


Abstract
Genome-wide maps of chromatin states have become a powerful representation of genome annotation and regulatory activity. We collected public and in-house plant epigenomic data sets and applied a Hidden Markov Model to define chromatin states, which included 290 553 (36 chromatin states), 831 235 (38 chromatin states) and 3 936 844 (26 chromatin states) segments across the whole genome of Arabidopsis thaliana, Oryza sativa and Zea mays, respectively. We constructed a Plant Chromatin State Database (PCSD, http://systemsbiology.cau.edu.cn/chromstates) to integrate detailed information about chromatin states, including the features and distribution of states, segments in states and related genes with segments. The self-organization mapping (SOM) results for these different chromatin signatures and UCSC Genome Browser for visualization were also integrated into the PCSD database. We further provided differential SOM maps between two epigenetic marks for chromatin state comparison and custom tools for new data analysis. The segments and related genes in SOM maps can be searched and used for motif and GO analysis, respectively. In addition, multi-species integration can be used to discover conserved features at the epigenomic level. In summary, our PCSD database integrated the identified chromatin states with epigenetic features and may be beneficial for communities to discover causal functions hidden in plant chromatin.
INTRODUCTION
Chromatin is a genome-organizing platform, regulating gene expression, cell division and differentiation, etc. Epigenetic regulation, such as DNA methylation, histone modifications and variants, plays a key role in controlling chromatin structure. A combination of multiple epigenetic marks exist at the whole genome level, and specific combinations of epigenetic marks are meaningful in biological function (1). Epigenomic data sets are a rich resource to identify regulatory elements and functional annotations in coding and non-coding regions (2). The computational integration of high-throughput epigenomic profiling, called chromatin state maps, has become a powerful representation of genome annotation and regulatory activity (3–5). Chromatin states with epigenetic features impact gene activity in developmental processes and in response to environmental cues (6).
Chromatin states are identified by computing multi-dimensional matrices to interpret a variety of epigenomic data sets (4). Many algorithms have been developed to identify chromatin states, such as post hoc combination, principal component analysis (PCA), clustering, ChromHMM, chromstaR and Segway (5). Among these algorithms, ChromHMM, which is based on a Hidden Markov Model (7), is frequently used and has been successfully applied in many animals and plants, such as human (8,9), Drosophila (10) and barley (11). Ernst et al. used the ChromHMM algorithm to identify and analyse 51 chromatin states, including promoter-associated, transcription-associated, active intergenic, large-scale repressed and repeat-associated states, and revealed the genome-wide locations of diverse classes of epigenetic function (8). In addition to human and Drosophila, chromatin states have been widely studied in other animals, such as mouse (12) and worm (13). Compared with studies of chromatin states in animals, especially human, studies of chromatin states in plants are limited. There are several studies on chromatin state identification in Arabidopsis, rice and barley using ChIP-seq or ChIP-chip data sets with different algorithms (11,14–18). In these studies, epigenomic data were very limited, either in the type of epigenetic marks or in the number of epigenomic data sets, thus resulting in an incomplete definition of chromatin states. Furthermore, it is difficult to search and compare these chromatin states in plants.
Recently, public plant epigenomic data sets are emerging quickly, including DNase-seq, ATAC-seq, meDIP-seq, ChIP-seq and MNase-seq data. DNase-seq and ATAC-seq data were used to identify the regulatory DNA elements in Arabidopsis and rice (19–23). ChIP-seq data for various histone modifications, histone variants and TFs have accumulated in public databases for studies on the regulation of gene expression in developmental processes and in response to environmental treatments in Arabidopsis, rice and maize (24–33). In addition, our group also accumulated in-house epigenomic data sets, such as DNase-seq and ChIP-seq data for histone modifications and variants in Arabidopsis and rice (34–37). The abundance of epigenomic data sets enabled construction of a plant chromatin state database to better decode chromatin states with epigenomic data sets and to discover causal functions hidden in plant chromatin.
Here, we collected public and in-house epigenomic data sets for diverse epigenetic modifications to identify chromatin states based on a Hidden Markov Model in Arabidopsis thaliana, Oryza sativa and Zea mays. We constructed a Plant Chromatin State Database (PCSD), which contains search tools, analysis tools and the UCSC Genome Browser for visualization. The self-organization mapping (SOM) results for the different chromatin signatures were integrated in our PCSD database. The discovery and characterization of plant chromatin states may offer insights into the locations and functions of regulatory regions and genes in response to developmental and environmental signals.
MATERIALS AND METHODS
Integration of epigenomic data sets
We collected plant epigenomic data sets with next generation sequencing technology, including ChIP-seq, DNase-seq, meDIP-seq and MNase-seq; these data sets contain important information about histone modification regions, TF binding sites, DNA methylation regions, and accessible DNA regions on chromatin. These epigenomic data sets represent three species, A. thaliana, O. sativa and Z. mays, which have an abundance of publicly available data sets of different types. The public data sets were downloaded from NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) (38) and Sequence Read Archive (http://www.ncbi.nlm.nih.gov/sra) (39). The in-house data sets of Arabidopsis and rice were previously published (34–37).
Epigenomic data processing
FastQC software (Version 0.10.1) (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) was used for quality control, and low-quality reads were filtered by FASTX Toolkit (Version 0.0.13) (http://hannonlab.cshl.edu/fastx_toolkit/). Adaptor sequences were cut by cutadapt software (Version 1.4.1) (http://cutadapt.readthedocs.io/en/stable/). Bowtie2 software (Version 2.1.0) (40) was used to align sequencing reads to the reference genomes (TAIR10 in Arabidopsis, TIGR6.1 in rice and AGPv3.27 in maize) with the default parameters. The enriched regions were called by MACS software (Version 1.4.1) (41) (Figure 1A). The nomodel parameter was set, and the d-value parameter was set as 200. CEAS software (Version 1.0.2) (42) was used to analyse the distribution of epigenetic marks in the genome and the distance between the transcription start sites (TSSs) and transcription termination sites (TTSs) of genes and the nearest called peaks.
