


Abstract
MicroRNAs (miRNAs) are small non-coding RNAs of ∼ 22 nucleotides that are involved in negative regulation of mRNA at the post-transcriptional level. Previously, we developed miRTarBase which provides information about experimentally validated miRNA-target interactions (MTIs). Here, we describe an updated database containing 422 517 curated MTIs from 4076 miRNAs and 23 054 target genes collected from over 8500 articles. The number of MTIs curated by strong evidence has increased ∼1.4-fold since the last update in 2016. In this updated version, target sites validated by reporter assay that are available in the literature can be downloaded. The target site sequence can extract new features for analysis via a machine learning approach which can help to evaluate the performance of miRNA-target prediction tools. Furthermore, different ways of browsing enhance user browsing specific MTIs. With these improvements, miRTarBase serves as more comprehensively annotated, experimentally validated miRNA-target interactions databases in the field of miRNA related research. miRTarBase is available at http://miRTarBase.mbc.nctu.edu.tw/.
INTRODUCTION
MicroRNAs (miRNAs) are, small non-coding RNAs with 18–25 nucleotides, which are central regulators at the post-transcriptional level in both animals and plants. Perfect or near-perfect complementary binding of miRNAs and their target mRNA negatively regulates gene expression by accelerating mRNA degradation or suppressing mRNA translation (1). miRNAs are involved in many biological processes like cell-cycle (2), cell differentiation (3), and apoptosis (4) as well as in different diseases including cancer (5). The study of miRNAs and their target interactions has been gaining interest among researchers due to their causal relationship with disease development. Importantly, dysregulated expression patterns of miRNA may serve as potential biomarkers for disease diagnosis and prognosis (6).
In order to analyze and identify miRNA-target interactions (MTIs), many web-based miRNA-related databases have been established. Of these, miRBase (5) is the primary miRNA sequence repository that facilitates searches for comprehensive miRNA nomenclature, sequence, and annotation data. The DIANA-TarBase (6) is a database that contains manually curated experimentally validated miRNA-gene interactions, with detailed meta-data, experimental methodologies, and conditions. HMDD (7) and miR2Disease (8) contain experimentally validated human miRNA and disease associated MTIs. Although MTIs databases are widely available, MTI-related research has been prolific in recent years. Thus, a centralized information repository of experimentally validated miRNA-target interactions that is easily accessible and updated over the long term must be developed.
Even though computational approaches, that rely on base pairing of miRNAs and their targets, are useful for identifying miRNAs and their target binding sites (9), perfect seed pairing may not always represent the actual interaction between an miRNA and its target (10). Thus, experimental verification must still be carried out to establish miRNAs and their target interactions to elucidate the functions of a given miRNA. MTIs can be experimentally verified in several ways (11). The expression levels of mRNA can be detected using techniques like qRT-PCR, whereas protein products can be measured by ELISA, immunohistochemistry, and Western blot. Reporter assay is the most reliable method to demonstrate direct interaction between an miRNA and its target by measuring reducing activity or the expression of reporter protein (12). Methods like microarray or high-throughput sequencing provide indirect relationships between miRNAs and their targets (13). The newly developed techniques such as CLIP (14), PAR-CLIP, (15) and CLASH (16) incorporate high-throughput sequencing that can also be used to identify miRNAs and their targets.
Recently, miRNA-target interactions related research has rapidly increased (Figure S1). To enhance the identification of miRNA targets and their roles in various biological processes, we previously developed miRTarBase (17–19). To date, it has been widely used for improving the accurate rate of target prediction, integrating miRNA related databases or web-based tools, reconstructing miRNA-gene regulatory networks for different diseases, as well as the incorporation into The Cancer Genome Atlas (TCGA) Research Network study. Herein, we provide an up-to-date repository of experimentally validated MTIs with improved annotations and visualization data to facilitate biological investigation, network-based studies and computational machine learning applications.
