


Abstract
Although thousands of pseudogenes have been annotated in the human genome, their transcriptional regulation, expression profiles and functional mechanisms are largely unknown. In this study, we developed dreamBase (http://rna.sysu.edu.cn/dreamBase) to facilitate the investigation of DNA modification, RNA regulation and protein binding of potential expressed pseudogenes from multidimensional high-throughput sequencing data. Based on ∼5500 ChIP-seq and DNase-seq datasets, we identified genome-wide binding profiles of various transcription-associated factors around pseudogene loci. By integrating ∼18 000 RNA-seq data, we analysed the expression profiles of pseudogenes and explored their co-expression patterns with their parent genes in 32 cancers and 31 normal tissues. By combining microRNA binding sites, we demonstrated complex post-transcriptional regulation networks involving 275 microRNAs and 1201 pseudogenes. We generated ceRNA networks to illustrate the crosstalk between pseudogenes and their parent genes through competitive binding of microRNAs. In addition, we studied transcriptome-wide interactions between RNA binding proteins (RBPs) and pseudogenes based on 458 CLIP-seq datasets. In conjunction with epitranscriptome sequencing data, we also mapped 1039 RNA modification sites onto 635 pseudogenes. This database will provide insights into the transcriptional regulation, expression, functions and mechanisms of pseudogenes as well as their roles in biological processes and diseases.
INTRODUCTION
Pseudogenes are defined as dysfunctional copies of protein-coding genes (1–4). Pseudogenes arise from genomic duplications or mRNA retro-transposition into the genome, which results in the loss of pseudogene expression due to loss of the transcription regulation region or accumulation of deleterious mutations (5–8). The human genome contains ∼15 000 pseudogenes, and this value is compatible to that of protein-coding genes (7,9,10). Importantly, accumulating evidence demonstrates that numerous pseudogenes exhibit transcriptional activity and are expressed as non-coding RNAs (ncRNAs) that play important roles in biological processes and diseases (5,9,11,12).
Transcribed pseudogenes have been validated to perform at least three functions: (i) pseudogenes are transcribed as sense or antisense products of their counterparts to regulate the expression of the functional gene (13,14); (ii) pseudogenes unite dsRNAs with their parent genes to generate endo-siRNAs and further inhibit gene expression via the RNAi pathway (15–18) or impact the stability of the protein-coding mRNA (19–23); (iii) pseudogenes function as competitive endogenous RNAs (ceRNAs) to absorb microRNAs from their cognate genes (24–28). Therefore, the transcribed products of pseudogenes critically impact their parent protein-coding genes via the ceRNA network (29). However, the number of expressed pseudogenes and their regulatory features are largely unknown and need to be investigated.
Given the high sequence similarity between pseudogenes and their parent genes, it is difficult to specifically detect the transcripts of an expressed pseudogene by traditional experiments. Currently, the development of high-throughput sequencing technologies makes the detection and analysis of expressed pseudogenes more efficient than ever before. RNA-Seq technology has been broadly used to identify and quantify expressed RNAs (30). Researchers have developed computational pipelines to inspect transcribed pseudogenes (5,9). In addition, chromatin immunoprecipitation followed by sequencing (ChIP-seq) is a technique for identifying transcription factor binding sites and histone modifications across pseudogenes (31,32). High-throughput sequencing of RNA isolated by crosslinking immunoprecipitation (CLIP-seq) is a technology developed to detect the relationships between RNA and RNA-binding proteins (RBPs) and is useful to investigate the functions and mechanisms of pseudogenes. Recently, various epitranscriptome sequencing technologies have been used to decode RNA modifications at the whole transcriptome-level (33,34), facilitating the analysis of different types of RNA modifications on pseudogenes. It is necessary to integrate these sequencing data for systematic investigations of expressed pseudogenes and their functions.
In this study, by assessing a large amount of high-throughput sequencing data, we established dreamBase to comprehensively study the regulatory features and functions of potentially expressed pseudogenes (Figure 1). Through integrating and analysing these multi-dimensional data, we identified millions of regulatory features, including both transcriptional regulation and post-transcriptional regulation of pseudogenes. All of these analysed data could be visualized in our dreamBase genome browser and freely downloaded. dreamBase will facilitate studies on the transcriptional regulation, expression, and modification of pseudogenes and their interactions with microRNAs and RBPs in human health and disease.
