


Abstract
De novo mutations (DNMs) have been shown to be a major cause of severe early-onset genetic disorders such as autism spectrum disorder and intellectual disability. Over one million DNMs have been identified in developmental disorders by next generation sequencing, but linking these DNMs to the genes that they impact remains a challenge, as the majority of them are embedded in non-coding regions. As most developmental diseases occur in the early stages of development or during childhood, it is crucial to clarify the details of epigenetic regulation in early development in order to interpret the mechanisms underlying developmental disorders. Here, we develop EpiDenovo, a database that is freely available at http://www.epidenovo.biols.ac.cn/, and which provides the associations between embryonic epigenomes and DNMs in developmental disorders, including several neuropsychiatric disorders and congenital heart disease. EpiDenovo provides an easy-to-use web interface allowing users rapidly to find the epigenetic signatures of DNMs and the expression patterns of the genes that they regulate during embryonic development. In summary, EpiDenovo is a useful resource for selecting candidate genes for further functional studies in embryonic development, and for investigating regulatory DNMs as well as other genetic variants causing or underlying developmental disorders.
INTRODUCTION
During early mammalian development, many significant epigenetic events occur, including the alteration of chromatin modification and chromatin accessibility, and the regulation of transcription factors (1,2). Epigenetic modifications, such as histone methylation and acetylation, can act as regulatory switches for gene transcription during embryonic development, and their dysfunction can give rise to developmental abnormalities (3,4). For example, altered epigenetic regulation in early development has been shown to be associated with schizophrenia (5). Thus, understanding the correlations among transcriptome and epigenome during early development will help in interpreting the underlying mechanisms that lead to neurodevelopmental disorders and to other developmental diseases (6).
De novo mutations (DNMs) in coding regions have already been shown to be a major cause of severe early-onset genetic disorders, such as autism spectrum disorder and intellectual disability (7). In addition, DNMs in regulatory elements can cause neurodevelopmental disorders (8), such as autism and schizophrenia (6–10). In congenital heart disease, another kind of developmental disease, a marked excess of DNMs was observed in the genes involved in the production, removal or reading of H3K4 methylation (H3K4me) (11). Furthermore, a non-coding genetic variant, a distal regulator of endothelin-1 gene expression, is associated with five vascular diseases (12). These studies indicate the crucial roles of DNMs in regulatory elements (13) in congenital heart disease. Additionally, high-resolution 3D maps of chromatin interactions during early human cortical development have identified hundreds of genes that interact physically with enhancers gained in humans, many of which are implicated in mediating the expression of quantitative trait loci (eQTL), and are associated with human cognitive function (7). Taking together, DNMs occurring in regulatory regions might interrupt gene regulation and induce developmental malformations (7). However, the association between epigenetic regulation and DNMs in developmental diseases is rarely understood.
Here, we present a database, named EpiDenovo (Figure 1), which is a platform for exploring the associations between embryonic epigenetic regulation and DNMs in developmental disorders, including neuropsychiatric disorders and congenital heart disease. The main intention of EpiDenovo is to investigate early developmental epigenomes and transcriptomes that are related to DNMs in developmental disorders, as certain DNMs could interrupt epigenetic regulation and gene expression during early embryonic development and consequently induce the symptoms of developmental diseases and disorders (14). Considering stage-specific gene activation is preserved during pre-implantation development in both humans and mice (15), we have also integrated mouse embryonic epigenomes to expand the interpretive information of genes associated with DNMs. The present study provides a framework to help understanding of the impact of DNMs on early development and highlighted the novel mechanisms underlying the onset of developmental disorders.
Figure 1.

Workflow to identify regulatory de novo mutations involved in embryonic epigenetic regulation by EpiDenovo.
