Mean nuclear to cytoplasmic ratio ± SD was calculated from ratios of nuclear intensities divided by cytoplasmic intensities.
Generation of genetically modified mice bearing a germline mTOR knock‐in S2159A allele using CRISPR‐Cas9 genome editing technology
A 20‐nucleotide guide sequence targeting genomic mTOR upstream of S2159 was subcloned into pX330. Forward sequence: 5′‐caccgtggttggggtcgtatgttcc; reverse sequence: 5′‐aaacggaacatacgaccccaaccac. The following single‐stranded oligonucleotide served as repair template, which includes the targeted sequence (underlined), left and right homology arms, and the serine 2159 to alanine mutation (also underlined; tct to Gct): 5′‐gttcaaagctcacatccctggagctgcagtatgtgtcccccaaacttctgatgtgccgagaccttgagttggctgtg[ccCggG] acataTgacccAaaccagccaatcattcgcattcaatccatagccccgGctttgcaagtcatcacatccaagcagaggcctcgg aagctgactctga‐3′.
The repair template also included several silent mutations (capitalized) to prevent re‐targeting of edited genomic signals, plus a new SmaI restriction site ([ccCggG]) to facilitate genotyping. The gRNA targeting plasmid (pX330‐mTOR) and the repair template were co‐microinjected into single‐cell fertilized mouse oocytes and implanted into a pseudo‐pregnant mouse. Heterozygous founders were identified through SmaI restriction of genomic DNA. To confirm co‐recombination of both the SmaI site and the S2159 to Ala mutation, the genomic region was TOPO cloned and sequenced.
The following PCR primers were used for TOPO cloning: Forward‐ 5′‐ATC CAG ACT CGC TTC TGC TGG AGA‐3′; reverse‐ 5′‐CTT TCT CAT CCA ACA GAC ATG GGG GAGT‐3′.
To generate mTORA/A homozygous mice, mice heterozygous for mTOR S2159A were mated (mTOR+/mTORA x mTOR+/mTORA). These mice were generated with the assistance of the Molecular Genetics Core of the MDRC (Michigan Diabetes Research Center) and the UM Transgenic Core.
For genotyping, a~700 nt fragment of genomic DNA surrounding the mTOR S2159 locus was PCR amplified and digested with SmaI. The following PCR primers were used for genotyping: Forward: 5′‐CTTCGTGACCCTCTCCTATCT‐3′; reverse: 5′‐AAGCCTGGGACCTCTACTATC‐3′.
Image editing
Adobe Photoshop was used for image preparation using levels, brightness, and contrast equivalently over the entire image. Thin dotted lines on images indicate where irrelevant lanes were excised and flanking lanes juxtaposed from the same image at the same exposure.
Statistical analysis
Results are presented as mean ± SD (or ± SEM, where indicated) or as 95% confidence intervals. Significance of the difference between two measurements was determined by paired or unpaired Student's t‐test (two‐tailed or one‐tailed, where indicated). Statistical test used and P‐values are cited in the figure legends.
Author contributions
DCF, CB, MGM, and CNL conceived the project, designed experiments, and/or contributed intellectually. CB, DK, KH, BEU, KAS, AST, IEG, DHF, HAA‐J, TMB, GKS, K‐WC, and SMR performed experiments. DCF and CB analyzed the data and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Acknowledgments
We thank Kate Fitzgerald (UMass Worcester), David Barbie (Dana Farber Cancer Institute), Alan R. Saltiel (University of California San Diego), and David Bridges (University of Michigan School of Public Health) for sharing reagents and/or advice. We also thank Daniel Lucas‐Alcarez (University of Michigan) for assistance with isolation of bone marrow‐derived macrophages from mice. This work was supported by grants to DCF from the American Diabetes Association (ADA) (Basic Science Grant no. 1‐12‐BS‐49) and the NIH (R01‐DK‐100722). The work was also supported by the Molecular Genetics Core of the Michigan Diabetes Research Center (MDRC) (#P30‐DK020572; NIH‐NIDDK).