Figure 1.

An overview of PCSD. (A) Workflow of PCSD construction. We collected public and in-house epigenomic data sets in Arabidopsis thaliana, Oryza sativa and Zea mays. After quality control, we mapped the high-quality reads to reference genomes with bowtie2 software. The mapped reads were used for genome segmentation and definition of the chromatin states with ChromHMM. All segments were put into 1,350 units of an initialized SOM map. The trained SOM map was applied with ERANGE software according to the signal density, which was calculated by MACS software. The chromatin states and trained SOM maps were used for PCSD construction. (B) Homepage of PCSD. Information on the chromatin state and SOM map for a species can be searched by clicking on the species on the homepage. Basic functions, including search, analysis, UCSC view, download and help, can be quickly accessed in the navigation bar. (C) Search function of PCSD. In PCSD, users can search chromatin states by submitting locus ID and search detailed information about chromatin states by a state search. Each state is represented by a colour. The active states are represented by warm colours, whereas the repressed states are represented by cool colours. SOM search was used to search and compare different SOM maps. Epigenetic features can be displayed after an epigenetic data search. Then, users can find detailed information on segments and segment-related genes. The states marked by different colours and epigenetic mark signals around segments and genes are shown in the UCSC Genome Browser. To further investigate the function of segments and related genes, motif analysis and GO analysis were provided in PCSD, respectively. (D) Analysis functions of PCSD. In PCSD, users can conduct custom analysis using bigwig (BW) files or BED files to calculate the average signals or number of genomic regions in each chromatin state, respectively. The correlation with our collected data can be shown after uploading a custom BW file for epigenetic marks. The signal density in the custom BW file can be mapped to the trained SOM maps. Two different SOM maps can be compared by means of subtraction, addition, maximum, and minimum. Users can obtain detailed information on segments and related genes in the units of SOM maps, and motif and GO analysis were provided for functional analysis of segments and genes, respectively.
Plant chromatin state definition
The Hidden Markov Model was applied to aggregate or collapse the multi-dimensional matrices into a small number of chromatin states (8,9). ChromHMM software (Version 1.12) was used for genome segmentation and chromatin state definition (Figure 1A) (7) with a bin size set to 200 bp. The LearnModel program of ChromHMM was used to learn the chromatin state model and genome segmentation, and the numstates were initially set as ten to fifty states. The CompareModels program of ChromHMM was used to compare these learned models with different numbers of states to choose the best models (11). The OverlapEnrichment program of ChromHMM was used to calculate fold enrichments for chromatin states relative to genomic features and genic annotations, including coding gene region, TE region, intergenic region, centromere region, promoter, 5′UTR, exon, intron and 3′UTR.
The segment-related genes in each state were identified by the intersect program in BEDTools software (Version 2.17.0) (43). Genes, including their promoter and downstream region (1 kb upstream and downstream of genes in Arabidopsis, 2 kb for rice and maize), that overlapped with segments were identified as segment-related genes.
Self-organization mapping (SOM) training
Average signal values of epigenomic data in each segment were used for SOM training and were calculated by the bigWigAverageOverBed program of UCSC Genome Browser (44). ERANGE software (Version 3.3) was used for self-organizing map training (Figure 1A). The size of SOM maps was set as 30 by 45. The trails parameter was set as 10, and the timestep parameter was set as one-third of numbers of segments (45).
In SOM analysis for two epigenomic data sets, the average signal values of the epigenomic data in each segment were calculated by bigwigAverageOverBed program after uploading the BW file. Then, the new data were mapped to the trained SOM map by the mapsom program in ERANGE software.
Identification of top hits between different species
We used a BLAST algorithm to compare the protein sequences between Arabidopsis, rice and maize. The e-value was set as 1e–3. The top three genes in the blast results are provided in our database.
Custom analysis of chromatin state distribution
Users can provide genome-wide intensities (bigWig files) or functional genomic regions (BED files) to analyse the chromatin state distribution. The overlap between regions in the BED file and segments in chromatin states are calculated by the intersect program in BEDTools software. The number of overlapped regions in each state is used to calculate the chromatin state distribution. The average signals of segments in chromatin states are calculated by the bigWigAverageOverBed program, and then the average signals for chromatin state distribution in each state are calculated by the groupby program in BEDTools software.
Correlation analysis for epigenomic data sets
The Spearman's rank correlation coefficient is calculated by applying the plotCorrelation program in Deeptools software (Version 2.2.4) (46), and the heatmap of correlation is drawn by the R package.
Motif analysis
To find significantly enriched motifs in the segments of interest, we built a motif analysis tool with in-house codes following public methods (47,48). A total of 1035 motifs was integrated from the Plant Cis-acting Regulatory DNA Elements (PLACE) database (49), PlantCARE database (50), AthaMap database (51) and publications (23,52–56).
The significance test for enriched motifs in segments of interest is based on Z-scores and P-values. The Z-score and P-value of the scanned motifi are calculated by following formulas:
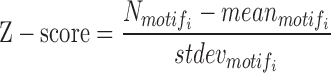 |
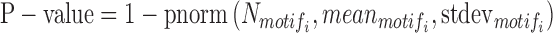 |
where 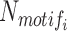 represents the number of occurrences of motifi in m segments of interest, and
represents the number of occurrences of motifi in m segments of interest, and 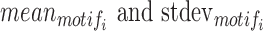 represent the mean and standard deviation of the number of detected occurrences of motifi in random 1000 surrogate sets of m segments in background, respectively, and pnorm() is the distribution function for the normal distribution in the R package (57–60). The motifs with P-value < 0.05 are considered significantly enriched in the segments of interest compared to background, blank state or all states, the choice of which can be selected by users.
represent the mean and standard deviation of the number of detected occurrences of motifi in random 1000 surrogate sets of m segments in background, respectively, and pnorm() is the distribution function for the normal distribution in the R package (57–60). The motifs with P-value < 0.05 are considered significantly enriched in the segments of interest compared to background, blank state or all states, the choice of which can be selected by users.