DATA COLLECTION AND PROCESSING
Data sources
EpiDenovo is a comprehensive, annotated resource of DNMs in developmental disorders, based on the epigenomes of publicly available chromatin immunoprecipitation sequencing (ChIP-seq) and chromatin accessibility data during the embryonic development of mammals, including humans and mice. Samples collected include DNMs from denovo-db (16) and epigenomes from Sequence Read Archive (SRA) in the NCBI Gene Expression Omnibus (GEO) database (17). The following metadata for each sample was systematically annotated: assay, factor, species, group, cell state, characteristics, Experiment Acc. ID, Run Acc. ID, library layout and PMID. In total, we curated 1415 high-throughput sequencing datasets for mammalian embryonic development from GEO, and 283 888 DNMs in developmental disorders from denovo-db (16). In terms of embryonic epigenomes, our database contained 875 RNA-seq (Supplementary Table S1), 181 ChIP-seq (Supplementary Table S2), 43 ATAC-seq (Supplementary Table S3), 19 DNase-seq (Supplementary Table S4) and 297 Hi-C (Supplementary Table S5) datasets. In terms of DNMs, we curated 283 888 DNMs, including 228,925 DNMs in autism (ASD), 17 717 DNMs in developmental disorders (DD), 3903 DNMs in congenital heart disease (CHD), 2575 DNMs in intellectual disability (ID), 1654 DNMs in schizophrenia (SCZ), 1035 DNMs in epilepsy (EE), 78 DNMs in neural tube defects (NTD) as well as DNMs in other diseases. The distribution of DNMs in different gene elements is listed in Supplementary Table S6. We found that 82 385 (35.98%) and 142 572 (62.28%) of DNMs were located in the intergenic and intron regions, respectively, indicating that, in developmental disorders, the vast majority (98.26%) of DNMs occur in non-coding regions.
Data downloading and preparation
All raw data deposited in SRA format were downloaded from GEO using Aspera and converted into the FASTQ format using the fastq-dump of SRAToolkit t from NCBI. Sequencing adapters and low quality sequences were trimmed using the Trim Galore program of Babraham Bioinformatics (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/), with default parameters.
Reads mapping and coverage
All RNA-seq data were mapped to the mm10 genome for mouse, and hg38 genome for human, using STAR (v2.5.3a) (18), which was shown to be highly effective in mapping RNA-seq reads containing SNPs (19). Then, duplicated reads for pair-end data were removed by SAMtools (v1.5) (20). All ChIP-seq, ATAC-seq and Dnase-seq (Mnase-seq or FAIRE-seq) data were mapped to the mm10 genome for mouse, and hg38 genome for humans, by using SpeedSeq (v0.1.2) (21), which is an open-source genome analysis platform that achieves alignment, variant detection and function with a low memory requirement. Then, we removed any duplicated reads for both pair-end and single-end data using SAMtools. For all sequencing datasets, the bigwig files for JBrowse visualization were generated from BAM files by using ‘bamCoverage’ from deepTools (22) with parameters ‘–ignoreDuplicates –normalizeUsingRPKM –skipNonCoveredRegions –binSize 25 –ignoreForNormalization chrX chrM’. Samples with too low coverage (mapped data < 1M) were filtered.
Peak calling and annotation
BAM files of mapping results were merged for the same sample using SAMtools and converted to BED format by using BEDTools (23). Peaks of regulatory regions were called for each sample by using MACS2 (24) from datasets of ChIP-seq, ATAC-seq and DNase-seq with parameters ‘-f BED -B -q 0.01 –fix-bimodal –extsize 147 –keep-dup auto’. In particular, the input signal was used as the control to call peaks for the ChIP-seq dataset which has a corresponding control (input) experiment (Supplementary Table S7). Peak annotation was performed by using HOMER (25) with default parameters. Motif analysis on peak regions was performed with HOMER function findMotifsGenome.pl with parameters ‘-size 50 -mask’. In addition, 74,060,441 peaks regions were curated from GTRD (26) to expand the annotation of transcription binding sites from other tissues or cells.
Hi-C data analysis and curation
Paired-end raw reads of Hi-C libraries were aligned, processed and corrected iteratively using HiCPro (v2.8.1) (27). A 40- or 200-kb bin size was chosen for the examination of the global interaction patterns of the genome. The binned interaction matrices were then normalized using the iterative correction method (27,28) to correct biases such as the GC content, mappability and effective fragment length in Hi-C data. In addition, we also curated 3 095 881 chromatin contact pairs from the 4DGeneome (29) in order to expand the annotation of chromatin interactions from other tissues or cells.
Gene expression and stage-specific genes
BAM files of RNA-seq data were merged for the same sample using SAMtools (20), and transcript reconstruction was performed by StringTie (version v1.3.3b) (30), based on the gene annotation from Ensembl GRCh38 (release 89). The Fragments Per Kilobase of transcript per Million mapped reads (FPKM) value of gene expression was also determined and normalized by Cufflinks (31,32). An ANOVA-like test was applied in order to screen for genes that were differentially expressed among all groups by using edgeR (33,34).