The EMBO Journal (2018) 37: 19–38
References
- Acosta‐Jaquez HA, Keller JA, Foster KG, Ekim B, Soliman GA, Feener EP, Ballif BA, Fingar DC (2009) Site‐specific mTOR phosphorylation promotes mTORC1‐mediated signaling and cell growth. Mol Cell Biol 29: 4308–4324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alain T, Lun X, Martineau Y, Sean P, Pulendran B, Petroulakis E, Zemp FJ, Lemay CG, Roy D, Bell JC, Thomas G, Kozma SC, Forsyth PA, Costa‐Mattioli M, Sonenberg N (2010) Vesicular stomatitis virus oncolysis is potentiated by impairing mTORC1‐dependent type I IFN production. Proc Natl Acad Sci USA 107: 1576–1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessi DR, Pearce LR, Garcia‐Martinez JM (2009) New insights into mTOR signaling: mTORC2 and beyond. Sci Signal 2: pe27 [DOI] [PubMed] [Google Scholar]
- Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E, Scholl C, Frohling S, Chan EM, Sos ML, Michel K, Mermel C, Silver SJ, Weir BA, Reiling JH, Sheng Q, Gupta PB et al (2009) Systematic RNA interference reveals that oncogenic KRAS‐driven cancers require TBK1. Nature 462: 108–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm JS, Zhao JJ, Yao J, Kim SY, Firestein R, Dunn IF, Sjostrom SK, Garraway LA, Weremowicz S, Richardson AL, Greulich H, Stewart CJ, Mulvey LA, Shen RR, Ambrogio L, Hirozane‐Kishikawa T, Hill DE, Vidal M, Meyerson M, Grenier JK et al (2007) Integrative genomic approaches identify IKBKE as a breast cancer oncogene. Cell 129: 1065–1079 [DOI] [PubMed] [Google Scholar]
- Boor PP, Metselaar HJ, Mancham S, van der Laan LJ, Kwekkeboom J (2013) Rapamycin has suppressive and stimulatory effects on human plasmacytoid dendritic cell functions. Clin Exp Immunol 174: 389–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown EJ, Beal PA, Keith CT, Chen J, Shin TB, Schreiber SL (1995) Control of p70 S6 kinase by kinase activity of FRAP in vivo . Nature 377: 441–446 [DOI] [PubMed] [Google Scholar]
- Cao W, Manicassamy S, Tang H, Kasturi SP, Pirani A, Murthy N, Pulendran B (2008) Toll‐like receptor‐mediated induction of type I interferon in plasmacytoid dendritic cells requires the rapamycin‐sensitive PI(3)K‐mTOR‐p70S6K pathway. Nat Immunol 9: 1157–1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Zheng XF, Brown EJ, Schreiber SL (1995) Identification of an 11‐kDa FKBP12‐rapamycin‐binding domain within the 289‐kDa FKBP12‐rapamycin‐associated protein and characterization of a critical serine residue. Proc Natl Acad Sci USA 92: 4947–4951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang SH, Bazuine M, Lumeng CN, Geletka LM, Mowers J, White NM, Ma JT, Zhou J, Qi N, Westcott D, Delproposto JB, Blackwell TS, Yull FE, Saltiel AR (2009) The protein kinase IKKepsilon regulates energy balance in obese mice. Cell 138: 961–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien Y, Kim S, Bumeister R, Loo YM, Kwon SW, Johnson CL, Balakireva MG, Romeo Y, Kopelovich L, Gale M Jr, Yeaman C, Camonis JH, Zhao Y, White MA (2006) RalB GTPase‐mediated activation of the IkappaB family kinase TBK1 couples innate immune signaling to tumor cell survival. Cell 127: 157–170 [DOI] [PubMed] [Google Scholar]
- Clark K, Plater L, Peggie M, Cohen P (2009) Use of the pharmacological inhibitor BX795 to study the regulation and physiological roles of TBK1 and IkappaB kinase epsilon: a distinct upstream kinase mediates Ser‐172 phosphorylation and activation. J Biol Chem 284: 14136–14146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark K, Peggie M, Plater L, Sorcek RJ, Young ER, Madwed JB, Hough J, McIver EG, Cohen P (2011) Novel cross‐talk within the IKK family controls innate immunity. Biochem J 434: 93–104 [DOI] [PubMed] [Google Scholar]
- Clement JF, Meloche S, Servant MJ (2008) The IKK‐related kinases: from innate immunity to oncogenesis. Cell Res 18: 889–899 [DOI] [PubMed] [Google Scholar]
- Colina R, Costa‐Mattioli M, Dowling RJ, Jaramillo M, Tai LH, Breitbach CJ, Martineau Y, Larsson O, Rong L, Svitkin YV, Makrigiannis AP, Bell JC, Sonenberg N (2008) Translational control of the innate immune response through IRF‐7. Nature 452: 323–328 [DOI] [PubMed] [Google Scholar]