Figure 1.

System overview of the dreamBase core framework. All results generated by dreamBase are deposited in MySQL relational databases and displayed in the visual browser and web page.
MATERIALS AND METHODS
Integration and annotation of pseudogenes from public databases
The human pseudogenes were retrieved from public resources, including Yale pseudogene database (version 83) (10), GENCODE database (version 26) (35), the Pseudogene Decoration Resource (psiDR) (7) and ENSEMBL database (version 89) (36). To guarantee a precise description of the pseudogenes, we only retained terms that were annotated in at least two of these resources. All the genomic coordinates of these pseudogenes were converted into the human GRCh38 assembly (also known as hg38) using the UCSC LiftOver tool (37).
Comprehensive identification of transcriptional regulation evidence of pseudogenes from ChIP-seq and DNase-seq experiments
The data from ChIP-seq and DNase-seq experiments performed in multiple human cell lines were retrieved from the Gene Expression Omnibus (GEO) (38) and ENCODE project (39,40). In total, we manually collected ∼5100 ChIP-seq experimental datasets of numerous DNA-binding proteins, including transcription factors (TFs), Pol II and histone modifications, and 385 DNase-seq experimental datasets of DNase I (Table 1). In dreamBase, we annotated examined transcriptional regulatory domains (from –4 kb to +4 kb around the TSSs) of pseudogenes by using these ChIP-seq and DNase-seq data. In addition, we also integrated the regulatory data from psiDR (7) into dreamBase, which analysed ∼500 ChIP-seq experiments (Table 1) and chromatin activity data from GENCODE project.
Table 1. Number of ChIP-seq datasets integrated in dreamBase.
| Active promoter | Active polII | Histone | DnaseI | Total | |
|---|---|---|---|---|---|
| Manually collected | 2490 | 200 | 2466 | 385 | 5541 |
| Download from psiDR | - | - | - | - | 500 |
Expression analysis of pseudogenes in multiple cancers and human tissues by RNA-Seq data
We manually collected a large number of expressed pseudogenes from ∼18 000 RNA-Seq experiments that were analysed using a pipeline especially designed for pseudogenes (5,9). In addition, the expression of pseudogenes and their parent genes, was assessed in 10 359 RNA-Seq experiments of 32 types of cancers derived from the TCGA (The Cancer Genome Atlas) project (41) (Supplementary Table S1) and 7834 RNA-Seq experiments of 31 normal tissues derived from GTEx (Genotype-Tissue Expression) project (42) (Supplementary Table S2). FPKM values (43) and log2-fold change (log2FC, scaled by FPKM + 0.01) were used to quantify the pseudogene's expression level and the expression change between tumour and normal samples, respectively. To ensure the accuracy of analysis results, we only retained pseudogenes that were expressed in at least 50% of the total samples for downstream analysis.
Exploration of co-expression patterns between pseudogenes and their parent genes
We developed a tool named Co-Pseudo to explore the co-expression patterns between pseudogenes and their parent genes. The Co-Pseudo tool allowed users to study co-expression patterns with both of pseudogenes and their parent genes expressed at least in 30%, 50%, 80% or 90% of RNA-seq samples. Co-expression patterns were determined by Pearson correlation coefficient and P-value using t-tests (44). We provided the visualization of these expression data in either FPKM or log2 (FPKM + 0.01) forms with scatter plots accompanied with regression lines and boxplots accompanied with outliers.
Study of RBP binding sites on pseudogenes from published CLIP-seq data
To investigate the binding sites of RBPs on pseudogenes, we manually collected the cluster/peak data generated from ∼500 CLIP-seq experiments, including HITS-CLIP, PAR-CLIP, iCLIP and CLASH. The clusters/peaks generated by these CLIP-seq data were firstly obtained from the public database (38,45) and then intersected with the coordinates of pseudogenes to investigate the potential binding regions on pseudogenes.