Gene co-expression and function enrichment analysis
Co-expression analysis represents a powerful tool for the identification of genes involved in the same molecular process. Weighted gene co-expression network analysis (WGCNA) (35) was performed to understand the co-expression relationships between genes at a transcriptome-wide level (35,36). One-step network construction workflow was employed with a soft-thresholding power value of six for human and eight for mouse, respectively. Genes with null expression <98% in all samples (n = 51 247 for humans; n = 45 008 for mice) were selected to perform WGCNA analysis; a kME > 0.3 was assigned to an eigengene module (36). Finally, these co-expression genes in certain networks were selected to perform function enrichment analysis, including Gene Ontology and the KEGG pathway by using R package clusterProfiler (37).
Scoring system to identify regulatory DNMs
Each mutation was scored, based on its annotated records in five regulatory categories: conservation score, histone modification state, transcription factor binding sites, chromatin interacting regions and chromatin accessible regions. In contrast to the scoring scheme of RBP-Var (38), which classified variants into classes with a heuristic scoring system, EpiDenovo employed a quantitative scoring system to evaluate the regulatory significance of a DNM in different categories.
For the conservation category (C), we used PhyloP scores in 100 vertebrate genomes to assign conservation scores to DNMs. PhyloP scores of all DNMs in a chromosome followed a Gaussian distribution. Considering that a DNM has a conservation score of c, μ and σ, as these are the fitted parameters of the corresponding Gaussian model, then the score of the DNM in the conservation category is defined as follows:
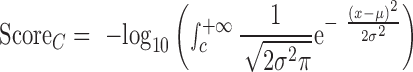 |
For the other four regulatory categories, we used the number of annotated hits (records) to assign a score to a DNM in the corresponding category. Specifically, the numbers of hits of all DNMs in each chromosome were fitted to a Poisson distribution model. Taking a DNM to have k hits in one regulatory category (F), λ is the fitted parameter of the corresponding Poisson model, and the score of the DNM in this category is defined as follows:
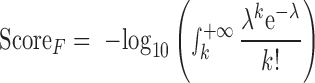 |
The total score of a DNM is the sum of scores of the five regulatory categories. The calculation of the scoring system was implemented by R and Perl.
Database architecture
All metadata in EpiDenovo were stored in a MySQL database while the network data, including the co-expression network and the regulatory network with do novo mutation, were deposited in neo4j, which is a high-performance graph database management system. The web interface of EpiDenovo was implemented in Cascading Style Sheets (CSS), Hyper Text Markup Language (HTML) and a Hypertext Preprocessor (PHP). The web design was derived from the free templates of Bootstrap (http://getbootstrap.com). Signal data visualization was implemented by using the JBrowse Genome Browser. The liftOver routine was employed, with a corresponding chain file from UCSC to convert genomic coordinates between different genome versions of humans.
DATABASE FEATURES AND APPLICATIONS
Database organization and web interface
As the EpiDenovo database contains embryonic epigenetic data from both humans and mice, these two species were both chosen as candidate species. Two reference versions were also provided for humans: hg19 and hg38. The rationale here being that hg19 is the most popular and hg38 is the most recent. Data retrieving in EpiDenovo could be achieved in three ways: ‘Gene symbols’, ‘Denovo mutations’ (only for humans) and ‘Regulatory regions’. A ‘Gene symbols’ search is very useful in terms of searching for gene expression and epigenetic regulation of genes of interest during embryonic development, and candidate regulatory regions or DNMs that are based on genes. ‘Denovo mutation’ retrieval is appropriate for analyzing the results of genetic studies into developmental disorders, and especially the results of high-throughput studies. This then gives support for further functional studies to identify the causal DNMs, and sheds light on the underlying molecular mechanisms of developmental disorders. In addition, EpiDenovo allows ‘Regulatory region’ retrieval, which could elucidate the potential roles of regulatory regions in providing genomic regions and locations of DNMs that were not deposited in the curated database. Further, the JBrowse Genome Browser (http://jbrowse.org) was applied in order to establish a well-organized ‘JBrowse’ page for visualizing genome-wide signals of expression and epigenetic data sets during embryonic development. Users could select and browse sequencing signals of any epigenetic type and any cell or tissue in the developmental stage across a genomic region of specific interest. The searching results for epilepsy related gene KCNMA1 in the EpiDenovo database were used as an example of the web-interface (Figure 2). EpiDenovo works well in all major web browsers including Google Chrome, Mozilla Firefox and Internet Explorer. In addition, a regulatory network was constructed and visualized based on regulatory information while the visualization interface of the co-expression network was developed based on netviewer in PoplarGene (39). In addition, a heatmap plot and functional enrichment of GO and KEGG for co-expressed genes, were shown in the sections that followed. Motif enrichment for each dataset of epigenetic factor was also provided. Finally, we built inner links between ‘Gene symbols’, ‘Denovo mutations’ and ‘Regulatory regions’, according to regulation information.