- Cornu M, Albert V, Hall MN (2013) mTOR in aging, metabolism, and cancer. Curr Opin Genet Dev 23: 53–62 [DOI] [PubMed] [Google Scholar]
- Dibble CC, Manning BD (2013) Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat Cell Biol 15: 555–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibble CC, Cantley LC (2015) Regulation of mTORC1 by PI3K signaling. Trends Cell Biol 25: 545–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekim B, Magnuson B, Acosta‐Jaquez HA, Keller JA, Feener EP, Fingar DC (2011) mTOR kinase domain phosphorylation promotes mTORC1 signaling, cell growth, and cell cycle progression. Mol Cell Biol 31: 2787–2801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson AK, Gale M Jr (2008) Regulation of interferon production and innate antiviral immunity through translational control of IRF‐7. Cell Res 18: 433–435 [DOI] [PubMed] [Google Scholar]
- Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, Maniatis T (2003) IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol 4: 491–496 [DOI] [PubMed] [Google Scholar]
- Grivennikov SI, Greten FR, Karin M (2010) Immunity, inflammation, and cancer. Cell 140: 883–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J, Brown M, Fitzgerald KJ, Sabatini DM (2006) Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt‐FOXO and PKCalpha, but not S6K1. Dev Cell 11: 859–871 [DOI] [PubMed] [Google Scholar]
- van der Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH (2007) Insulin signaling to mTOR mediated by the AKT/PKB substrate PRAS40. Nat Cell Biol 9: 316–323 [DOI] [PubMed] [Google Scholar]
- Hacker H, Karin M (2006) Regulation and function of IKK and IKK‐related kinases. Sci STKE 2006: re13 [DOI] [PubMed] [Google Scholar]
- Hagiwara A, Cornu M, Cybulski N, Polak P, Betz C, Trapani F, Terracciano L, Heim MH, Ruegg MA, Hall MN (2012) Hepatic mTORC2 activates glycolysis and lipogenesis through Akt, glucokinase, and SREBP1c. Cell Metab 15: 725–738 [DOI] [PubMed] [Google Scholar]
- Hara K, Yonezawa K, Kozlowski MT, Sugimoto T, Andrabi K, Weng QP, Kasuga M, Nishimoto I, Avruch J (1997) Regulation of eIF‐4E BP1 phosphorylation by mTOR. J Biol Chem 272: 26457–26463 [DOI] [PubMed] [Google Scholar]
- Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, Tokunaga C, Avruch J, Yonezawa K (2002) Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 110: 177–189 [DOI] [PubMed] [Google Scholar]
- Hasan M, Gonugunta VK, Dobbs N, Ali A, Palchik G, Calvaruso MA, DeBerardinis RJ, Yan N (2017) Chronic innate immune activation of TBK1 suppresses mTORC1 activity and dysregulates cellular metabolism. Proc Natl Acad Sci USA 114: 746–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helgason E, Phung QT, Dueber EC (2013) Recent insights into the complexity of Tank‐binding kinase 1 signaling networks: the emerging role of cellular localization in the activation and substrate specificity of TBK1. FEBS Lett 587: 1230–1237 [DOI] [PubMed] [Google Scholar]
- Hiscott J (2007) Triggering the innate antiviral response through IRF‐3 activation. J Biol Chem 282: 15325–15329 [DOI] [PubMed] [Google Scholar]
- Howell JJ, Ricoult SJ, Ben‐Sahra I, Manning BD (2013) A growing role for mTOR in promoting anabolic metabolism. Biochem Soc Trans 41: 906–912 [DOI] [PubMed] [Google Scholar]
- Huang K, Fingar DC (2014) Growing knowledge of the mTOR signaling network. Semin Cell Dev Biol 36: 79–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutti JE, Shen RR, Abbott DW, Zhou AY, Sprott KM, Asara JM, Hahn WC, Cantley LC (2009) Phosphorylation of the tumor suppressor CYLD by the breast cancer oncogene IKKepsilon promotes cell transformation. Mol Cell 34: 461–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikushima H, Negishi H, Taniguchi T (2013) The IRF family transcription factors at the interface of innate and adaptive immune responses. Cold Spring Harb Symp Quant Biol 78: 105–116 [DOI] [PubMed] [Google Scholar]