SYSTEM OVERVIEW AND DATABASE CONTENT
Since the first release of miRTarBase in 2011 until the present, the body of experimentally supported data content has not only dramatically increased but the various functions of the web interface have also been extended. Here, we briefly describe the data content and web-based functions. The database aims to provide the more comprehensive collection of experimentally supported MTIs either in data content or in the web-based function, to accelerate miRNA research. Figure 1 illustrates the current design and system flow of miRTarBase. First, MTI related articles are downloaded from PubMed. Second, enhanced text mining systems are used to extract MTIs. Third, our curator group manually curates the articles to retrieve the important information regarding experimentally verified miRNA–target interactions. We integrated many databases and standalone tools including miRBase (5) and HMDD (7) for miRNA and disease information, and NCBI Entrez gene (20) and RefSeq (21) for target gene information and 3’ untranslated region of target sequences. Likewise, we integrated PubMed, to provide article information, TargetScan (22) and miRanda (23) for target prediction, Gene Expression Omnibus (GEO) (24) and The Cancer Genome Atlas (TCGA) (25,26) for gene and miRNA expression profiling, and KEGG (27) and DAVID (28) for functional annotations of miRNA target genes. Independent studies of MTIs usually describe miRNA using old versions of their name or do not clearly describe the mature form of miRNA. To overcome this problem, our curators checked for the mature form of sequence or confirmed the miRNA version of each study. We also uniformly mapped the miRNA accession to the miRBase v21; unmapped miRNAs were excluded from our database. In addition, gene names were also uniformly mapped by Entrez gene symbol or by Entrez gene alias name (downloaded August 2017). Datasets from the GEO and TCGA with miRNA and mRNA expression profiles were incorporated into miRTarBase. Currently, the database includes 21 human datasets from the NCBI GEO database with at least nine matched mRNA and miRNA samples containing 1596 samples. In addition, a total of 571 samples of 19 cancers from two platforms of TCGA were integrated into the current database. As a result, correlations of expression between miRNA and mRNA in matched samples provide further evidence for direct targets of miRNA.
Figure 1.

Overall system flow of miRTarBase. miRTarBase aims to be a more comprehensive database of experimentally verified miRNA-targets interaction. This database was released in 2011. The current database contains more than 420,000 MTIs.
UPDATED DATABASE CONTENT AND STATISTICS
The improvements and updated database content that are provided by the miRTarBase 2018 are presented in Table 1. The major improvements in this version include (i) a significant increase in the number of curated articles and MTIs, supported by both strong and limited experimental evidences; (ii) the extraction of miRNA-target interactions using an enhanced automatic text-mining system for further manual curation and (iii) the establishment of an enhanced web interface for miRNA-target sites and disease information. The improvements in this version are described in detail as follows.
Table 1. Advances, improvements and the number of miRNA-target interactions with different validation methods provided by miRTarBase 7.0.