Establishment of the microRNA-mediated post-transcriptional regulation network of pseudogenes and other genes
Based on CLIP-seq data of AGO proteins and the binding sites of microRNAs, we inferred thousands of regulatory relationships between microRNAs and pseudogenes. The binding sites of microRNAs on pseudogenes, long non-coding RNAs (lncRNAs) and 3′UTR regions of protein-coding genes were predicted by miRanda (46) using the ‘-strict’ parameter. The binding sites of microRNAs that shared over 95% regions with data of AGO CLIP-seq data were retained for downstream analysis. To construct the ceRNA network, the AGO-binding evidence for pseudogenes and parent genes or lncRNAs should be observed in the same CLIP-seq experiments simultaneously. A hypergeometric test (47) was performed to evaluate the significance of each pseudogene-gene/lncRNA pair. The test calculated the P-value using the following formula:
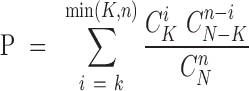 |
where N represented the total number of miRNAs used to predict targets, K represented the number of miRNAs that interacted with the pseudogenes, n represented the number of miRNAs that interacted with the parent genes/lncRNAs, and k represented the common miRNA number between these two genes. In addition, Pearson correlation analysis was performed between each pair of pseudogene-gene/lncRNA, microRNA-pseudogene and microRNA-gene/lncRNA. Cytoscape.js (48), a fully featured graph library that is written in javascript, was used to visualize the complex ceRNA networks.
Mapping of RNA modification sites on pseudogenes using high-throughput modification sequencing data
We curated RNA modification data derived from ∼400 epitranscriptome sequencing experiments of 18 studies from public resources (49), including m6A-Seq, pseudo-seq, CeU-Seq data, Aza-IP data and RiboMeth-Seq data. After calling the peak distribution for the entire genome, bedtools (50) was used to characterize RNA modification sites on pseudogenes by mapping these data onto the respective RNA molecules.
Construction of dreamBase genome browser
We used Jbrowse (51) to construct the dreamBase genome browser to display all of the binding sites generated from these high-throughput sequencing data. We integrated all of our curated peak data, including the binding sites of transcription factors, pol II, DNase I, histone modification, microRNAs, RNA binding proteins, RNA modifications and the sequences of pseudogenes and displayed this information in the dreamBase genome browser.
DATABASE CONTENT AND WEB INTERFACE
The genome-wide landscape of transcriptional regulatory elements around pseudogenes
dreamBase provides comprehensive annotation and identification of the relationships between the transcriptional regulatory elements and pseudogenes based on a large amount of ChIP-seq and DNase-seq experiments. These experiments reflect the binding sites of transcription-related factors and other chromatin-associated proteins (including transcription factors, pol II, DNase I and histone modifications) distribution around the TSSs of pseudogenes.
The pseudogene of ATP binding cassette subfamily A member 17 (ABCA17P) is a ubiquitously expressed pseudogene in various human tissues (52). In dreamBase, our results revealed strong transcriptional evidence for ABCA17P. Around the TSS of ABCA17P, we observed active binding signals of pol II, TFs, histone modifications and DNase I hypersensitivity sites, which were supported by 3, 81, 298 and 39 experiments, respectively (Supplementary Figure S1A). The information of sample details and binding site locations could be inspected by clicking the corresponding number of supporting experiments (Supplementary Figure S1B&C).
Users can use comparison operators to search multiple columns simultaneously with flexible parameters. For example, by setting all the transcriptional-related columns with ‘> = 10′ parameter, dreamBase selects ∼2000 pseudogenes, which contained transcriptional signals of four types of transcription-associated factors, and each signal was supported by at least 10 high-throughput sequencing experiments (Supplementary Figure S2).
Expression profiles of pseudogenes across multiple cancer types and human tissues
Increasing evidence demonstrated that expressed pseudogenes have tissue- and cancer-specific characteristics and play important roles in cancers and biological processes. On our website, we provide 3 sub-modules of the ‘Expression’ module, including ‘Joint-analysis’, ‘Pan-Cancer’ and ‘Normal-Tissues’ (Materials and methods), to explore the expression profiles of pseudogenes from RNA-Seq data in different cancers and human tissues (Supplementary Figure S3).
In the Pan-cancer page, log2FC of pseudogenes between tumour and normal samples in all of the 32 types of cancers were visualized through heatmap tables. Red colour represents the expression of pseudogenes that are upregulated in tumour samples, whereas blue colour represents downregulated expression. A recent study reported that double homeobox A pseudogene 8 (DUXAP8) was upregulated in non-small-cell lung cancer cell tissues, and this gene had the potential to be a new candidate prognostic marker for NSCLC patients (53). Using the ‘Pan-Cancer’ sub-module, which was developed based on the TCGA Pan-Cancer project, we clearly observed that DUXAP8 was upregulated approximately 3.3-fold in lung squamous cell carcinoma as demonstrated by expression heatmap tables. In addition, DUXAP8 was up regulated not only in lung cancer but also in most other cancer types, which suggesting that DUXAP8 may serve as an oncogene in many cancers (Figure 2).