GO analysis
GO (Gene Ontology) analysis for genes of interest was provided by agriGOv2 (61) with suggested background and calculation methods. Three statistical methods were included: Fisher's exact (default), hypergeometric and χ2 tests. Multiple comparison correction methods can be used, including Benjamini–Yekutieli (default), Benjamini–Hochberg, Storey q-value and Holm methods (62).
Database implementation
The PCSD database was constructed on a standard LAMP (Linux+Apache+MySQL+PHP) system. The data set was stored in MySQL (www.mysql.com), and the web interface was built by PHP scripts (www.php.net) on Red Hat Linux powered by an Apache server (www.apache.org).
RESULTS
Database resources
We collected 216, 100 and 95 public and in-house epigenomic data sets and applied a Hidden Markov Model to define chromatin states in A. thaliana, O. sativa and Z. mays, respectively (Table 1 and Supplementary Table S1). These data sets included 19 in-house data sets comprised of 2 DNase-seq data sets under normal light condition and extended darkness in Arabidopsis and 17 histone modification and histone variant ChIP-seq data sets (H3K4me3, H3K4me2, H3K27ac, H3K27me3, H3K9ac and H2A.Z in callus or seedling) in rice (34–37). Before defining the chromatin states, we selected high confidence data to accurately build the chromatin state model: (i) we filtered out the data with low mapping rates; (ii) we performed a correlation analysis for these epigenomic data and filtered out the data located in an incorrect cluster that may not be consistent with their function (Supplementary Figure S1) and (iii) we trained the chromatin state model and deleted data with weak signals and inconsistent replication data. Finally, we define chromatin states, which included 290 553 (36 chromatin states from 216 data sets), 831 235 (38 chromatin states from 100 data sets) and 3 936 844 (26 chromatin states from 95 data sets) segments across the whole genome in A. thaliana, O. sativa and Z. mays, respectively (Figure 2 and Supplementary Figure S2). Different chromatin states contained different epigenetic features. In A. thaliana, chromatin states 16–24 are preferentially located in promoter and 5′UTR regions, which are enriched in DHSs and active histone modifications, such as H3K27ac and H3K4me3; in contrast, chromatin states 32–34 are preferentially located in TE regions, which are enriched in DNA methylation and repressed histone modifications, such as H3K9me2 and H3K27me1. The related epigenetic marks were classified into similar states. For example, LHP1 has high emission parameters in states where H3K27me3 also has high emission parameters (Figure 2A). Our chromatin state analysis results show that LHP1 binds to the genomic regions associated with H3K27me3, reflecting the interaction of LHP1 with H3K27me3 (63).
Table 1. Statistics on the epigenomic data used in PCSD.
| Data type | Arabidopsis thaliana | Oryza sativa | Zea mays |
|---|---|---|---|
| DNA methylation | 5 | 9 | 4 |
| Histone acetylation | 17 | 12 | 12 |
| Histone 3 lysine 36 methylation | 1 | 7 | 4 |
| Histone 3 lysine 4 trimethylation | 13 | 12 | 13 |
| Histone 3 lysine 4 dimethylation | 5 | 2 | 0 |
| Histone 3 lysine 4 monomethylation | 7 | 0 | 0 |
| Histone 3 lysine 27 methylation | 8 | 11 | 7 |
| Histone 3 lysine 9 methylation | 13 | 3 | 4 |
| Histone variant | 14 | 6 | 0 |
| MNase | 0 | 5 | 25 |
| Accessible DNA regions | 44 | 19 | 0 |
| Chromatin associated factor | 27 | 3 | 14 |
| Transcription factor | 62 | 11 | 12 |
Figure 2.

Heatmaps for emission parameters (A) and fold enrichment of genomic elements (B) in Arabidopsis thaliana.
In the chromatin state model, we can find the association between preferential location and preferential epigenetic marks of a state. For example, state 1 and state 2 in Arabidopsis are preferentially located at 3′UTR; state 1 is preferentially enriched in H3.3 and state 2 is enriched in not only H3.3 but also histone acetylation (Figure 2). This observation suggests that H3.3 and histone acetylation may influence the 3′UTR region. To investigate these epigenetic features, we divided the genome into six subclasses, namely, promoter, 5′UTR, 3′UTR, coding exon, intron, and intergenic regions, to analyse the distribution of epigenomic data. Furthermore, average signal profiles around TSS and TTS and meta-gene profiles along generic regions for every epigenetic mark were also generated using the normalized density. In addition, we investigated the distance between the called peaks of epigenomic data and TSS/TTS to explore the related genes for epigenetic marks.
To further effectively integrate, visualize and mine diverse epigenomic data sets, the self-organization mapping (SOM) results for these different chromatin signatures were integrated into our plant chromatin state database (PCSD). The segments with different chromatin signatures were mapped to trained SOM maps with 1350 units. Each unit contained segments that were defined as similar states with similar chromatin signatures.
Database construction
We constructed a plant chromatin state database (PCSD) to integrate information on chromatin states, SOM maps and epigenomic data features of three species, namely, A. thaliana, O. sativa and Z. mays. The basic information on chromatin states in these species can be searched by clicking the species name on the homepage (Figure 1B). On each species page, emission parameters of epigenetic marks and fold enrichment of genomic features in every state are shown (Figure 2 and Supplementary Figure S2). In addition, a table showing preferential epigenetic marks and the preferential location of each state on the species pages was marked with a specific colour for each state. The active states are represented by warm colours, whereas the repressed states are represented by cool colours. The segment counts and nucleotide counts of SOM maps in each species are also shown on the species pages.