Figure 2.

Web-interface of EpiDenovo. The snapshot of searching result for epilepsy related gene KCNMA in EpiDenovo database.
Implications and applications
We identified 86 109 DNMs (33.87% of all DNMs) that were embedded in the regulatory elements involved in embryonic development, and 9340 genes that were regulated by these regulatory elements (Supplementary Table S8). We found 25 390, 43 939 and 9513 DNMs located in potential regulatory regions of chromatin states, from pachytene spermatocytes, round spermatids and mature sperm, respectively, indicating that germline DNMs originate from errors in DNA replication during gametogenesis, particularly in sperm cells and their precursors (7). Among these DNMs, 538, 172, 27 164, 347, 55 735, 2153 DNMs are associated with chromatin factors H3F3B, H3K27ac, H3K27me3, H3K4me1, H3K4me3 and PolII. So, most of them were associated with H3K4me3 and H3K27me3, indicating that DNMs could primarily occur not only in active enhancers, but also in poised enhancers (40,41). Interestingly, we also observed de novo mutation hotspots occurred simultaneously with high density of active histone modifications, permissive state of chromatin accessibility and intense chromatin interaction in non-coding regions nearby gene cluster (Figure 3).
Figure 3.

Circos plot of chr5–8 to show the relationship between DNMs and epigenetic regulation. The regions covered by a transparent sector is an example of DNMs hotspots occurred simultaneously with active genetic markers in non-coding regions nearby gene cluster.
To illuminate the applications of EpiDenovo, we enumerated six disease associated genes including NOTCH2, LMX1A, CHD5, SCN3A, HDAC4 and BCL11A. All of these genes were involved in the embryonic epigenetic regulation which may be mediated by regulatory DNMs (Figure 4).
Figure 4.

To illuminate the applications of EpiDenovo, we enumerated six disease associated genes including NOTCH2, LMX1A, CHD5, SCN3A, HDAC4 and BCL11.
DISCUSSION AND PERSPECTIVES
To our knowledge, there are several ChIP-seq databases (ENCODE (42), GTRD (26), ChIPBase (43), Cistrome DB (44), Roadmap Epigenomics (45), Factorbook (46), ChIP-Atlas (http://chip-atlas.org), GeneProf (47), NGS-QC (48) and DBTMEE (49)), but all of these have curated little embryonic epigenetic datasets (except DBTMEE), and were unlinked to genetic variants or human disease. RegulomeDB (50) and 3DSNP (51) are databases that undertake attempts at decoding the roles of SNPs embedded in DNA regulatory elements; however, RegulomeDB was not sensitive enough to decipher the functions of DNMs in neuropsychiatric disorders, although it has a high specificity, according to our recent study which demonstrated that DNMs involved in post-transcriptional dysregulation contribute to six neuropsychiatric disorders (52). So, it remains a challenge to investigate the roles of DNMs in DNA regulatory elements in developmental disorders, such as neuropsychiatric disorders. This study represents the first attempt at using the integrated analysis of both epigenetic regulation and gene expression during embryonic development to interpret the formation and function of DNMs.
The principal advantages of EpiDenovo, compared to other databases, for the annotation of regulatory variants are as follows:
It contains the most comprehensive collection of ChIP-seq, ATAC-seq, DNase-seq and Hi-C data with respect to the chromatin state during embryonic development for both humans and mice.
It has the potential to contribute to research, not only on developmental diseases, but also on embryonic development, as it provides the association of DNMs with the transcriptome and epigenome during embryonic development in human developmental disorders.