Figure 2.

The boxplot expression patterns of DUXAP8 in 32 types of cancers.
Transcriptome-wide co-expression analysis and ceRNA network construction between pseudogenes and their parent genes
Transcribed pseudogenes can regulate their parent genes via co-expression. Moreover, increasing evidence showed that pseudogenes and their parent genes form regulatory pairs and function by influencing each other. Therefore, the co-expression networks of pseudogenes and their parent genes could be used to predict functions of pseudogenes based on the annotation of their co-expressed parental protein-coding genes. We developed a web-based tool named Co-Pseudo to explore the co-expression patterns between pseudogenes and their parent genes in multiple human cancers and normal tissues. A previous study demonstrated that ATP binding cassette subfamily C member 6 (ABCC6) and its transcribed pseudogene ABCC6P1 were co-expressed across a variety of human tissues (54). Using our Co-Pseudo tool to inspect this event in 31 normal tissues derived from the GTEx project, significantly positive co-expression relationships between ABCC6P1 and ABCC6 were clearly illustrated across numerous tissues (Supplementary Figure S4).
One of the important roles of an expressed pseudogene is distracting microRNAs from their parent genes and establishing ceRNA networks (27,29,55). In dreamBase, we constructed a tool named CeRNA-Pseudo to visualize the crosstalk between pseudogenes and their parent genes based on competitive microRNA binding evidence. A recent study reported a potential ceRNA pair composed of a putative tumour suppressor a-catenin (CTNNA1) and its pseudogene CTNNA1P1 (9). In our study, we inspected the relationship between CTNNA1 and CTNNA1P1 using CeRNA-Pseudo. Our results presented that these two genes shared seven common microRNAs (Supplementary Figure S5). Detailed information, including average expression value, the correlation coefficient and P-value for each set of pseudogenes, parent genes and microRNAs, were also calculated in every cancer (Supplementary Figure S6).
Interactions between RBPs and pseudogenes
Accumulated evidence has demonstrated that various RBPs play important roles in regulating ncRNAs at the post-transcription level (56,57). However, research on the relationships between these RBPs and the transcribed pseudogenes is lacking. We identified 112 RBPs that had thousands of binding sites on RNA molecules of pseudogenes by analyzing peak data from 458 CLIP-seq experiments. On average, each pseudogene contained 2.06 RBP binding sites which were supported by 1.15 experiments. Some RBPs were significantly enriched in pseudogenes. For instance, fused in sarcoma (FUS) protein has interaction sites with 5106 distinct pseudogenes (Supplementary Table S3). Among these sites, RP11-958N24.1 contains 94 distinctive binding sites that were supported by four CLIP-seq experiments. Detailed information regarding RBPs and the corresponding number of supporting CLIP-seq experiments were also recorded (Supplementary Figure S7).
The distribution of different types of RNA modifications on pseudogenes
In eukaryotes, >100 different types of RNA modifications have been identified on various RNA molecules. However, the characteristics and distribution of these modifications on pseudogenes remain largely unknown. In our study, we provided transcriptome-wide RNA modification maps of pseudogenes based on RNA modification sites identified from a large amount of high-throughput epitranscriptome sequencing experiments. Our results demonstrated 1423 RNA modification sites distributed on 1237 pseudogenes, and most of these site (∼88%) involved N6-Methyladenosine (m6A) modification. In addition, pseudouridine (Ψ) modification and 2′-O-methylation (2′-O-Me) modification were also identified on pseudogene RNA molecules. Our analysis results will further the study of the post-transcriptional modification mechanisms of expressed pseudogenes.
DISCUSSION AND CONCLUSIONS
Compared with other pseudogene resources (7,10), which mainly focus on the annotation of pseudogenes, dreamBase has some distinct and important promotions (Figure 1). dreamBase advances this field of study based on the following features:
We annotated genome-wide transcribed signatures on the transcriptional regulatory domains of pseudogenes using a large amount of ChIP-seq data and DNase-seq data. To summarize, we annotated active promoter and enhancer regions of pseudogenes based on millions of binding sites of 475 different TFs and 48 histone modifications. In addition, we employed the binding sites of Pol II to characterize the transcriptional signals of pseudogenes.