A variety of search and analysis tools are provided in PCSD. Users can obtain chromatin states, SOM maps, and epigenetic features by ‘Locus ID search’, ‘State search’, ‘SOM search’ and ‘Epigenetic data search’ tools. The obtained segments and related genes can be used for functional analysis, including motif and GO enrichment analysis, respectively (Figure 1C). Users can conduct custom analyses using genome-wide intensities (bigWig files, BW files) or functional genomic regions (BED files) for the chromatin state distribution, correlations with our collected data, and comparative SOM training maps by ‘BED file analysis’, ‘BW file analysis’, and ‘SOM analysis’ tools (Figure 1D). Functional analysis tools, including GO analysis by agriGOv2 (61) and motif analysis, are also integrated. A BLAST tool is provided to find the similar sequences in Arabidopsis, rice and maize. Also, visualization of the chromatin states and associations with genes and epigenetic marks in the genome are shown in the UCSC Genome Browser (44). From the download page, users can obtain all locations of segments, information for related genes in each state, and the BW file that is used in our database.
Search function
A Locus ID search can be used to search the associated states in promoters, gene bodies and downstream of query genes. The top hits between A. thaliana, O. sativa and Z. mays are provided in the result of the locus ID search to compare chromatin features among different species. The associated epigenetic marks and distance between query genes and marks are also shown in the results of the locus ID search. The searched genes can link to UCSC Genome Browser for detailed visualization (Figure 3F, Supplementary Figures S3 and S5E). Taking the AGO4 gene as an example, we found that the chromatin states around Arabidopsis AGO4 were similar to those of rice AGO4 through visualization in the UCSC Genome Browser (Supplementary Figure S3A and B). The promoter region of the AGO4 genes are both located in states with active marks, such as DHS, H3K4me3, H3K9ac and H3K27ac, and the gene body region of the AGO4 genes are both located in H3K36me3.
Figure 3.

An example of analysis and comparison for H3K27me3 (SRX853860) and LHP1 (SRX1519404) in chromatin states and SOM maps. (A) Emission parameters of H3K27me3 and LHP1 in 36 states. (B) SOM maps of H3K27me3 and LHP1 by SOM search. (C) SOM comparison between the two SOM maps in (B). The compared SOM map is the result of the minimum between H3K27me3 and LHP1 to explore the overlapping units. The red ellipse encompasses the main units with common signals between H3K27me3 and LHP1. (D) Functional analysis for segments in units with a common signal form the SOM map in (C). Selected enriched motifs (‘GAGAGAGAGAGAGAGA’ and ‘CCTCGT’) in the segments are shown. (E) Functional analysis of related genes in units with a common signal from the SOM map in (C). Selected enriched GO terms (GO terms related to flower development) in related genes are shown. (F) Selected genes (FLC and AP1) used for GO analysis associated with chromatin states and epigenetic marks in the UCSC Genome Browser.
State search is for searching the features of chromatin states, including emission parameters of epigenetic marks and fold enrichment of genomic features. Epigenetic data search is provided to search the results of the processed epigenomic data sets, including the data name and source, and the emission parameters in every state (Figure 3A).
SOM map search can be used to search the SOM maps for epigenetic marks (Figure 3B and Supplementary Figures S4A and S5A). In the SOM maps of epigenetic marks, the segments with chromatin states and related genes in every unit can be obtained by clicking the scores of the corresponding unit in the SOM map table, which is shown in the results of the SOM search. SOM compare is also provided for users to compare the common and different units between two SOM maps (Figure 3C and Supplementary Figures S4B and S5B). Four comparative operations can be selected, i.e.: subtraction (SUB), addition (ADD), maximum (MAX) and minimum (MIN).
The state search can provide segments that belong to one state, and the SOM map search can provide segments in one unit. The segments can be sent to motif analysis by clicking the ‘motif analysis’ button on the pages. The state search, SOM search and epigenetic data search can also provide the related genes, which can be sent to GO analysis by clicking the ‘GO analysis’ button on the pages.
Analysis tools
In PCSD, we provided custom analysis tools for new epigenomic data sets based on chromatin state information, including analysis for distribution and correlation with the collected data (Figure 1D). Different calculation methods and different analysis results are provided according to the type of uploaded data. When a BED file with functional genomic regions is uploaded, the number of regions in each state is calculated and the result is shown in a heatmap, which displays the distribution of chromatin states for the BED file. When a BW file with genome-wide intensities is uploaded, the average signal in each state is calculated and the result is also shown in a heatmap. In addition, the correlation with these collected data can be calculated with this BW file. Users can select one or more types of epigenomic data to analyse the correlation with custom data.
We also provided custom SOM analysis by uploading BW files in PCSD. The signal density from the custom BW file can be mapped to our trained SOM map to show units with high signals. The custom SOM map can be compared with these trained SOM maps in PCSD. Users can also compare two custom SOM maps or compare a custom SOM map with an existing SOM map in PCSD. The operations for comparison are subtraction (SUB), addition (ADD), maximum (MAX) and minimum (MIN). The segments of interest and related genes in the compared SOM maps can be sent for motif analysis and GO analysis, respectively.
Functional analysis tools, including motif analysis and GO enrichment analysis, are provided for users to explore the potential functions of segments and related genes. A BLAST tool is also provided for users to analyse sequence similarity in Arabidopsis, rice and maize.
Visualization in UCSC Genome Browser
In PCSD, visualization of chromatin states and associations with genes and epigenetic marks in the genome are shown in the UCSC Genome Browser (Figure 1C, Figure 3F and Supplementary Figure S3). In every search result, the segments and related genes are shown with direct links to their display in the UCSC Genome Browser. The segments are marked by the defined colors for chromatin states to show the relationship among chromatin states, genes and epigenetic marks.
Functional applications
Here, we show several examples to demonstrate how to use PCSD for epigenomic data analysis. In the Arabidopsis chromatin states, we found that LHP1 and H3K27me3 were located in similar states (Figure 2A). In the epigenetic data search, we found that both LHP1 and H3K27me3 have high emission parameters in states 11–15 (Figure 3A). These results are consistent with studies on the interactions between the LHP1 protein and H3K27me3 (63). The units with higher signals were also similar in the SOM maps of H3K27me3 and LHP1 (Figure 3B). The common units were generated by the MIN operation in the SOM compare tools based on the minimum score between H3K27me3 and LHP1, and they were displayed in the compared SOM map (Figure 3C). Motif analysis of the segments in common units showed that the ‘GAGAGAGAGAGAGAGAGA’ (GAGAMGSA1) motif was significantly enriched (Figure 3D and Supplementary Table S2). This result is consistent with a previous study that found that GAGAGA motifs are enriched in LHP1 and H3K27me3 target loci (64). In addition, some motifs related to flower development are also enriched, such as the ‘CCTCGT’ motif (TOE1–1/TOE2–1) (53) (Figure 3D and Supplementary Table S2). The GO terms related to flower development were significantly enriched in the genes within common units (Figure 3E and Supplementary Table S3). The genes associated with flower development, such as FLC and AP1, were located in states that were preferentially enriched in H3K27me3 (Figure 3F), which is consistent with a previous study that maintenance of FLC repression is associated with the deposition of H3K27me3 (65).