It allows ‘Regulatory region’ retrieval, which could elucidate the potential roles of regulatory regions by providing genomic regions and locations of novel DNMs as well as genetic variants that were not deposited in the current database.
It provides a well-organized visualization using JBrowse to show the epigenetic signals of each sample in user defined genomic regions.
It employs a statistical scoring system to annotate and prioritize the DNMs involved in epigenetic regulation.
It provides an in-depth annotation of the genes of interest by performing weighted gene co-expression network analysis and functional enrichment analysis.
It provides motif enrichment in peaks of epigenetic factor for each experiment to predict the potential binding of transcription factors by similarity of binding motif.
EpiDenovo contains all currently available epigenetic datasets, and we will continue to update the database with new epigenetic datasets from early development, especially from brain development. As more regulatory DNMs will be validated, we aim to assess and improve the current scoring system. We are fully dedicated to the maintenance and improvement of EpiDenovo and making it to be a useful database for the research on embryonic development and developmental diseases.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Chenghang Du in the Beijing Institutes of Life Science, Chinese Academy of Sciences and Dr Jiansong Li in the Beijing Institute of Heart Lung and Blood Vessel Diseases for their help in maintaining the high performance computing systems. It is greatly appreciated for the initial discussion with Dr Yu-Cheng T. Yang in the Department of Statistics, University of California, Los Angeles.
Authors’ contributions: F.B.M., Z.S.S. and Y.L.D. conceived and designed the database; S.G. collected the SRA information from GEO. FBM downloaded and analyzed the sequencing data. H.N.Y. and L.Y.X. constructed the preliminary website. Q.L. and F.B.M. accomplished and maintained the full functional database; X.F.L. tested and debugged the database. X.L.Z. and H.J.T. wrote and revised the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Key R&D Program of China [2016YFC0900400 to Z.S.S.]. Funding for open access charge: National Key R&D Program of China [2016YFC0900400 to Z.S.S.].
Conflict of interest statement. None declared.
REFERENCES
- 1. Saitou M., Kagiwada S., Kurimoto K.. Epigenetic reprogramming in mouse pre-implantation development and primordial germ cells. Development. 2012; 139:15–31. [DOI] [PubMed] [Google Scholar]
- 2. Burton A., Torres-Padilla M.E.. Chromatin dynamics in the regulation of cell fate allocation during early embryogenesis. Nat. Rev. Mol. Cell. Biol. 2014; 15:722–734. [DOI] [PubMed] [Google Scholar]
- 3. Tordjman S., Somogyi E., Coulon N., Kermarrec S., Cohen D., Bronsard G., Bonnot O., Weismann-Arcache C., Botbol M., Lauth B. et al. Gene x environment interactions in autism spectrum disorders: role of epigenetic mechanisms. Front. Psychiatry. 2014; 5:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shi L., Wu J.. Epigenetic regulation in mammalian preimplantation embryo development. Reprod. Biol. Endocrinol. 2009; 7:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Malkki H. Neurodevelopmental disorders. Altered epigenetic regulation in early development associated with schizophrenia. Nat. Rev. Neurol. 2016; 12:1. [DOI] [PubMed] [Google Scholar]
- 6. Won H., de la Torre-Ubieta L., Stein J.L., Parikshak N.N., Huang J., Opland C.K., Gandal M.J., Sutton G.J., Hormozdiari F., Lu D. et al. Chromosome conformation elucidates regulatory relationships in developing human brain. Nature. 2016; 538:523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Acuna-Hidalgo R., Veltman J.A., Hoischen A.. New insights into the generation and role of de novo mutations in health and disease. Genome Biol. 2016; 17:241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Short P.J., McRae J.F., Gallone G., Sifrim A., Won H., Geschwind D.H., Wright C.F., Firth H.V., FitzPatrick D.R., Barrett J.C. et al. De novo mutations in regulatory elements cause neurodevelopmental disorders. doi:10.1101/112896. [DOI] [PMC free article] [PubMed]
- 9. Takata A., Ionita-Laza I., Gogos J.A., Xu B., Karayiorgou M.. De novo synonymous mutations in regulatory elements contribute to the genetic etiology of autism and Schizophrenia. Neuron. 2016; 89:940–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sun W., Poschmann J., Cruz-Herrera Del Rosario R., Parikshak N.N., Hajan H.S., Kumar V., Ramasamy R., Belgard T.G., Elanggovan B., Wong C.C. et al. Histone acetylome-wide association study of autism spectrum disorder. Cell. 2016; 167:1385–1397. [DOI] [PubMed] [Google Scholar]
