


Abstract
Exosomes, which are nanosized endocytic vesicles that are secreted by most cells, contain an abundant cargo of different RNA species that can modulate the behavior of recipient cells and may be used as circulating biomarkers for diseases. Here, we develop a web-accessible database (http://www.exoRBase.org), exoRBase, which is a repository of circular RNA (circRNA), long non-coding RNA (lncRNA) and messenger RNA (mRNA) derived from RNA-seq data analyses of human blood exosomes. Experimental validations from the published literature are also included. exoRBase features the integration and visualization of RNA expression profiles based on normalized RNA-seq data spanning both normal individuals and patients with different diseases. exoRBase aims to collect and characterize all long RNA species in human blood exosomes. The first release of exoRBase contains 58 330 circRNAs, 15 501 lncRNAs and 18 333 mRNAs. The annotation, expression level and possible original tissues are provided. exoRBase will aid researchers in identifying molecular signatures in blood exosomes and will trigger new exosomal biomarker discovery and functional implication for human diseases.
INTRODUCTION
Exosomes are specialized membranous, nanosized endocytic vesicles that are secreted by most cells. They contain a variety of protein and RNA species that can modulate the behavior of recipient cells and may be used as biomarkers for diagnosis of human diseases (1,2). They have been found in the blood of healthy individuals and patients with malignancies, and they are valuable sources of biomarkers due to their selective cargo loading and resemblance to their parental cells. A rich cargo of different RNA species is present and stabilized in exosomes. microRNA (miRNA) has been well characterized and studied in exosomes. Nevertheless, the small quantity and lack of specific expression of miRNA limit its extensive application in biomarker discovery. Increasing evidence shows that long RNA species, including mRNA, lncRNA and circRNA, are also found in human blood exosomes with clinical implications (3–7). For instance, androgen receptor splice variant 7 (AR-V7) is detectable in plasma-derived exosomal RNA of patients with castration-resistant prostate cancer (CRPC) and may be a predictive biomarker of resistance to hormonal therapy in CRPC (4). Human telomerase reverse transcriptase (TERT) mRNA was found to be absent in serum-derived exosomes from normal persons but was variably detected in patients with different cancer types (3). The detection of serum exosomal HOTAIR lncRNA may be a potential biomarker for diagnosing laryngeal squamous cell carcinoma (LSCC) or rheumatoid arthritis (8,9). Recently, our research also showed the presence of abundant circRNAs in exosomes (10) and detected many circRNA candidates with specific expression patterns in different normal and cancerous tissues (11). Furthermore, Barzin et al. found that unshielded RN7SL1 could transfer to breast cancer cells via exosomes and activate PRR RIG-I to promote the aggressive features of cancer after exosome transfer (12). The presence of unshielded RN7SL1 in exosomes from patient blood also highlights its potential clinical relevance. Therefore, there is a substantial number of long RNA species in human blood exosomes that are potential regulators or biomarkers. A compendium of these RNA species will aid researchers in identifying molecular signatures in blood exosomes and will trigger new circulating biomarker discovery and functional implication for human diseases.
Currently, a database named ExoCarta (www.exocarta.org) contains the proteins/RNA/lipids in exosomes in multiple organisms that were identified from manually curated literature. This database is mainly a collection of protein-coding genes (protein and mRNA) and miRNA species in diverse cell types from different organisms. Here, we developed the web-accessible exoRBase, aiming to collect all long RNA species derived from RNA-seq data analyses of human blood exosomes. It features the integration and visualization of RNA expression profiles based on normalized RNA-seq data spanning both normal individuals and patients with different diseases. The first release of exoRBase contains 58 330 circRNAs, 15 501 lncRNAs and 18 333 mRNAs from 87 blood exosomal RNA-seq datasets. Furthermore, 77 experimental validations from 18 published articles have been included. The RNA-seq expression profiles of 30 tissues from the GTEx project (13) have been used to annotate possible original tissues of exosomal RNAs. The whole dataset can be easily browsed, queried and downloaded through the webpage. In addition, exoRBase allows researchers to submit new exosomal RNAs and their expression profiles in human blood exosomes.