We discovered an interesting result about the distribution of histone variant H2A.Z in the Arabidopsis genome through a similar SOM map comparison tool. As shown in Supplementary Figure S4A, the left regions of H2A.Z’s SOM map have strong signals for H3K4me3, and the right top regions of H2A.Z’s SOM maps have strong signals for H3K27me3. By the MIN operation in the SOM compare tools, we obtained the common units between H2A.Z and these two histone modifications, H3K4me3 (Supplementary Figure S4B) and H3K27me3 (Supplementary Figure S4C). These new findings suggested that H2A.Z might have relationships with both active mark H3K4me3 and repressive mark H3K27me3 in different genome regions. Though GO analysis, we found that the segment-related genes in main common units between H2A.Z and H3K4me3 were enriched in GO terms related to meiotic chromosome segregation, DNA-dependent DNA replication, histone H3-K9 methylation and DNA methylation (Supplementary Figure S4D and Table S3) and that the segment-related genes in the main common units between H2A.Z and H3K27me3 were enriched in GO terms related to oxidation-reduction process, cell wall modification and anatomical structure morphogenesis (Supplementary Figure S4E and Table S3).
In addition to comparing the common units between two SOM maps, users can also compare the differential units between two SOM maps. For example, two SOM maps, DHSs under normal condition and extended darkness, have similar units with high DHSs signals (Supplementary Figure S5A). We can investigate the differential units by the SUB operation in the SOM compare tools (Supplementary Figure S5B). Photosynthesis-associated motifs and GO terms are enriched in main differential units by functional analysis (Supplementary Figure S5C–E and Tables S2, S3), which is consistent with our previous study (37).
DISCUSSION
The epigenome is a complete set of epigenetic marks at every genomic position. Epigenomic data sets are a rich resource for understanding genome activity in both genes and regulatory regions of chromatin. Here, we applied established procedures using ChromHMM (7–9) and SOM maps (45) to combine epigenetic modifications and define 36, 38 and 26 chromatin states in A. thaliana, O. sativa and Z. mays, respectively. Then, we employed SOM maps with 1350 units to integrate, visualize, and mine diverse epigenomic data sets to cluster segments with different chromatin signatures. We further constructed a plant chromatin state database (PCSD) to integrate these epigenomic signatures.
In the chromatin states, genes with different degrees of activity were identified according to these collected epigenetic marks distinguished by different colours. It is convenient to identify the activity of genes and nearby regions with visualization in the UCSC Genome Browser either for homologous genes in one species (Supplementary Figure S3A, C and D) or for orthologous genes in different species (Supplementary Figure S3A and B). Through a comparison of chromatin states between paralogues/orthologues, the conservation and divergence of evolution can be revealed at an epigenetic level.
In addition to the chromatin state explorer, PCSD also provides tools to compare epigenomic data sets. The same epigenetic mark can be compared in different conditions. For example, DNase-seq data in extended darkness and control conditions can be compared by the SUB operation in SOM compare tools (Supplementary Figure S5B). In addition, the common units can also be compared in SOM maps between two epigenetic marks located in similar positions. For example, the interaction between LHP1 and H3K27me3 (63) shows that the two epigenetic marks are located in similar units in the SOM maps (Figure 3B). The common units can be obtained by the MIN operation in SOM compare tools (Figure 3C). Furthermore, the SOM comparison tool in PCSD might allow us to discover something new. Histone variant H2A.Z is known to be one of the most conserved, but enigmatic, histone variants, and it has been implicated in a variety of chromosomal processes. In plants, the relationship between H2A.Z at the TSS and transcription appears to be roughly parabolic, and H2A.Z deposited in the bodies of genes negatively correlates with transcription (66,67). Interestingly, we found that the SOM map of H2A.Z shows strong overlap with both active histone methylation mark H3K4me3 and repressive marker H3K27me3 (Supplementary Figure S4). Our SOM comparison results might be related to the dual function of plant H2A.Z in transcriptional regulation.
There are still some limitations to PCSD. The type and number of publicly available epigenomic data sets for rice and maize are insufficient. We have supplemented in-house data for histone modifications and variants in rice, but many more epigenomic data sets in maize are still needed, such as DNase-seq and histone variant data. In recent studies, the chromatin interactions between genes and regulatory elements were associated with epigenetic marks (68). New data types to study chromatin structure, such as chromosome conformation capture (3C) and Hi-C technologies, have been developed, and they should be integrated into our database in the future. We will continue to update PCSD with more types of data and greater amounts of data sets and will add other plants with an accumulation of epigenomic data sets, such as cotton (69,70), and tomato (71,72).
In summary, we have identified chromatin states in plants and present a plant chromatin state database, PCSD (http://systemsbiology.cau.edu.cn/chromstates). In PCSD, users can search chromatin states, SOM maps, and epigenetic features with the UCSC Genome Browser for detailed visualization. Functional analysis tools, such as motif and GO analysis, are provided for annotating segments and related genes, respectively. In addition, analysis tools are provided for custom data sets, including distributions of chromatin states, correlations with epigenomic data, SOM map analysis and comparisons, and BLAST tools. The decoded chromatin states could provide a powerful approach for functional annotation of the plant genome, and they might reveal detailed regions of diverse classes of epigenetic functions. Identification of the activity of genes and regions may guide genome editing and epigenome editing technologies, such as CRISPR/Cas9. Multi-species integration can be used to compare epigenetic patterns across plant species and to discover conservation and divergence between species at an epigenomic level. Our PCSD database might be used as a template for chromatin states in other plants, and it contributes to the construction of a plant ENCODE system, such as the pENCODE system (73). We hope that our PCSD database will be beneficial to plant and epigenetic research communities.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [31371291, 31771467, 31571360]; Ministry of Science and Technology of China [2013CBA01400]. Funding for open access charge: National Natural Science Foundation of China [31371291, 31771467, 31571360]; Ministry of Science and Technology of China [2013CBA01400].