- Inoki K, Li Y, Zhu T, Wu J, Guan KL (2002) TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 4: 648–657 [DOI] [PubMed] [Google Scholar]
- Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, Huang Q, Qin J, Su B (2006) SIN1/MIP1 maintains rictor‐mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 127: 125–137 [DOI] [PubMed] [Google Scholar]
- Jacinto E, Lorberg A (2008) TOR regulation of AGC kinases in yeast and mammals. Biochem J 410: 19–37 [DOI] [PubMed] [Google Scholar]
- Joung SM, Park ZY, Rani S, Takeuchi O, Akira S, Lee JY (2011) Akt contributes to activation of the TRIF‐dependent signaling pathways of TLRs by interacting with TANK‐binding kinase 1. J Immunol 186: 499–507 [DOI] [PubMed] [Google Scholar]
- Karin M (2009) NF‐kappaB as a critical link between inflammation and cancer. Cold Spring Harb Perspect Biol 1: a000141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshwani MM, von Daake S, Newton AC, Harris TK, Taylor SS (2011) Hydrophobic motif phosphorylation is not required for activation loop phosphorylation of p70 Ribosomal protein S6 kinase 1 (S6K1). J Biol Chem 286: 23552–23558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument‐Bromage H, Tempst P, Sabatini DM (2002) mTOR interacts with raptor to form a nutrient‐sensitive complex that signals to the cell growth machinery. Cell 110: 163–175 [DOI] [PubMed] [Google Scholar]
- Kim JK, Jung Y, Wang J, Joseph J, Mishra A, Hill EE, Krebsbach PH, Pienta KJ, Shiozawa Y, Taichman RS (2013) TBK1 regulates prostate cancer dormancy through mTOR inhibition. Neoplasia 15: 1064–1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitajima S, Thummalapalli R, Barbie DA (2016) Inflammation as a driver and vulnerability of KRAS mediated oncogenesis. Semin Cell Dev Biol 58: 127–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinert M, Parker BL, Fritzen AM, Knudsen JR, Jensen TE, Kjobsted R, Sylow L, Ruegg M, James DE, Richter EA (2017) Mammalian target of rapamycin complex 2 regulates muscle glucose uptake during exercise in mice. J Physiol 595: 4845–4855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Harris TE, Keller SR, Choi KM, Magnuson MA, Lawrence JC Jr (2008) Muscle‐specific deletion of rictor impairs insulin‐stimulated glucose transport and enhances Basal glycogen synthase activity. Mol Cell Biol 28: 61–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Lawrence JC Jr, Jung DY, Ko HJ, Keller SR, Kim JK, Magnuson MA, Harris TE (2010) Fat cell‐specific ablation of rictor in mice impairs insulin‐regulated fat cell and whole‐body glucose and lipid metabolism. Diabetes 59: 1397–1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM (2012) mTOR signaling in growth control and disease. Cell 149: 274–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PS, Wilhelmson AS, Hubner AP, Reynolds SB, Gallacchi DA, Chiou TT, Kwiatkowski DJ (2010) mTORC1‐S6K activation by endotoxin contributes to cytokine up‐regulation and early lethality in animals. PLoS One 5: e14399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PL, Tang Y, Li H, Guertin DA (2016) Raptor/mTORC1 loss in adipocytes causes progressive lipodystrophy and fatty liver disease. Mol Metab 5: 422–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real‐time quantitative PCR and the 2ΔΔCT . Methods 25: 402–408 [DOI] [PubMed] [Google Scholar]
- Ma XM, Blenis J (2009) Molecular mechanisms of mTOR‐mediated translational control. Nat Rev Mol Cell Biol 10: 307–318 [DOI] [PubMed] [Google Scholar]
- Ma X, Helgason E, Phung QT, Quan CL, Iyer RS, Lee MW, Bowman KK, Starovasnik MA, Dueber EC (2012) Molecular basis of Tank‐binding kinase 1 activation by transautophosphorylation. Proc Natl Acad Sci USA 109: 9378–9383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnuson B, Ekim B, Fingar DC (2012) Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks. Biochem J 441: 1–21 [DOI] [PubMed] [Google Scholar]
- Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC (2002) Identification of the tuberous sclerosis complex‐2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3‐kinase/akt pathway. Mol Cell 10: 151–162 [DOI] [PubMed] [Google Scholar]