MATERIALS AND METHODS
Data collection
We manually curated RNA-seq data for samples derived from human blood exosomes from the NCBI Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) (14) or Short Read Archive (SRA, http://www.ncbi.nlm.nih.gov/sra/) (15). Currently, exoRBase includes 87 exosome samples from six experiments (GSE100206, GSE99985, GSE100063, GSE100207, GSE100232, GSE93078). These samples are from different biological conditions, including normal persons (NP), coronary heart disease (CHD), colorectal cancer (CRC), hepatocellular carcinoma (HCC), pancreatic adenocarcinoma (PAAD) and Breast Cancer (BC). In addition, the current collections include whole blood experiments (five samples, GSE73570). To complement the exoRBase database, we also searched PubMed for exosomal RNAs with experimental validation, which resulted in 18 literature records involving 77 exosomal RNAs. The RNA-seq expression profiles of 30 tissues from the GTEx project (13) were used to annotate possible original tissues of exosomal RNAs. In addition, resource of all human circRNAs were downloaded from circBase database (16). The genomic coordinates of circRNAs were converted to hg38 using the UCSC liftover tool. Then these circRNAs were combined with our collections. We also integrated the tissue types of expressed circRNAs from the results of our previous research, where 27 296 circRNAs were detected from six tissues (11).
Analysis of RNA-seq data
All collected datasets were processed through a consistent bioinformatics pipeline (Figure 1). The raw sequencing reads were first trimmed by removing adapters and low-quality bases using Trimmomatic (Version 0.36) (17). Then, the trimmed reads were mapped to the human reference genome (GRCh38) by HISAT2 (version 2.0.4) (18). featureCounts (version 1.5.0-p1) (19) was employed to quantify the number of reads aligned to regions of protein-coding RNAs (mRNAs) or long non-coding RNAs (lncRNAs). Read counts for each gene were normalized to the TPM values (Transcripts Per Million) (20). Annotations of protein-coding and long non-coding RNAs in the human genome were retrieved from GENCODE (v25) (21). Additionally, circular RNAs were identified and quantified by using ACFS (version 2.2.1) (22) and find_circ (v1.2) (23) for each sample. Circular RNAs that were detected by both tools were used in our analysis. Considering the various library sizes, the read counts of each circular RNAs were normalized by calculating the RPM values (Read counts per Million mapped reads).
Figure 1.

Overall design of the exoRBase database. exoRBase curates all available RNA-seq experiments and reported validations for exosomal RNAs. All RNA-seq raw data was processed using a consistent bioinformatics pipeline. All identified circRNAs, lncRNAs and mRNAs were quantified and annotated in exoRBase. Contents of the exoRBase database include ‘Browse’, ‘Search and Visualization’, ‘Downloads’ and ‘Submission’.
Calculation of tissue specificity for lncRNA and mRNA
To assess tissue specificity of lncRNA and mRNA across human tissues, we downloaded the expression atlas from GTEx (Genotype-Tissue Expression project) (13), which profiled gene expression across 30 human tissues. Genes that were lowly expressed (<0.1 RPKM) in all tissues were removed. We defined the tissue specificity score as the difference between the logarithm of the total number of tissues and the Shannon entropy of the expression values for a gene (24). The score was calculated as follows:
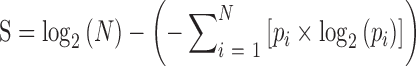 |
where 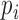 stands for the relative frequency, which was computed as
stands for the relative frequency, which was computed as 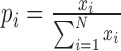 ,
, 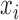 is the gene expression level for gene in tissue i, and N is the total number of all tissues. For each gene, one specificity score and 30 frequency scores (
is the gene expression level for gene in tissue i, and N is the total number of all tissues. For each gene, one specificity score and 30 frequency scores (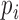 ) were calculated. A gene was defined as tissue-specific when its max frequency score was more than double the second largest one and its specificity score was no less than 1.
) were calculated. A gene was defined as tissue-specific when its max frequency score was more than double the second largest one and its specificity score was no less than 1.
Assigning circRNAs to their related linear RNAs
CircRNAs are transcribed from intra- or inter-genic loci. A circRNA was assigned to be related to a gene when this circRNA was transcribed from regions within the gene or when this gene was located nearest to the circRNA.
Database implementation
The exoRBase database was built using Netty 4.0 (web server) and MySQL (database server). All data were processed and organized into a MySQL Database Management System (version 5.7.18). The interface components of the website were designed and implemented using the Twirl template engine. The database query was based on Scala script. Plots of query results in Search and Browse pages were produced by Highcharts 5.0.12. The website has been tested in several popular web browsers, including Internet Explorer, Google Chrome and Firefox.
RESULTS
Data summary
exoRBase currently contains data from 92 RNA-seq experiments and all available validated cases reported in recent publications. Except for the whole blood samples, all data were derived from blood exosome samples. Additionally, these samples were obtained from diverse groups, including normal persons (NP, 32 samples) and patients with coronary heart disease (CHD, 6 samples), colorectal cancer (CRC, 12 samples), hepatocellular carcinoma (HCC, 21 samples), pancreatic adenocarcinoma (PAAD, 14 samples) or breast cancer (BC, 2 samples) (Table 1). All RNA-seq data were processed to identify and quantify circRNAs, lncRNAs and mRNAs (Figure 1). The numbers of detected RNAs in different sample groups are summarized in Table 1.