Conflict of interest statement. None declared.
REFERENCES
- 1. Strahl B.D., Allis C.D.. The language of covalent histone modifications. Nature. 2000; 403:41–45. [DOI] [PubMed] [Google Scholar]
- 2. Consortium E.P. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cuvier O., Fierz B.. Dynamic chromatin technologies: from individual molecules to epigenomic regulation in cells. Nat. Rev. Genet. 2017; 18:457–472. [DOI] [PubMed] [Google Scholar]
- 4. Stricker S.H., Koferle A., Beck S.. From profiles to function in epigenomics. Nat. Rev. Genet. 2017; 18:51–66. [DOI] [PubMed] [Google Scholar]
- 5. Taudt A., Colome-Tatche M., Johannes F.. Genetic sources of population epigenomic variation. Nat. Rev. Genet. 2016; 17:319–332. [DOI] [PubMed] [Google Scholar]
- 6. Xiao J., Jin R., Wagner D.. Developmental transitions: integrating environmental cues with hormonal signaling in the chromatin landscape in plants. Genome Biol. 2017; 18:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ernst J., Kellis M.. ChromHMM: automating chromatin-state discovery and characterization. Nat. Methods. 2012; 9:215–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ernst J., Kellis M.. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat. Biotechnol. 2010; 28:817–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ernst J., Kheradpour P., Mikkelsen T.S., Shoresh N., Ward L.D., Epstein C.B., Zhang X., Wang L., Issner R., Coyne M. et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011; 473:43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kharchenko P.V., Alekseyenko A.A., Schwartz Y.B., Minoda A., Riddle N.C., Ernst J., Sabo P.J., Larschan E., Gorchakov A.A., Gu T. et al. Comprehensive analysis of the chromatin landscape in Drosophila melanogaster. Nature. 2011; 471:480–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Baker K., Dhillon T., Colas I., Cook N., Milne I., Milne L., Bayer M., Flavell A.J.. Chromatin state analysis of the barley epigenome reveals a higher-order structure defined by H3K27me1 and H3K27me3 abundance. Plant J. 2015; 84:111–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Eng D., Vogel W.K., Flann N.S., Gross M.K., Kioussi C.. Genome-wide mapping of chromatin state of mouse forelimbs. Open Access Bioinformatics. 2014; 6:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu T., Rechtsteiner A., Egelhofer T.A., Vielle A., Latorre I., Cheung M.S., Ercan S., Ikegami K., Jensen M., Kolasinska-Zwierz P. et al. Broad chromosomal domains of histone modification patterns in C. elegans. Genome Res. 2011; 21:227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li X., Wang X., He K., Ma Y., Su N., He H., Stolc V., Tongprasit W., Jin W., Jiang J. et al. High-resolution mapping of epigenetic modifications of the rice genome uncovers interplay between DNA methylation, histone methylation, and gene expression. Plant cell. 2008; 20:259–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Luo C., Sidote D.J., Zhang Y., Kerstetter R.A., Michael T.P., Lam E.. Integrative analysis of chromatin states in Arabidopsis identified potential regulatory mechanisms for natural antisense transcript production. Plant J. 2013; 73:77–90. [DOI] [PubMed] [Google Scholar]
- 16. Roudier F., Ahmed I., Berard C., Sarazin A., Mary-Huard T., Cortijo S., Bouyer D., Caillieux E., Duvernois-Berthet E., Al-Shikhley L. et al. Integrative epigenomic mapping defines four main chromatin states in Arabidopsis. EMBO J. 2011; 30:1928–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sequeira-Mendes J., Araguez I., Peiro R., Mendez-Giraldez R., Zhang X., Jacobsen S.E., Bastolla U., Gutierrez C.. The functional topography of the Arabidopsis genome is organized in a reduced number of linear motifs of chromatin states. Plant Cell. 2014; 26:2351–2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang C., Liu C., Roqueiro D., Grimm D., Schwab R., Becker C., Lanz C., Weigel D.. Genome-wide analysis of local chromatin packing in Arabidopsis thaliana. Genome Res. 2015; 25:246–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lu Z., Hofmeister B.T., Vollmers C., DuBois R.M., Schmitz R.J.. Combining ATAC-seq with nuclei sorting for discovery of cis-regulatory regions in plant genomes. Nucleic Acids Res. 2017; 45:e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sullivan A.M., Arsovski A.A., Lempe J., Bubb K.L., Weirauch M.T., Sabo P.J., Sandstrom R., Thurman R.E., Neph S., Reynolds A.P. et al. Mapping and dynamics of regulatory DNA and transcription factor networks in A. thaliana. Cell Rep. 2014; 8:2015–2030. [DOI] [PubMed] [Google Scholar]
- 21. Wilkins O., Hafemeister C., Plessis A., Holloway-Phillips M.M., Pham G.M., Nicotra A.B., Gregorio G.B., Jagadish S.V., Septiningsih E.M., Bonneau R. et al. EGRINs (Environmental Gene Regulatory Influence Networks) in rice that function in the response to water deficit, high temperature, and agricultural environments. Plant Cell. 2016; 28:2365–2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang W., Wu Y., Schnable J.C., Zeng Z., Freeling M., Crawford G.E., Jiang J.. High-resolution mapping of open chromatin in the rice genome. Genome Res. 2012; 22:151–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang W., Zhang T., Wu Y., Jiang J.. Genome-wide identification of regulatory DNA elements and protein-binding footprints using signatures of open chromatin in Arabidopsis. Plant Cell. 2012; 24:2719–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brusslan J.A., Bonora G., Rus-Canterbury A.M., Tariq F., Jaroszewicz A., Pellegrini M.. A genome-wide chronological study of gene expression and two histone modifications, H3K4me3 and H3K9ac, during developmental leaf senescence. Plant Physiol. 