We collected and analysed expression data from ∼18 000 RNA-seq experiments that were processed by a bioinformatics pipeline especially designed for pseudogenes (5,9) and investigated the expression level of pseudogenes across cancer tissues and cell lines. We also performed pan-cancer and co-expression analysis on expressed pseudogenes ground on expression data from ∼10,000 RNA-seq experiments of 32 tumor tissues and 31 normal tissues.
We combined predicted binding sites of microRNAs with evidence supported by AGO CLIP-seq data to study the relevance of microRNAs against RNA molecules of pseudogenes. In addition, we combined these interaction and expression data of pseudogenes, and therefore constructed ceRNA networks that consisted of pseudogenes and other RNAs. These features will make dreamBase a valuable resource for understanding the complex networks and mechanisms mediated by pseudogenes.
We integrated the binding sites of RBPs derived from 458 CLIP-seq datasets and systematically study the binding patterns of RBPs on RNA products of pseudogenes.
We provided transcriptome-wide profiling of RNA modifications on expressed pseudogenes based on epitranscriptome sequencing technologies.
In conclusion, dreamBase was assembled with a large amount of data derived from ChIP-seq, DNase-seq, RNA-Seq and CLIP-seq experiments and provided regulatory evidence of the transcription, RNA regulation and protein binding features of potentially expressed pseudogenes. We also identified thousands of post-transcriptional regulatory relationships between pseudogenes and their parent genes, lncRNAs, microRNAs, and RBPs. dreamBase will provide researchers with a comprehensive and powerful platform to decode the transcription, expression, regulation, and modification of pseudogenes in multiple cancers and normal tissues.
FURTHER DIRECTIONS
The amount of high-throughput sequencing data is growing rapidly and applied to a broader set of tissues, cell lines, and conditions. Moreover, new technologies, such as single-cell sequencing, single-molecule sequencing, and direct RNA sequencing become increasingly important in identifying variations in sequences and sub-cell types and can be applied to more accurately demonstrate the various characteristics of expressed pseudogenes and other types of RNAs. We will continuously maintain and update the database with the integration of more aspects of the data to improve our understanding of expressed pseudogenes and other types of genes in human health and diseases.
Supplementary Material
Footnotes
Present address: Liang-Hu Qu, Biotechnology Research Center, Sun Yat-sen University, Guangzhou 510275, PR China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR online.
FUNDING
National Key R&D Program of China [2017YFA0504400]; National Natural Science Foundation of China [31771459, 31770879, 31401975, 31370791, 91440110, 30900820, 31230042, 31471223]; Guangdong Province [S2012010010510, S2013010012457]; Science and Technology New Star in ZhuJiang Guangzhou city [2012J2200025]; Fundamental Research Funds for the Central Universities [2011330003161070, 14lgjc18, 16lgjc73]; China Postdoctoral Science Foundation [200902348]; Guangdong Province Key Laboratory of Computational Science and the Guangdong Province Computational Science Innovative Research Team (in part). Funding for open access charge: National Key R&D Program of China [2017YFA0504400]; National Natural Science Foundation of China [31771459, 31770879, 31401975, 31370791, 91440110, 30900820, 31230042, 31471223]; Guangdong Province [S2012010010510, S2013010012457]; Science and Technology New Star in ZhuJiang Guangzhou city [2012J2200025]; Fundamental Research Funds for the Central Universities [2011330003161070, 14lgjc18, 16lgjc73]; China Postdoctoral Science Foundation [200902348]; Guangdong Province Key Laboratory of Computational Science and the Guangdong Province Computational Science Innovative Research Team (in part).
Conflict of interest statement. None declared.