| Features | miRTarBase 6.0 | miRTarBase 7.0 |
|---|---|---|
| Release date | 2015/09/15 | 2017/09/15 |
| Known miRNA entry | miRBase v20 | miRBase v21 |
| Known Gene entry | Entrez 2015 | Entrez 2017 |
| Species | 18 | 23 |
| Curated articles | 4966 | 8510 |
| miRNAs | 3786 | 4076 |
| Target genes | 22 563 | 23 054 |
| CLIP-seq datasets | 138 | 231 |
| Curated miRNA-target interactions | 366 181 | 422 517 |
| Text-mining technique to prescreen literature | NLP | Enhanced NLP |
| Download by validated miRNA-target sites | None | Yes |
| Browse by miRNA, gene, and disease | None | Yes |
| MTIs Supported by strong experimental evidences | ||
| Number of MTIs validated by ‘Reporter assay’ | 6694 | 9489 |
| Number of MTIs validated by ‘Western blot’ | 4580 | 7258 |
| Number of MTIs validated by ‘qPCR’ | 4645 | 8210 |
| Number of MTIs validated by ‘Reporter assay and Western blot’ | 3854 | 6032 |
| Number of MTIs validated by ‘Reporter assay or Western blot’ | 7439 | 10 581 |
This version has added a significant number of highly curated articles and expanded its content. The current release (September 2017, version 7) includes a total 422 517 curated MTIs between 4076 miRNAs and 23 054 target genes, which were collected from 8573 articles. The number of MTIs has increased ∼1.4-fold curated by strong evidence (Reporter assay or Western blot) since the 2016 miRTarBase update. MTIs which are supported by different experimental approaches are currently available. The update has significantly increased the number of MTIs in comparison to miRTarBase 6.0, as shown in Table 1. The top 10 miRNAs with different target genes on strong evidence are shown in Figure 2A. The top 10 genes, which were regulated by different miRNA on strong evidence, are shown in Figure 2B. The top 10 human disease of different literatures are shown in Figure 2C. The relevant literature was manually surveyed to ensure collection of strong evidence for MTIs. As a manually curated, experimentally supported database, this updated version retrieves MTIs that are supported by strong evidence. For example, we have curated roughly 3,000 new MTIs supported by reporter assay and/or Western blot. In addition, many MTIs were supported by more than one independent study. The CLIP-seq datasets include HITS-CLIP and PAR-CLIP, which provide limited experimental evidence and were analyzed as described below.
Figure 2.

Top 10 Human miRNAs, genes, and diseases with strong evidence of MTIs in miRTarBase.
Collection and analysis of the newly published CLIP-seq data sets
The previous version of database contained a total of 138 CLIP-seq datasets from 21 independent studies. This updated version contains additional 93 CLIP-seq data from four independent studies (29–32). The samples were annotated using the same categories as used previously: source of dataset, types of NGS approach and RNA binding protein (RBP), target species, dataset accession number, tissue or cell line, condition (Supplementary Table S1). CLIP-seq raw data were analyzed using miRTarCLIP (33). In addition, Piranha (34) was used for peak calling and TargetScan (Release 7.1) (22) was used to identify miRNA target sites. The detailed miRTarCLIP process involves trimming adapter reads, removing low-quality reads (phred quality score 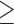 20 were retained), cytosine (C) to thymine (T) conversion (only in PAR-CLIP data), aligning reads against the reference 3’UTR sequence with TargetScan, clustering target sites and peak calling with Piranha, and finally analyzing miRNA-target sites. As a result, the number of MTIs has been significantly increased; there was an ∼1.4-fold increase in MTIs that were validated by NGS, and these are shown in Table 1.
20 were retained), cytosine (C) to thymine (T) conversion (only in PAR-CLIP data), aligning reads against the reference 3’UTR sequence with TargetScan, clustering target sites and peak calling with Piranha, and finally analyzing miRNA-target sites. As a result, the number of MTIs has been significantly increased; there was an ∼1.4-fold increase in MTIs that were validated by NGS, and these are shown in Table 1.
Literature associated with disease
Most of the literature, especially more recent studies, indicates that MTIs are associated with human diseases. Some studies have reported that circulating miRNAs function as biomarkers for disease diagnosis and provide indications of potential target therapeutic agents. In previous version, we integrated data from HMDD v2.0 (7) to represent dynamic miRNA-disease relationships with word clouds. Here, we provide an ‘Evidence’ section containing disease information from the literature on each MTI. The browser page also provides a ‘browse by disease’ option. This extensive disease information enables researchers to search for miRNA-associated diseases more quickly and conveniently. For the disease annotation task, here we collected a list of disease name based on MeSH tree and adopted a partial matching algorithm. After proofreading, all of the extracted articles contained full or part of the disease name.