Table 1. Numbers of long RNAs detected in different sample groups.
| Groups | circRNA | lncRNA | mRNA |
|---|---|---|---|
| Normal | 40 645 | 10 626 | 16 932 |
| CHD | 3914 | 1628 | 13 848 |
| CRC | 16 115 | 9756 | 16 605 |
| HCC | 26 625 | 9725 | 17 221 |
| PAAD | 19 540 | 11 376 | 17 427 |
| WhB | 46 516 | 13 128 | 16 230 |
| BC | – | 12 136 | 17 102 |
| Validations | 4 | 31 | 38 |
Browse the database
Users can browse the exoRBase database with various options. The ‘Sample types’ option will allow users to browse RNAs that were detected in single or multiple sample types. Users can also filter the results with ‘Detection frequency’, ‘Expression rank’ and ‘Validation’ options. ‘Detection frequency’ denotes how frequently the RNA was detected in all included samples. The frequency value ranges from 0 to 1. A value of 0 means that the selected RNA was not detected in any sample, and 1 means that it was detected in all samples. ‘Expression rank’ denotes the rank of expression level among all RNAs of the same type (circRNA, lncRNA or mRNA). RNAs were categorized into ten units according to their expression levels. For example, ‘0–10%’ means the expression level of a selected RNA ranks in the first tenth. ‘Validation’ denotes whether the RNAs were validated in reported publications. These options provide users with reference scores for selecting candidate RNAs.
Furthermore, exoRBase provides additional options specific to circRNAs, lncRNAs or mRNAs. For circRNAs, users can select circRNAs that are or are not annotated in the circBase database. Additionally, users can filter their browsing by the hosting genes in ‘Gene symbol’. Users can also filter browse results with genomic locations. For lncRNAs and mRNAs, we provide the ‘Tissue specificity’ option for selecting lncRNAs and/or mRNAs that were specifically expressed in one tissue. Some RNAs detected in a blood exosome may come from specific tissues. The ‘Tissue specificity’ option will help users select RNA candidates that are produced from some specific tissues into blood exosomes. These specific RNAs might be meaningful in the clinical diagnosis of some diseases.
Search and downloads
Users can query RNAs of interest by genomic loci for circRNAs or by gene name for lncRNAs and mRNAs. CircRNA is an emerging RNA type, so many circRNAs do not have accordant names. Therefore, we designated the exosomal circRNAs as exo-circ-XXX. Users can search a circRNA by genomic location or new circRNA names. In addition, to facilitate the search function, searching by the name of their hosting genes is also allowed. The search results will show basic information about the queried RNAs in a table. When searching by gene name, users enter one or multiple official gene symbols in the search box. Multiple gene names should be separated by commas. In addition, the expression levels in different sample groups will be shown as a line plot or a heatmap. In a line plot, users can hide the expression curves of one or multiple RNAs by clicking the RNA names on the left. Clicking the RNA names again will bring back the corresponding expression curves. Clicking on the gene name hyperlinks will lead users to a page containing detailed information about the corresponding genes and boxplots showing expression levels in each sample groups. Additionally, all of these plots can be saved to local disks.
The expression profiles of circRNA, lncRNA and mRNA in each sample group and combined samples can be downloaded for customized analyses. Furthermore, annotation files describing detailed information about RNAs can be downloaded on the ‘Downloads’ page. The manually curated information of validated RNAs can also be accessed by users.
Submitting data to exoRBase
The collections of exoRBase may not cover all long RNAs derived from blood exosomes. Hence, we provide a submission interface to invite the community to upload novel RNA or RNA-seq expression profiles derived from blood exosome samples. Notably, before submitting, users should check whether their study meets the standards proposed by ISEV (International Society for Extracellular Vesicles) (25). The minimal requirements for exosome study contain five different features, including the biophysical methods (e.g. transmission electron microscopy) to characterize exosomes, the detection of cytosolic exosomal enriched proteins (e.g. Alix, TSG101), detection of membrane proteins (e.g. CD9, CD63), nonexosomal proteins (e.g. gp96) and particle analysis (e.g. nanoparticle tracking analysis). The raw RNA-seq data should be processed via the pipeline that was particularly described in Methods.