REFERENCES
- 1. Balakirev E.S., Ayala F.J.. Pseudogenes: are they “junk” or functional DNA?. Annu. Rev. Genet. 2003; 37:123–151. [DOI] [PubMed] [Google Scholar]
- 2. Echols N., Harrison P., Balasubramanian S., Luscombe N.M., Bertone P., Zhang Z., Gerstein M.. Comprehensive analysis of amino acid and nucleotide composition in eukaryotic genomes, comparing genes and pseudogenes. Nucleic Acids Res. 2002; 30:2515–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Harrison P.M., Echols N., Gerstein M.B.. Digging for dead genes: an analysis of the characteristics of the pseudogene population in the Caenorhabditis elegans genome. Nucleic Acids Res. 2001; 29:818–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mighell A.J., Smith N.R., Robinson P.A., Markham A.F.. Vertebrate pseudogenes. Febs Lett. 2000; 468:109–114. [DOI] [PubMed] [Google Scholar]
- 5. Kalyana-Sundaram S., Kumar-Sinha C., Shankar S., Robinson D.R., Wu Y.M., Cao X., Asangani I.A., Kothari V., Prensner J.R., Lonigro R.J. et al. Expressed pseudogenes in the transcriptional landscape of human cancers. Cell. 2012; 149:1622–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sasidharan R., Gerstein M.. Genomics: protein fossils live on as RNA. Nature. 2008; 453:729–731. [DOI] [PubMed] [Google Scholar]
- 7. Pei B., Sisu C., Frankish A., Howald C., Habegger L., Mu X.J., Harte R., Balasubramanian S., Tanzer A., Diekhans M. et al. The GENCODE pseudogene resource. Genome Biol. 2012; 13:R51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Milligan M.J., Harvey E., Yu A., Morgan A.L., Smith D.L., Zhang E., Berengut J., Sivananthan J., Subramaniam R., Skoric A. et al. Global intersection of long non-coding RNAs with processed and unprocessed seudogenes in the human genome. Front. Genet. 2016; 7:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Han L., Yuan Y., Zheng S., Yang Y., Li J., Edgerton M.E., Diao L., Xu Y., Verhaak R.G., Liang H.. The Pan-Cancer analysis of pseudogene expression reveals biologically and clinically relevant tumour subtypes. Nat. Commun. 2014; 5:3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Karro J.E., Yan Y., Zheng D., Zhang Z., Carriero N., Cayting P., Harrrison P., Gerstein M.. Pseudogene.org: a comprehensive database and comparison platform for pseudogene annotation. Nucleic Acids Res. 2007; 35:D55–D60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wen Y.Z., Zheng L.L., Qu L.H., Ayala F.J., Lun Z.R.. Pseudogenes are not pseudo any more. RNA Biol. 2012; 9:27–32. [DOI] [PubMed] [Google Scholar]
- 12. Poliseno L., Marranci A., Pandolfi P.P.. Pseudogenes in human cancer. Front. Medi. 2015; 2:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhou B.S., Beidler D.R., Cheng Y.C.. Identification of antisense RNA transcripts from a human DNA topoisomerase I pseudogene. Cancer Res. 1992; 52:4280–4285. [PubMed] [Google Scholar]
- 14. Korneev S.A., Park J.H., O'Shea M.. Neuronal expression of neural nitric oxide synthase (nNOS) protein is suppressed by an antisense RNA transcribed from an NOS pseudogene. J. Neurosci. 1999; 19:7711–7720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Watanabe T., Totoki Y., Toyoda A., Kaneda M., Kuramochi-Miyagawa S., Obata Y., Chiba H., Kohara Y., Kono T., Nakano T. et al. Endogenous siRNAs from naturally formed dsRNAs regulate transcripts in mouse oocytes. Nature. 2008; 453:539–543. [DOI] [PubMed] [Google Scholar]
- 16. Tam O.H., Aravin A.A., Stein P., Girard A., Murchison E.P., Cheloufi S., Hodges E., Anger M., Sachidanandam R., Schultz R.M. et al. Pseudogene-derived small interfering RNAs regulate gene expression in mouse oocytes. Nature. 2008; 453:534–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wen Y.Z., Zheng L.L., Liao J.Y., Wang M.H., Wei Y., Guo X.M., Qu L.H., Ayala F.J., Lun Z.R.. Pseudogene-derived small interference RNAs regulate gene expression in African Trypanosoma brucei. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:8345–8350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gong C., Tang Y., Maquat L.E.. mRNA-mRNA duplexes that autoelicit Staufen1-mediated mRNA decay. Nat. Struct. Mol. Biol. 2013; 20:1214–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hirotsune S., Yoshida N., Chen A., Garrett L., Sugiyama F., Takahashi S., Yagami K., Wynshaw-Boris A., Yoshiki A.. An expressed pseudogene regulates the messenger-RNA stability of its homologous coding gene. Nature. 2003; 423:91–96. [DOI] [PubMed] [Google Scholar]