ENHANCED TEXT MINING SYSTEM
Accumulating knowledge regarding MTIs in the literature can expedite research aimed at understanding the complex physiological processes between miRNAs and their targets. However, the large volume of published work poses a formidable challenge. We devised an automatic MTI detection pipeline comprised of various text processing components. In essence, the MTI system inspects the literature, typically obtained from large databases such as PubMed, and identifies pairs of miRNA and the target gene pairs therein. Our system encompasses two key components: named entity recognition (NER) and relation extraction (RE), as well as other common natural language processing (NLP) procedures. More specifically, the NER module identifies entities mentioned in the text and labels them with appropriate tags. Subsequently, the RE module detects potential relations among those entities using information provided in the previous step. Existing work regarding these tasks can be broadly distinguished into two categories: rule-based and machine-learning-based approaches, and have been widely adopted to conduct MTI on public miRNA databases including DIANA-TarBase v7.0 (6), miRCancer (35) and miRSel (36).
However, there are some limitations and weaknesses inherent to both approaches. In rule-based methods, explicit rules must be developed by domain experts but it is hard to cover all the possible variations with manually defined rules. Thus, inflexible rules make it difficult for approaches to deal with unpredictable exceptions, which may result in some types of word mutations, such as insertion, deletion, or substitution (IDS), in the target named entities. In contrast, machine learning (ML) models can automatically learn implicit patterns for generating principles. Nevertheless, the ML-based model is not as intuitive as rule-based methods and the results cannot be easily interpreted by humans. Consequently, we utilized the statistical-principle-based approach (SPBA) (37) to implement our MTI detection system. Figure 3 illustrates the details of the processing in our MTI detection system.
Figure 3.

Natural language processing (NLP) techniques for identifying MTI articles. (1) Collecting Articles from public literature database, PubMed; (2) detecting sentence boundary of each abstract; (3) preprocessing articles in the following steps; split sentences, tokenization, stemming and stopword removal; (4) Named Entity Recognition (NER) based on statistical principle-based approach (SPBA), Conditional Random Fields (CRFs) and dictionary-based approach; (5) all sentences will be labeled as labeled sentences for principle matching; (6) following construction and evaluation of model, an inference engine for MTI extraction was developed in SPBA; (7) the inference engine is constructed for assist domain exports to manually check the generated positive principles (the * element is not required, e.g., experiment); (8) all curated articles were manually evaluated by biological domain experts.
On the whole, the developed approach, SPBA, can automatically elicit labeled sequences and incorporate them into more representative principles by observing dominated principles. Afterwards, a partial matching algorithm is employed in harnessing the advantages of both rule- and machine learning-based approaches while surpassing their limitations. The method is proved to outperform in several distinct tasks such as topic detection and sentimental analysis (37–40).
In this updated version, a set of principles generated in the previous version was manually curated by domain experts to enhance the recognition of miRNA-target interactions (MTIs). Afterwards, the proposed SPBA-based approach can successfully extract the relation between miRNA and their targets from articles by the partial matching algorithm. All of the selected articles in this update are validated by several curators. Through the screening mechanism based on SPBA, the enhanced text mining pipeline singled out 3371 papers, which can be referred to as a summary of MTIs. After manual proofreading, 2617 papers were confirmed to contain MTIs, indicating that precision of 78% was achieved by our system. The enhanced SPBA-based MTI detector improved on the precision of the previous version by 7%.