- Marion JD, Roberts CF, Call RJ, Forbes JL, Nelson KT, Bell JE, Bell JK (2013) Mechanism of endogenous regulation of the type I interferon response by Suppressor of IKK{epsilon} (SIKE), a novel substrate of TANK binding kinase 1 (TBK1). J Biol Chem 288: 18612–18623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin TD, Chen XW, Kaplan RE, Saltiel AR, Walker CL, Reiner DJ, Der CJ (2014) Ral and Rheb GTPase activating proteins integrate mTOR and GTPase signaling in aging, autophagy, and tumor cell invasion. Mol Cell 53: 209–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWhirter SM, Fitzgerald KA, Rosains J, Rowe DC, Golenbock DT, Maniatis T (2004) IFN‐regulatory factor 3‐dependent gene expression is defective in Tbk1‐deficient mouse embryonic fibroblasts. Proc Natl Acad Sci USA 101: 233–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogensen TH (2009) Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev 22: 240–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori M, Yoneyama M, Ito T, Takahashi K, Inagaki F, Fujita T (2004) Identification of Ser‐386 of interferon regulatory factor 3 as critical target for inducible phosphorylation that determines activation. J Biol Chem 279: 9698–9702 [DOI] [PubMed] [Google Scholar]
- Mowers J, Uhm M, Reilly SM, Simon J, Leto D, Chiang SH, Chang L, Saltiel AR (2013) Inflammation produces catecholamine resistance in obesity via activation of PDE3B by the protein kinases IKKepsilon and TBK1. Elife 2: e01119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill LA, Golenbock D, Bowie AG (2013) The history of Toll‐like receptors ‐ redefining innate immunity. Nat Rev Immunol 13: 453–460 [DOI] [PubMed] [Google Scholar]
- Ou YH, Torres M, Ram R, Formstecher E, Roland C, Cheng T, Brekken R, Wurz R, Tasker A, Polverino T, Tan SL, White MA (2011) TBK1 directly engages Akt/PKB survival signaling to support oncogenic transformation. Mol Cell 41: 458–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panne D, McWhirter SM, Maniatis T, Harrison SC (2007) Interferon regulatory factor 3 is regulated by a dual phosphorylation‐dependent switch. J Biol Chem 282: 22816–22822 [DOI] [PubMed] [Google Scholar]
- Pearce LR, Komander D, Alessi DR (2010) The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol 11: 9–22 [DOI] [PubMed] [Google Scholar]
- Peters RT, Liao SM, Maniatis T (2000) IKKepsilon is part of a novel PMA‐inducible IkappaB kinase complex. Mol Cell 5: 513–522 [DOI] [PubMed] [Google Scholar]
- Polak P, Cybulski N, Feige JN, Auwerx J, Ruegg MA, Hall MN (2008) Adipose‐specific knockout of raptor results in lean mice with enhanced mitochondrial respiration. Cell Metab 8: 399–410 [DOI] [PubMed] [Google Scholar]
- Reilly SM, Chiang SH, Decker SJ, Chang L, Uhm M, Larsen MJ, Rubin JR, Mowers J, White NM, Hochberg I, Downes M, Yu RT, Liddle C, Evans RM, Oh D, Li P, Olefsky JM, Saltiel AR (2013) An inhibitor of the protein kinases TBK1 and IKK‐varepsilon improves obesity‐related metabolic dysfunctions in mice. Nat Med 19: 313–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricoult SJ, Manning BD (2013) The multifaceted role of mTORC1 in the control of lipid metabolism. EMBO Rep 14: 242–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, Carr SA, Sabatini DM (2007) PRAS40 is an insulin‐regulated inhibitor of the mTORC1 protein kinase. Mol Cell 25: 903–915 [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument‐Bromage H, Tempst P, Sabatini DM (2004) Rictor, a novel binding partner of mTOR, defines a rapamycin‐insensitive and raptor‐independent pathway that regulates the cytoskeleton. Curr Biol 14: 1296–1302 [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM (2005) Phosphorylation and regulation of Akt/PKB by the rictor‐mTOR complex. Science 307: 1098–1101 [DOI] [PubMed] [Google Scholar]
- Saxton RA, Sabatini DM (2017) mTOR signaling in growth, metabolism, and disease. Cell 169: 361–371 [DOI] [PubMed] [Google Scholar]