- 20. Lee J.T. Molecular biology: complicity of gene and pseudogene. Nature. 2003; 423:26–28. [DOI] [PubMed] [Google Scholar]
- 21. Podlaha O., Zhang J.. Nonneutral evolution of the transcribed pseudogene Makorin1-p1 in mice. Mol. Biol. Evol. 2004; 21:2202–2209. [DOI] [PubMed] [Google Scholar]
- 22. Yano Y., Saito R., Yoshida N., Yoshiki A., Wynshaw-Boris A., Tomita M., Hirotsune S.. A new role for expressed pseudogenes as ncRNA: regulation of mRNA stability of its homologous coding gene. J. Mol. Med. (Berlin, Germany). 2004; 82:414–422. [DOI] [PubMed] [Google Scholar]
- 23. Han Y.J., Ma S.F., Yourek G., Park Y.D., Garcia J.G.. A transcribed pseudogene of MYLK promotes cell proliferation. FASEB J. 2011; 25:2305–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gray T.A., Wilson A., Fortin P.J., Nicholls R.D.. The putatively functional Mkrn1-p1 pseudogene is neither expressed nor imprinted, nor does it regulate its source gene in trans. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:12039–12044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brunetti A., Manfioletti G., Chiefari E., Goldfine I.D., Foti D.. Transcriptional regulation of human insulin receptor gene by the high-mobility group protein HMGI(Y). FASEB J. 2001; 15:492–500. [DOI] [PubMed] [Google Scholar]
- 26. Chiefari E., Iiritano S., Paonessa F., Le Pera I., Arcidiacono B., Filocamo M., Foti D., Liebhaber S.A., Brunetti A.. Pseudogene-mediated posttranscriptional silencing of HMGA1 can result in insulin resistance and type 2 diabetes. Nat. Commun. 2010; 1:40. [DOI] [PubMed] [Google Scholar]
- 27. Poliseno L., Salmena L., Zhang J., Carver B., Haveman W.J., Pandolfi P.P.. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature. 2010; 465:1033–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Salmena L., Carracedo A., Pandolfi P.P.. Tenets of PTEN tumor suppression. Cell. 2008; 133:403–414. [DOI] [PubMed] [Google Scholar]
- 29. An Y., Furber K.L., Ji S.. Pseudogenes regulate parental gene expression via ceRNA network. J. Cell. Mol. Med. 2017; 21:185–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zheng L.L., Li J.H., Wu J., Sun W.J., Liu S., Wang Z.L., Zhou H., Yang J.H., Qu L.H.. deepBase v2.0: identification, expression, evolution and function of small RNAs, LncRNAs and circular RNAs from deep-sequencing data. Nucleic Acids Res. 2016; 44:D196–D202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Park P.J. ChIP-seq: advantages and challenges of a maturing technology. Nat. Rev. Genet. 2009; 10:669–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhou K.R., Liu S., Sun W.J., Zheng L.L., Zhou H., Yang J.H., Qu L.H.. ChIPBase v2.0: decoding transcriptional regulatory networks of non-coding RNAs and protein-coding genes from ChIP-seq data. Nucleic Acids Res. 2017; 45:D43–D50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Helm M., Motorin Y.. Detecting RNA modifications in the epitranscriptome: predict and validate. Nat. Rev. Genet. 2017; 18:275–291. [DOI] [PubMed] [Google Scholar]
- 34. Li X., Xiong X., Yi C.. Epitranscriptome sequencing technologies: decoding RNA modifications. Nat. Methods. 2016; 14:23–31. [DOI] [PubMed] [Google Scholar]
- 35. Harrow J., Frankish A., Gonzalez J.M., Tapanari E., Diekhans M., Kokocinski F., Aken B.L., Barrell D., Zadissa A., Searle S. et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012; 22:1760–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Aken B.L., Ayling S., Barrell D., Clarke L., Curwen V., Fairley S., Fernandez Banet J., Billis K., García Girón C., Hourlier T. et al. The Ensembl gene annotation system. Database. 