ENHANCED WEB INTERFACE
The previous version provided a user friendly, graphical visualization interface for biologists to investigate MTIs in detail. We provide all of the possible relevant information, including miRNA secondary structure, miRNA-related diseases, putative target sites, target sites provided by the authors, evidence of MTIs from original descriptions in each article, expression profiling of miRNAs and their target genes using GEO and TCGA datasets, CLIP-seq evidence for MTIs, target gene set enrichment analysis, and the MTI networks. However, improvements were required to identify easier ways to find the relevant MTIs. Here, we provide three different ways to browse for specific MTIs: ‘browse by miRNA’, ‘browse by target genes’, and ‘browse by disease’. Upon browsing, these three categories present the top 50 results. Based on text-mining, a new column for disease description has been added to each MTI in the ‘Evidence’ page, which provides information regarding the experimentally supported literature. The most important new element is that the browser provides the target site in web interface and also downloads pages for further study. In the era of artificial intelligence, especially with regards to machine learning and deep learning applications, we believe that the datasets retrieved from experimentally validated miRNA-target sites improve the accuracy of current target prediction tools and reduce the chances of false-positive predictions.
DATA AVAILABILITY
The Download page of miRTarBase provides the more comprehensive data for download that include: (1) All published miRNA target interaction data. (2) MTI PubMed Abstract Manually Curation Corpus (3) Catalog by Species of MTIs. (4) Catalog by Experimental Evidences of MTIs. We also provide all the previous version release in the ‘Previous Release’ page.
DISCUSSION
We aimed to provide more convenient and accurate information regarding MTIs, speeding up miRNA research in biomedical science, and provide assistance to researchers in the field. Since 2011, many researchers have used miRTarBase to conduct fruitful research, including improving the accuracy rate of target prediction, integrating miRNA related databases and web-based tools, and reconstructing miRNA-gene regulatory networks in different diseases. In addition, the database was incorporated into The Cancer Genome Atlas Research Network study. The main applications of the database are described below as well as in Supplementary DATA and Table S2 in detail.
miRTarBase has recently been used to conduct integrative analysis in many high quality miRNA related studies, such as clarifying the super-enhancer-mediated RNA processing mechanism (41), informing the miRNA dysregulation in autism spectrum disorder (ASD) and neurological disease (42), supporting the design of tumor type-specific treatments (43), and expanding miRNA therapeutics, which is crucial to disease treatment and cure (44). Moreover, our database also functions significantly in research on different types of cancer, including liver cancer (45), ovarian cancer (46) and gastric cancer (47). Remarkably, in The Cancer Genome Atlas (TCGA) project, the research team integrated high-throughput experimental data and miRTarBase's MTI information to accomplish important research work and reveal a complex molecular landscape of cervical cancers and Pancreatic Ductal Adenocarcinoma (PDAC) to provide a roadmap for precision medicine (48,49).
SUMMARY AND PERSPECTIVES
Since the miRTarBase debuted in 2011, MTIs and related databases have been continually updated. Moreover, the collection of its datasets has been expanded. We have curated over 8,500 experimentally supported articles regarding miRNA-target interactions. As a result, ∼10 000 MTIs that have been validated by strong evidence have been included. Our improved text mining pipeline and a set of patterns which was generated automatically by PBA enhance the extraction of MTIs. With the addition of newly published CLIP-seq datasets, the collection of MTIs in the new miRTarBase is over 420 000. The enhanced web interface can browse the top 50 ranking miRNAs, genes, and diseases. A newly added download feature facilitates to download the target sites validated by reporter assay available in the literature. We believe that the target site sequence could extract new features for analysis via a machine learning approach that can help to evaluate the performance of miRNA-target prediction tools. We will extend and expand the miRTarBase continuously to the more comprehensive database by improving NLP technology, recruiting more curators, and enhancing the web function and annotation. To conclude, we provide an extensive repository of experimental information that clearly makes a continuous and worthwhile contribution to miRNA research related to cancer mechanisms, disease diagnosis and treatment.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Ministry of Science and Technology, Taiwan [MOST 103-2628-B-009-001-MY3, MOST 106-2627-M-009-002, MOST 106-2319-B-400-001, MOST 106-2633-B-009-001]; ‘Aiming for the Top University Program’ of the National Chiao Tung University and Ministry of Education, Taiwan [MOHW106-TDU-B-212-144005, MMH-HB-10602]. Funding for open access charge: Ministry of Science and Technology, Taiwan [MOST 106-2627-M-009-002, MOST 106-2319-B-400-001, MOST 106-2633-B-009-001].