- Schmitz F, Heit A, Dreher S, Eisenacher K, Mages J, Haas T, Krug A, Janssen KP, Kirschning CJ, Wagner H (2008) Mammalian target of rapamycin (mTOR) orchestrates the defense program of innate immune cells. Eur J Immunol 38: 2981–2992 [DOI] [PubMed] [Google Scholar]
- Schneider WM, Chevillotte MD, Rice CM (2014) Interferon‐stimulated genes: a complex web of host defenses. Annu Rev Immunol 32: 513–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen RR, Hahn WC (2011) Emerging roles for the non‐canonical IKKs in cancer. Oncogene 30: 631–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soliman GA, Acosta‐Jaquez HA, Dunlop EA, Ekim B, Maj NE, Tee AR, Fingar DC (2010) mTOR Ser‐2481 autophosphorylation monitors mTORC‐specific catalytic activity and clarifies rapamycin mechanism of action. J Biol Chem 285: 7866–7879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stan R, McLaughlin MM, Cafferkey R, Johnson RK, Rosenberg M, Livi GP (1994) Interaction between FKBP12‐rapamycin and TOR involves a conserved serine residue. J Biol Chem 269: 32027–32030 [PubMed] [Google Scholar]
- TenOever BR, Sharma S, Zou W, Sun Q, Grandvaux N, Julkunen I, Hemmi H, Yamamoto M, Akira S, Yeh WC, Lin R, Hiscott J (2004) Activation of TBK1 and IKKepsilon kinases by vesicular stomatitis virus infection and the role of viral ribonucleoprotein in the development of interferon antiviral immunity. J Virol 78: 10636–10649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- TenOever BR, Ng SL, Chua MA, McWhirter SM, Garcia‐Sastre A, Maniatis T (2007) Multiple functions of the IKK‐related kinase IKKepsilon in interferon‐mediated antiviral immunity. Science 315: 1274–1278 [DOI] [PubMed] [Google Scholar]
- Thoreen CC, Sabatini DM (2009) Rapamycin inhibits mTORC1, but not completely. Autophagy 5: 725–726 [DOI] [PubMed] [Google Scholar]
- Tu D, Zhu Z, Zhou AY, Yun CH, Lee KE, Toms AV, Li Y, Dunn GP, Chan E, Thai T, Yang S, Ficarro SB, Marto JA, Jeon H, Hahn WC, Barbie DA, Eck MJ (2013) Structure and ubiquitination‐dependent activation of TANK‐binding kinase 1. Cell Rep 3: 747–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhm M, Bazuine M, Zhao P, Chiang SH, Xiong T, Karunanithi S, Chang L, Saltiel AR (2017) Phosphorylation of the exocyst protein Exo84 by TBK1 promotes insulin‐stimulated GLUT4 trafficking. Sci Signal 10: eaah5085 [DOI] [PubMed] [Google Scholar]
- Wang F, Alain T, Szretter KJ, Stephenson K, Pol JG, Atherton MJ, Hoang HD, Fonseca BD, Zakaria C, Chen L, Rangwala Z, Hesch A, Chan ES, Tuinman C, Suthar MS, Jiang Z, Ashkar AA, Thomas G, Kozma SC, Gale M Jr et al (2016) S6K‐STING interaction regulates cytosolic DNA‐mediated activation of the transcription factor IRF3. Nat Immunol 17: 514–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, Richter B, Korac J, Waidmann O, Choudhary C, Dotsch V, Bumann D, Dikic I (2011) Phosphorylation of the autophagy receptor optineurin restricts salmonella growth. Science 333: 228–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Zhang D, Zhao B, Lu MK, You M, Condorelli G, Wang CY, Guan KL (2011) I{kappa}B kinase varepsilon and TANK‐binding kinase 1 activate AKT by direct phosphorylation. Proc Natl Acad Sci USA 108: 6474–6479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu T, Yi YS, Yang Y, Oh J, Jeong D, Cho JY (2012) The pivotal role of TBK1 in inflammatory responses mediated by macrophages. Mediators Inflamm 2012: 979105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Benakanakere MR, Hosur KB, Galicia JC, Martin M, Kinane DF (2010) Mammalian target of rapamycin (mTOR) regulates TLR3 induced cytokines in human oral keratinocytes. Mol Immunol 48: 294–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Golay HG, Barbie DA (2014) Targeting pathways downstream of KRAS in lung adenocarcinoma. Pharmacogenomics 15: 1507–1518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoncu R, Efeyan A, Sabatini DM (2011) mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 12: 21–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2