2016; 2016:baw093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rosenbloom K.R., Armstrong J., Barber G.P., Casper J., Clawson H., Diekhans M., Dreszer T.R., Fujita P.A., Guruvadoo L., Haeussler M. et al. The UCSC Genome Browser database: 2015 update. Nucleic Acids Res. 2015; 43:D670–D681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Barrett T., Wilhite S.E., Ledoux P., Evangelista C., Kim I.F., Tomashevsky M., Marshall K.A., Phillippy K.H., Sherman P.M., Holko M. et al. NCBI GEO: archive for functional genomics data sets—update. Nucleic Acids Res. 2013; 41:D991–D995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. ENCODE Project Consortium A User's Guide to the Encyclopedia of DNA Elements (ENCODE). PLoS Biol. 2011; 9:e1001046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cancer Genome Atlas Research, N. Weinstein J.N., Collisson E.A., Mills G.B., Shaw K.R., Ozenberger B.A., Ellrott K., Shmulevich I., Sander C., Stuart J.M.. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013; 45:1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. GTEx Consortium The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013; 45:580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Trapnell C., Williams B.A., Pertea G., Mortazavi A., Kwan G., van Baren M.J., Salzberg S.L., Wold B.J., Pachter L.. Transcript assembly and abundance estimation from RNA-Seq reveals thousands of new transcripts and switching among isoforms. Nat. Biotechnol. 2010; 28:511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kim T.K. T test as a parametric statistic. Korean J. Anesthesiol. 2015; 68:540–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Li J.-H., Liu S., Zhou H., Qu L.-H., Yang J.-H.. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein–RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014; 42:D92–D97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. John B., Enright A.J., Aravin A., Tuschl T., Sander C., Marks D.S.. Human microRNA targets. PLOS Biol. 2004; 2:e363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sumazin P., Yang X., Chiu H.S., Chung W.J., Iyer A., Llobet-Navas D., Rajbhandari P., Bansal M., Guarnieri P., Silva J. et al. An extensive microRNA-mediated network of RNA-RNA interactions regulates established oncogenic pathways in glioblastoma. Cell. 2011; 147:370–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Franz M., Lopes C.T., Huck G., Dong Y., Sumer O., Bader G.D.. Cytoscape.js: a graph theory library for visualisation and analysis. Bioinformatics (Oxford, England). 2016; 32:309–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sun W.J., Li J.H., Liu S., Wu J., Zhou H., Qu L.H., Yang J.H.. RMBase: a resource for decoding the landscape of RNA modifications from high-throughput sequencing data. Nucleic Acids Res. 2016; 44:D259–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Quinlan A.R. BEDTools: the Swiss-Army Tool for genome feature analysis. Curr. Protoc. Bioinformatics. 2014; 47, doi:10.1002/0471250953.bi1112s47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Skinner M.E., Uzilov A.V., Stein L.D., Mungall C.J., Holmes I.H.. JBrowse: a next-generation genome browser. Genome Res. 2009; 19:1630–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Piehler A.P., Wenzel J.J., Olstad O.K., Haug K.B., Kierulf P., Kaminski W.E.. The human ortholog of the rodent testis-specific ABC transporter Abca17 is a ubiquitously expressed pseudogene (ABCA17P) and shares a common 5΄ end with ABCA3. BMC Mol. Biol. 2006; 7:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sun M., Nie F.Q., Zang C., Wang Y., Hou J., Wei C., Li W., He X., Lu K.H.. The pseudogene DUXAP8 promotes non-small-cell lung cancer cell proliferation and invasion by epigenetically silencing EGR1 and RHOB. Mol. Ther. 2017; 25:739–751. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 54. Piehler A.P., Hellum M., Wenzel J.J., Kaminski E., Haug K.B., Kierulf P., Kaminski W.E.. The human ABC transporter pseudogene family: Evidence for transcription and gene-pseudogene interference. BMC Genomics. 2008; 9:165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Salmena L., Poliseno L., Tay Y., Kats L., Pandolfi P.P.. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language?. Cell. 2011; 146:353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gerstberger S., Hafner M., Tuschl T.. A census of human RNA-binding proteins. Nat. Rev. Genet. 2014; 15:829–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Glisovic T., Bachorik J.L., Yong J., Dreyfuss G.. RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett. 2008; 582:1977–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.