Conflict of interest statement. None declared.
REFERENCES
- 1. Bartel D.P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004; 116:281–297. [DOI] [PubMed] [Google Scholar]
- 2. Chen D., Farwell M.A., Zhang B.. MicroRNA as a new player in the cell cycle. J. Cell. Physiol. 2010; 225:296–301. [DOI] [PubMed] [Google Scholar]
- 3. Shivdasani R.A. MicroRNAs: regulators of gene expression and cell differentiation. Blood. 2006; 108:3646–3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lima R.T., Busacca S., Almeida G.M., Gaudino G., Fennell D.A., Vasconcelos M.H.. MicroRNA regulation of core apoptosis pathways in cancer. Eur. J. Cancer. 2011; 47:163–174. [DOI] [PubMed] [Google Scholar]
- 5. Kozomara A., Griffiths-Jones S.. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014; 42:D68–D73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vlachos I.S., Paraskevopoulou M.D., Karagkouni D., Georgakilas G., Vergoulis T., Kanellos I., Anastasopoulos I.L., Maniou S., Karathanou K., Kalfakakou D. et al. . DIANA-TarBase v7.0: indexing more than half a million experimentally supported miRNA:mRNA interactions. Nucleic Acids Res. 2015; 43:D153–D159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li Y., Qiu C., Tu J., Geng B., Yang J., Jiang T., Cui Q.. HMDD v2.0: a database for experimentally supported human microRNA and disease associations. Nucleic Acids Res. 2014; 42:D1070–D1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jiang Q., Wang Y., Hao Y., Juan L., Teng M., Zhang X., Li M., Wang G., Liu Y.. miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 2009; 37:D98–D104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rajewsky N. microRNA target predictions in animals. Nat. Genet. 2006; 38(Suppl):S8–S13. [DOI] [PubMed] [Google Scholar]
- 10. Didiano D., Hobert O.. Perfect seed pairing is not a generally reliable predictor for miRNA-target interactions. Nat. Struct. Mol. Biol. 2006; 13:849–851. [DOI] [PubMed] [Google Scholar]
- 11. Thomson D.W., Bracken C.P., Goodall G.J.. Experimental strategies for microRNA target identification. Nucleic Acids Res. 2011; 39:6845–6853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kuhn D.E., Martin M.M., Feldman D.S., Terry A.V. Jr, Nuovo G.J., Elton T.S.. Experimental validation of miRNA targets. Methods. 2008; 44:47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lee Y.J., Kim V., Muth D.C., Witwer K.W.. Validated MicroRNA Target Databases: an evaluation. Drug Dev. Res. 2015; 76:389–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ule J., Jensen K., Mele A., Darnell R.B.. CLIP: a method for identifying protein-RNA interaction sites in living cells. Methods. 2005; 37:376–386. [DOI] [PubMed] [Google Scholar]
- 15. Hafner M., Landthaler M., Burger L., Khorshid M., Hausser J., Berninger P., Rothballer A., Ascano M. Jr, Jungkamp A.C., Munschauer M. et al. . Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010; 141:129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Helwak A., Kudla G., Dudnakova T., Tollervey D.. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell. 2013; 153:654–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hsu S.D., Lin F.M., Wu W.Y., Liang C., Huang W.C., Chan W.L., Tsai W.T., Chen G.Z., Lee C.J., Chiu C.M. et al. . miRTarBase: a database curates experimentally validated microRNA-target interactions. Nucleic Acids Res. 2011; 39:D163–D169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hsu S.D., Tseng Y.T., Shrestha S., Lin Y.L., Khaleel A., Chou C.H., Chu C.F., Huang H.Y., Lin C.M., Ho S.Y. et al. . miRTarBase update 2014: an information resource for experimentally validated miRNA-target interactions. Nucleic Acids Res. 2014; 42:D78–D85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chou C.H., Chang N.W., Shrestha S., Hsu S.D., Lin Y.L., Lee W.H., Yang C.D., Hong H.C., Wei T.Y., Tu S.J. et al. . miRTarBase 2016: updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Res. 2016; 44:D239–D247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Maglott D., Ostell J., Pruitt K.D., Tatusova T.. Entrez Gene: gene-centered information at NCBI. Nucleic Acids Res. 2011; 39:D52–D57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. O’Leary N.A., Wright M.W., Brister J.R., Ciufo S., Haddad D., McVeigh R., Rajput B., Robbertse B., Smith-White B., Ako-Adjei D. et al. . Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016; 44:D733–D745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Agarwal V., Bell G.W., Nam J.W., Bartel D.P.. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015; 4, doi:10.7554/eLife.05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Betel D., Koppal A., Agius P., Sander C., Leslie C.. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol. 2010; 11:R90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Barrett T., Wilhite S.E., Ledoux P., Evangelista C., Kim I.F., Tomashevsky M., Marshall K.A., Phillippy K.H., Sherman P.M., Holko M. et al. . NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. 2013; 41:D991–D995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Deng M., Bragelmann J., Schultze J.L., Perner S.. Web-TCGA: an online platform for integrated analysis of molecular cancer data sets. BMC Bioinformatics. 2016; 17:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tomczak K., Czerwinska P., Wiznerowicz M.. The Cancer Genome Atlas (TCGA): an immeasurable source of knowledge. Contemp Oncol (Pozn). 2015; 19:A68–A77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kanehisa M., Furumichi M., Tanabe M., Sato Y., Morishima K.. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017; 45:D353–D361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Huang da W., Sherman B.T., Lempicki R.A.. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009; 4:44–57. [DOI] [PubMed] [Google Scholar]
- 29. Luna J.M., Barajas J.M., Teng K.Y., Sun H.L., Moore M.J., Rice C.M., Darnell R.B., Ghoshal K.. Argonaute CLIP defines a deregulated miR-122-bound transcriptome that correlates with patient survival in human liver cancer. Mol. Cell. 2017; 67:400–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hamilton M.P., Rajapakshe K.I., Bader D.A., Cerne J.Z., Smith E.A., Coarfa C., Hartig S.M., McGuire S.E.. The landscape of microRNA targeting in prostate cancer defined by AGO-PAR-CLIP. Neoplasia (New York, N.Y.). 2016; 18:356–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Krell J., Stebbing J., Carissimi C., Dabrowska A.F., de Giorgio A., Frampton A.E., Harding V., Fulci V., Macino G., Colombo T. et al. . TP53 regulates miRNA association with AGO2 to remodel the miRNA-mRNA interaction network. Genome Res. 2016; 26:331–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Spengler R.M., Zhang X., Cheng C., McLendon J.M., Skeie J.M., Johnson F.L., Davidson B.L., Boudreau R.L.. Elucidation of transcriptome-wide microRNA binding sites in human cardiac tissues by Ago2 HITS-CLIP. Nucleic Acids Res. 2016; 44:7120–7131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chou C.H., Lin F.M., Chou M.T., Hsu S.D., Chang T.H., Weng S.L., Shrestha S., Hsiao C.C., Hung J.H., Huang H.D.. A computational approach for identifying microRNA-target interactions using high-throughput CLIP and PAR-CLIP sequencing. BMC Genomics. 2013; 14(Suppl. 1):S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Uren P.J., Bahrami-Samani E., Burns S.C., Qiao M., Karginov F.V., Hodges E., Hannon G.J., Sanford J.R., Penalva L.O., Smith A.D.. Site identification in high-throughput RNA-protein interaction data. Bioinformatics. 2012; 28:3013–3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Xie B., Ding Q., Han H., Wu D.. miRCancer: a microRNA-cancer association database constructed by text mining on literature. Bioinformatics. 2013; 29:638–644. [DOI] [PubMed] [Google Scholar]