- 11. Brind’Amour J., Liu S., Hudson M., Chen C., Karimi M.M., Lorincz M.C.. An ultra-low-input native ChIP-seq protocol for genome-wide profiling of rare cell populations. Nat. Commun. 2015; 6:6033. [DOI] [PubMed] [Google Scholar]
- 12. Gupta R.M., Hadaya J., Trehan A., Zekavat S.M., Roselli C., Klarin D., Emdin C.A., Hilvering C.R.E., Bianchi V., Mueller C. et al. A genetic variant associated with five vascular diseases is a distal regulator of endothelin-1 gene expression. Cell. 2017; 170:522–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jia Z., Mao F.B., Wang L., Li M.Z., Shi Y.Y., Zhang B.R., Gao G.L.. Whole-exome sequencing identifies a de novo mutation in TRPM4 involved in pleiotropic ventricular septal defect. Int. J. Clin. Exp. Pathol. 2017; 10:5092–5104. [Google Scholar]
- 14. Gregor A., Oti M., Kouwenhoven E.N., Hoyer J., Sticht H., Ekici A.B., Kjaergaard S., Rauch A., Stunnenberg H.G., Uebe S. et al. De novo mutations in the genome organizer CTCF cause intellectual disability. Am. J. Hum. Genet. 2013; 93:124–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xue Z., Huang K., Cai C., Cai L., Jiang C.Y., Feng Y., Liu Z., Zeng Q., Cheng L., Sun Y.E. et al. Genetic programs in human and mouse early embryos revealed by single-cell RNA sequencing. Nature. 2013; 500:593–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Turner T.N., Yi Q., Krumm N., Huddleston J., Hoekzema K., HA F.S., Doebley A.L., Bernier R.A., Nickerson D.A., Eichler E.E.. denovo-db: a compendium of human de novo variants. Nucleic Acids Res. 2017; 45:D804–D811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Barrett T., Wilhite S.E., Ledoux P., Evangelista C., Kim I.F., Tomashevsky M., Marshall K.A., Phillippy K.H., Sherman P.M., Holko M. et al. NCBI GEO: archive for functional genomics data sets-update. Nucleic Acids Res. 2013; 41:D991–D995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wu J., Huang B., Chen H., Yin Q., Liu Y., Xiang Y., Zhang B., Liu B., Wang Q., Xia W. et al. The landscape of accessible chromatin in mammalian preimplantation embryos. Nature. 2016; 534:652–657. [DOI] [PubMed] [Google Scholar]
- 20. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., Proc G.P.D.. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chiang C., Layer R.M., Faust G.G., Lindberg M.R., Rose D.B., Garrison E.P., Marth G.T., Quinlan A.R., Hall I.M.. SpeedSeq: ultra-fast personal genome analysis and interpretation. Nat. Methods. 2015; 12:966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ramirez F., Dundar F., Diehl S., Gruning B.A., Manke T.. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014; 42:W187–W191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Feng J.X., Liu T., Qin B., Zhang Y., Liu X.S.. Identifying ChIP-seq enrichment using MACS. Nat. Protoc. 2012; 7:1728–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K.. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010; 38:576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yevshin I., Sharipov R., Valeev T., Kel A., Kolpakov F.. GTRD: a database of transcription factor binding sites identified by ChIP-seq experiments. Nucleic Acids Res. 2017; 45:D61–D67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Servant N., Varoquaux N., Lajoie B.R., Viara E., Chen C.J., Vert J.P., Heard E., Dekker J., Barillot E.. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015; 16:259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Imakaev M., Fudenberg G., McCord R.P., Naumova N., Goloborodko A., Lajoie B.R., Dekker J., Mirny L.A.. Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat. Methods. 2012; 9:999–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Teng L., He B., Wang J.H., Tan K.. 4DGenome: a comprehensive database of chromatin interactions. Bioinformatics. 2015; 31:2560–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pertea M., Pertea G.M., Antonescu C.M., Chang T.C., Mendell J.T., Salzberg S.L.. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015; 33:290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Trapnell C., Hendrickson D.G., Sauvageau M., Goff L., Rinn J.L., Pachter L.. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013; 31:46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Trapnell C., Williams B.A., Pertea G., Mortazavi A., Kwan G., van Baren M.J., Salzberg S.L., Wold B.J., Pachter L.. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010; 28:511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Robinson M.D., McCarthy D.J., Smyth G.K.. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010; 26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McCarthy D.J., Chen Y., Smyth G.K.. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012; 40:4288–4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Langfelder P., Horvath S.. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008; 9:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. van Dam S., Vosa U., van der Graaf A., Franke L., de Magalhaes J.P.. Gene co-expression analysis for functional classification and gene-disease predictions. Brief. Bioinform. 2017; doi:10.1093/bib/bbw139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yu G., Wang L.G., Han Y., He Q.Y.. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012; 16:284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mao F., Xiao L., Li X., Liang J., Teng H., Cai W., Sun Z.S.. RBP-Var: a database of functional variants involved in regulation mediated by RNA-binding proteins. Nucleic Acids Res. 2016; 44:D154–D163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liu Q., Ding C.J., Chu Y.G., Chen J.F., Zhang W.X., Zhang B.Y., Huang Q.J., Su X.H.. PoplarGene: poplar gene network and resource for mining functional information for genes from woody plants. Sci. Rep. 2016; 6:31356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Heinz S., Romanoski C.E., Benner C., Glass C.K.. The selection and function of cell type-specific enhancers. Nat. Rev. Mol. Cell Biol. 2015; 16:144–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhu Y., Sun L., Chen Z., Whitaker J.W., Wang T., Wang W.. Predicting enhancer transcription and activity from chromatin modifications. Nucleic Acids Res. 2013; 41:10032–10043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Consortium E.P. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhou K.R., Liu S., Sun W.J., Zheng L.L., Zhou H., Yang J.H., Qu L.H.. ChIPBase v2.0: decoding transcriptional regulatory networks of non-coding RNAs and protein-coding genes from ChIP-seq data. Nucleic Acids Res. 2017; 45:D43–D50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mei S.L., Qin Q., Wu Q., Sun H.F., Zheng R.B., Zang C.Z., Zhu M.Y., Wu J.X., Shi X.H., Taing L. et al. Cistrome Data Browser: a data portal for ChIP-Seq and chromatin accessibility data in human and mouse. Nucleic Acids Res. 2017; 45:D658–D662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bernstein B.E., Stamatoyannopoulos J.A., Costello J.F., Ren B., Milosavljevic A., Meissner A., Kellis M., Marra M.A., Beaudet A.L., Ecker J.R. et al. The NIH roadmap epigenomics mapping consortium. Nat. Biotechnol. 2010; 28:1045–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang J., Zhuang J., Iyer S., Lin X.Y., Greven M.C., Kim B.H., Moore J., Pierce B.G., Dong X., Virgil D. et al. Factorbook.org: a Wiki-based database for transcription factor-binding data generated by the ENCODE consortium. Nucleic Acids Res. 2013; 41:D171–D176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Halbritter F., Kousa A.I., Tomlinson S.R.. GeneProf data: a resource of curated, integrated and reusable high-throughput genomics experiments. Nucleic Acids Res. 2014; 42:D851–D858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mendoza-Parra M.A., Saravaki V., Cholley P.E., Blum M., Billore B., Gronemeyer H.. Antibody performance in ChIP-sequencing assays: From quality scores of public data sets to quantitative certification. F1000Res. 2016; 5:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Park S.J., Shirahige K., Ohsugi M., Nakai K.. DBTMEE: a database of transcriptome in mouse early embryos. Nucleic Acids Res. 2015; 43:D771–D776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Boyle A.P., Hong E.L., Hariharan M., Cheng Y., Schaub M.A., Kasowski M., Karczewski K.J., Park J., Hitz B.C., Weng S. et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012; 22:1790–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lu Y.M., Quan C., Chen H.B., Bo X.C., Zhang C.G.. 3DSNP: a database for linking human noncoding SNPs to their three-dimensional interacting genes. Nucleic Acids Res. 2017; 45:D643–D649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mao F., Wang L., Xiao L., Liu Q., Li X., He X., Rao R.C., Li J., Teng H., Dou Y. et al. De novo mutations involved in post-transcriptional dysregulation contribute to six neuropsychiatric disorders. 2017; doi:10.1101/175844.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.