2015; 168:1246–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. He G., Chen B., Wang X., Li X., Li J., He H., Yang M., Lu L., Qi Y., Wang X. et al. Conservation and divergence of transcriptomic and epigenomic variation in maize hybrids. Genome Biol. 2013; 14:R57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. He G., Zhu X., Elling A.A., Chen L., Wang X., Guo L., Liang M., He H., Zhang H., Chen F. et al. Global epigenetic and transcriptional trends among two rice subspecies and their reciprocal hybrids. Plant cell. 2010; 22:17–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Inagaki S., Takahashi M., Hosaka A., Ito T., Toyoda A., Fujiyama A., Tarutani Y., Kakutani T.. Gene-body chromatin modification dynamics mediate epigenome differentiation in Arabidopsis. EMBO J. 2017; 36:970–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ko D.K., Rohozinski D., Song Q., Taylor S.H., Juenger T.E., Harmon F.G., Chen Z.J.. Temporal shift of circadian-mediated gene expression and carbon fixation contributes to biomass heterosis in maize hybrids. PLoS Genet. 2016; 12:e1006197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu X., Zhou S., Wang W., Ye Y., Zhao Y., Xu Q., Zhou C., Tan F., Cheng S., Zhou D.X.. Regulation of histone methylation and reprogramming of gene expression in the rice inflorescence meristem. Plant Cell. 2015; 27:1428–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pedmale U.V., Huang S.S., Zander M., Cole B.J., Hetzel J., Ljung K., Reis P.A., Sridevi P., Nito K., Nery J.R. et al. Cryptochromes interact directly with PIFs to control plant growth in limiting blue light. Cell. 2016; 164:233–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang X., Elling A.A., Li X., Li N., Peng Z., He G., Sun H., Qi Y., Liu X.S., Deng X.W.. Genome-wide and organ-specific landscapes of epigenetic modifications and their relationships to mRNA and small RNA transcriptomes in maize. Plant Cell. 2009; 21:1053–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wollmann H., Holec S., Alden K., Clarke N.D., Jacques P.E., Berger F.. Dynamic deposition of histone variant H3.3 accompanies developmental remodeling of the Arabidopsis transcriptome. PLoS Genet. 2012; 8:e1002658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zong W., Tang N., Yang J., Peng L., Ma S., Xu Y., Li G., Xiong L.. Feedback regulation of ABA signaling and biosynthesis by a bZIP transcription factor targets drought-resistance-related genes. Plant Physiol. 2016; 171:2810–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang K., Xu W., Wang C., Yi X., Zhang W., Su Z.. Differential deposition of H2A.Z in combination with histone modifications within related genes in Oryza sativa callus and seedling. Plant J. 2017; 89:264–277. [DOI] [PubMed] [Google Scholar]
- 35. Zhang K., Xu W., Wang C., Yi X., Su Z.. Differential deposition of H2A.Z in rice seedling tissue during the day-night cycle. Plant Signal. Behav. 2017; 12:e1286438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Du Z., Li H., Wei Q., Zhao X., Wang C., Zhu Q., Yi X., Xu W., Liu X.S., Jin W. et al. Genome-wide analysis of histone modifications: H3K4me2, H3K4me3, H3K9ac, and H3K27ac in Oryza sativa L. Japonica. Mol. Plant. 2013; 6:1463–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liu Y., Zhang W., Zhang K., You Q., Yan H., Jiao Y., Jiang J., Xu W., Su Z.. Genome-wide mapping of DNase I hypersensitive sites reveals chromatin accessibility changes in Arabidopsis euchromatin and heterochromatin regions under extended darkness. Scientific Rep. 2017; 7:4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Barrett T., Wilhite S.E., Ledoux P., Evangelista C., Kim I.F., Tomashevsky M., Marshall K.A., Phillippy K.H., Sherman P.M., Holko M. et al. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. 2013; 41:D991–D995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kodama Y., Shumway M., Leinonen R., International Nucleotide Sequence Database C.. The Sequence Read Archive: explosive growth of sequencing data. Nucleic Acids Res. 2012; 40:D54–D56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Langmead B., Salzberg S.L.. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008; 9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shin H., Liu T., Manrai A.K., Liu X.S.. CEAS: cis-regulatory element annotation system. Bioinformatics. 2009; 25:2605–2606. [DOI] [PubMed] [Google Scholar]
- 43. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D.. The human genome browser at UCSC. Genome Res. 2002; 12:996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mortazavi A., Pepke S., Jansen C., Marinov G.K., Ernst J., Kellis M., Hardison R.C., Myers R.M., Wold B.J.. Integrating and mining the chromatin landscape of cell-type specificity using self-organizing maps. Genome Res. 2013; 23:2136–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ramirez F., Dundar F., Diehl S., Gruning B.A., Manke T.. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014; 42:W187–W191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nemhauser J.L., Mockler T.C., Chory J.. Interdependency of brassinosteroid and auxin signaling in Arabidopsis. PLoS Biol. 2004; 2:E258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Michael T.P., Mockler T.C., Breton G., McEntee C., Byer A., Trout J.D., Hazen S.P., Shen R., Priest H.D., Sullivan C.M. et al. Network discovery pipeline elucidates conserved time-of-day-specific cis-regulatory modules. PLoS Genet. 2008; 4:e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Higo K., Ugawa Y., Iwamoto M., Korenaga T.. Plant cis-acting regulatory DNA elements (PLACE) database: 1999. Nucleic Acids Res. 1999; 27:297–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rombauts S., Dehais P., Van Montagu M., Rouze P.. PlantCARE, a plant cis-acting regulatory element database. Nucleic Acids Res. 1999; 27:295–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hehl R., Bulow L.. AthaMap web tools for the analysis of transcriptional and posttranscriptional regulation of gene expression in Arabidopsis thaliana. Methods Mol. Biol. 2014; 1158:139–156. [DOI] [PubMed] [Google Scholar]