- 36. Naeem H., Kuffner R., Csaba G., Zimmer R.. miRSel: automated extraction of associations between microRNAs and genes from the biomedical literature. BMC Bioinformatics. 2010; 11:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chang N.-W., Dai H.-J., Hsieh Y.-L., Hsu W.-L.. Bioinformatics and Bioengineering (BIBE), 2016 IEEE 16th International Conference on. 2016; IEEE; 79–86. [Google Scholar]
- 38. Chang Y.-C., Hsieh Y.-L., Chen C.-C., Hsu W.-L.. International Conference on Industrial, Engineering and Other Applications of Applied Intelligent Systems. 2014; Springer; 339–348. [Google Scholar]
- 39. Chang Y.-C., Chen C.-C., Hsieh Y.-L., Chen C.C., Hsu W.-L.. ACL (2). 2015; 775–780. [Google Scholar]
- 40. Chang Y.-C., Hsieh Y.-L., Chen C.-C., Hsu W.-L.. A semantic frame-based intelligent agent for topic detection. Soft Comput. 2017; 21:391–401. [Google Scholar]
- 41. Suzuki H.I., Young R.A., Sharp P.A.. Super-enhancer-mediated RNA processing revealed by integrative MicroRNA network analysis. Cell. 2017; 168:1000–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wu Y.E., Parikshak N.N., Belgard T.G., Geschwind D.H.. Genome-wide, integrative analysis implicates microRNA dysregulation in autism spectrum disorder. Nat. Neurosci. 2016; 19:1463–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Khan F.M., Marquardt S., Gupta S.K., Knoll S., Schmitz U., Spitschak A., Engelmann D., Vera J., Wolkenhauer O., Putzer B.M.. Unraveling a tumor type-specific regulatory core underlying E2F1-mediated epithelial-mesenchymal transition to predict receptor protein signatures. Nat. Commun. 2017; 8:198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rupaimoole R., Slack F.J.. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017; 16:203–222. [DOI] [PubMed] [Google Scholar]
- 45. Chen Q.L., Lu Y.Y., Peng J.H., Dong S., Wei B., Song Y.N., Zhou Q.M., Zhang H., Liu P., Su S.B.. Dynamical Regulation Analysis Identifies Molecular Mechanisms of Fuzheng-Huayu Formula against Hepatitis B-Caused Liver Cirrhosis. Evidence-based Complement. Alternative Med.: eCAM. 2015; 2015:238495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schmeier S., Schaefer U., Essack M., Bajic V.B.. Network analysis of microRNAs and their regulation in human ovarian cancer. Bmc Syst. Biol. 2011; 5: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yan W.Y., Qian L.J., Chen J.J., Chen W.C., Shen B.R.. Comparison of prognostic MicroRNA biomarkers in blood and tissues for gastric cancer. J. Cancer. 2016; 7:95–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cancer Genome Atlas Research, N., Albert Einstein College of, M., Analytical Biological, S., Barretos Cancer, H., Baylor College of, M., Beckman Research Institute of City of, H., Buck Institute for Research on, A., Canada's Michael Smith Genome Sciences, C., Harvard Medical, S. Helen F.G.C.C., et al. Integrated genomic and molecular characterization of cervical cancer. Nature. 2017; 543:378–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cancer Genome Atlas Research Network. Electronic address, a.a.d.h.e. and Cancer Genome Atlas Research, N. Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell. 2017; 32:185–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The Download page of miRTarBase provides the more comprehensive data for download that include: (1) All published miRNA target interaction data. (2) MTI PubMed Abstract Manually Curation Corpus (3) Catalog by Species of MTIs. (4) Catalog by Experimental Evidences of MTIs. We also provide all the previous version release in the ‘Previous Release’ page.