- 52. Bolduc N., Yilmaz A., Mejia-Guerra M.K., Morohashi K., O’Connor D., Grotewold E., Hake S.. Unraveling the KNOTTED1 regulatory network in maize meristems. Genes Dev. 2012; 26:1685–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Franco-Zorrilla J.M., Lopez-Vidriero I., Carrasco J.L., Godoy M., Vera P., Solano R.. DNA-binding specificities of plant transcription factors and their potential to define target genes. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:2367–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Konishi M., Yanagisawa S.. Identification of a nitrate-responsive cis-element in the Arabidopsis NIR1 promoter defines the presence of multiple cis-regulatory elements for nitrogen response. Plant J. 2010; 63:269–282. [DOI] [PubMed] [Google Scholar]
- 55. Ramireddy E., Brenner W.G., Pfeifer A., Heyl A., Schmulling T.. In planta analysis of a cis-regulatory cytokinin response motif in Arabidopsis and identification of a novel enhancer sequence. Plant Cell Physiol. 2013; 54:1079–1092. [DOI] [PubMed] [Google Scholar]
- 56. Xuan Y.H., Priatama R.A., Huang J., Je B.I., Liu J.M., Park S.J., Piao H.L., Son D.Y., Lee J.J., Park S.H. et al. Indeterminate domain 10 regulates ammonium-mediated gene expression in rice roots. New Phytol. 2013; 197:791–804. [DOI] [PubMed] [Google Scholar]
- 57. Tian T., You Q., Zhang L., Yi X., Yan H., Xu W., Su Z.. SorghumFDB: sorghum functional genomics database with multidimensional network analysis. Database. 2016; 2016:baw099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. You Q., Xu W., Zhang K., Zhang L., Yi X., Yao D., Wang C., Zhang X., Zhao X., Provart N.J. et al. ccNET: database of co-expression networks with functional modules for diploid and polyploid Gossypium. Nucleic Acids Res. 2017; 45:D1090–D1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. You Q., Zhang L., Yi X., Zhang K., Yao D., Zhang X., Wang Q., Zhao X., Ling Y., Xu W. et al. Co-expression network analyses identify functional modules associated with development and stress response in Gossypium arboreum. Scientific Rep. 2016; 6:38436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yu J., Zhang Z., Wei J., Ling Y., Xu W., Su Z.. SFGD: a comprehensive platform for mining functional information from soybean transcriptome data and its use in identifying acyl-lipid metabolism pathways. BMC Genomics. 2014; 15:271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Tian T., Liu Y., Yan H., You Q., Yi X., Du Z., Xu W., Su Z.. agriGO v2.0: a GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017; doi:10.1093/nar/gkx382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Du Z., Zhou X., Ling Y., Zhang Z., Su Z.. agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res. 2010; 38:W64–W70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Veluchamy A., Jegu T., Ariel F., Latrasse D., Mariappan K.G., Kim S.K., Crespi M., Hirt H., Bergounioux C., Raynaud C. et al. LHP1 regulates H3K27me3 spreading and shapes the three-dimensional conformation of the Arabidopsis genome. PLoS One. 2016; 11:e0158936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hecker A., Brand L.H., Peter S., Simoncello N., Kilian J., Harter K., Gaudin V., Wanke D.. The Arabidopsis GAGA-binding factor basic pentacysteine6 recruits the polycomb-repressive complex1 component like heterochromatin protein1 to GAGA DNA motifs. Plant Physiol. 2015; 168:1013–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Buzas D.M., Robertson M., Finnegan E.J., Helliwell C.A.. Transcription-dependence of histone H3 lysine 27 trimethylation at the Arabidopsis polycomb target gene FLC. Plant J. 2011; 65:872–881. [DOI] [PubMed] [Google Scholar]
- 66. Coleman-Derr D., Zilberman D.. Deposition of histone variant H2A.Z within gene bodies regulates responsive genes. PLOS Genet. 2012; 8:e1002988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zilberman D., Coleman-Derr D., Ballinger T., Henikoff S.. Histone H2A.Z and DNA methylation are mutually antagonistic chromatin marks. Nature. 2008; 456:125–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Liu C., Wang C., Wang G., Becker C., Zaidem M., Weigel D.. Genome-wide analysis of chromatin packing in Arabidopsis thaliana at single-gene resolution. Genome Res. 2016; 26:1057–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wang M., Tu L., Lin M., Lin Z., Wang P., Yang Q., Ye Z., Shen C., Li J., Zhang L. et al. Asymmetric subgenome selection and cis-regulatory divergence during cotton domestication. Nat. Genet. 2017; 49:579–587. [DOI] [PubMed] [Google Scholar]
- 70. Zheng D., Ye W., Song Q., Han F., Zhang T., Chen Z.J.. Histone modifications define expression bias of homoeologous genomes in allotetraploid cotton. Plant Physiol. 2016; 172:1760–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ricardi M.M., Gonzalez R.M., Zhong S., Dominguez P.G., Duffy T., Turjanski P.G., Salgado Salter J.D., Alleva K., Carrari F., Giovannoni J.J. et al. Genome-wide data (ChIP-seq) enabled identification of cell wall-related and aquaporin genes as targets of tomato ASR1, a drought stress-responsive transcription factor. BMC Plant Biol. 2014; 14:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zhong S., Fei Z., Chen Y.R., Zheng Y., Huang M., Vrebalov J., McQuinn R., Gapper N., Liu B., Xiang J. et al. Single-base resolution methylomes of tomato fruit development reveal epigenome modifications associated with ripening. Nat. Biotechnol. 2013; 31:154–159. [DOI] [PubMed] [Google Scholar]
- 73. Lane A.K., Niederhuth C.E., Ji L., Schmitz R.J.. pENCODE: a plant encyclopedia of DNA elements. Annu. Rev. Genet. 2014; 48:49–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.