Applications of exoRBase: a utility case
The exoRBase database provides users with a function to extract RNA candidates of interest. The user could investigate, for example, any potential blood biomarkers for patients with liver cancer. The exploration is initiated from the ‘Browse’ page, where multiple options are provided. Clicking the ‘lncRNA and mRNA’ section of the ‘Browse’ page will direct users to the option page of lncRNA and mRNA (Figure 2A). First, the user chooses ‘HCC’ from the drop-down list of the ‘Sample types’ option (Figure 2B). Second, the user selects RNAs that showed different expression levels in HCC samples by choosing ‘HCC(up)’ in the ‘Different Group’ option. Users can further filter the results with the options ‘Tissue Specificity’, ‘Detection frequency’, ‘Expression rank’ and/or ‘Validation’ (Figure 2B). Finally, the user clicks the ‘Search’ button on the bottom of the option section. For instance, if ‘liver’ is selected in the ‘Tissue Specificity’ option, the browsing result will list a table that contains lncRNAs and mRNAs showing upregulated expression in HCC samples and specific expression in liver tissue (Figure 2C). These RNAs may come from rupturing liver cells and can be detected at a higher level, specifically in HCC patients. These RNAs might be considered blood biomarker candidates for HCC patients. More information on each RNA can be retrieved in the database. Taking lncRNA ‘HULC’ and mRNA ‘FGG’ as an example, the user inputs the gene name ‘HULC, FGG’ into the ‘Search’ box (Figure 3A). The search result will show a plot displaying the expression levels across all collected samples and a table that list brief descriptions of ‘HULC and FGG’ (Figure 3B and D). Additionally, users can switch the plot to a heatmap by clicking the ‘Heatmap’ button (Figure 3C). exoRBase also provides a boxplot that can be accessed by directly clicking the gene name or the last column in the table (Figure 3E). Clicking the gene name will also lead to a detailed description of the selected gene below the boxplot (Figure 3F). Through these plots, users can get a quick overview of the expression levels of HULC or FGG in all exosomal samples or sample groups. Furthermore, users can retrieve information about the circRNAs that are located in or near the gene by clicking the ‘Related circRNA’ column in the table.
Figure 2.

Screenshot of database browse. (A) Navigation bar of the database, ‘circRNA’ and ‘lncRNA and mRNA’ could be separately browsed in the ‘Browse’ section. (B) Options for customized browsing of lncRNA and mRNA. (C) Examples of customized browsing results.
Figure 3.

Overview of searching the database. (A) ‘Search’ section of the database. Searching ‘HULC’ and ‘FGG’ as an example. (B) Line charts of queried genes. (C) Heatmap charts of queried genes. (D) Brief information of queried genes. (E) Boxplot charts of queried genes. (F) Detailed description of the queried gene.
DISCUSSION AND FURTHER DIRECTIONS
Recent years have witnessed the rapid advances in high-throughput technologies and in purifying extracellular vesicles. An attractive approach that is currently being developed is ‘liquid biopsy’, which includes exosomes for biomarker discovery. exoRBase is the first resource containing all available long RNAs (circRNA, lncRNA and mRNA) derived from RNA-seq data of human blood exosomes. Related long RNAs that were reported in experimental validations are also included in the database. Users can explore exoRBase and visualize of RNA expression profiles spanning normal individuals and patients with different diseases. The database provides users with operations to extract RNAs of interest through customized browsing options, such as RNAs that have different expression levels in certain diseases and/or come from specific tissues. The exoRBase database will assist the community in identifying molecular signatures in blood exosomes.
Most circRNAs are expressed in low abundance and have specific spatiotemporal expression patterns (11,16,26). Hence, it is not surprising to see low expression levels and varying distributions of RNA quantity among different groups in the circRNA expression section of the exoRBase. The CHD samples in the current collection were from aged patients, where a relatively low number of circRNAs was identified. In addition, RNA-seq data on breast cancer by Barzin et al. (12) were generated from RNA samples with no fragmentation and low sequencing depth. Therefore, the data were only used to annotate lncRNAs and mRNAs with expressed sample groups.
We will update exoRBase regularly by providing more exosomal RNA expression profiles, as additional RNA-seq datasets become available. exoRBase will offer a more comprehensive collection of experimentally supported data and lead to the further development of integrative and visual tools.
ACKNOWLEDGEMENTS
We thank the contributors for providing the RNA-seq datasets. We thank InfinityBio and yunbios platform for the support on website development. We also thank NovelBioinformatics Ltd., Co. for the support of bioinformatics analysis with their NovelBio Cloud Analysis Platform.
FUNDING
National Natural Science Foundation of China [81672779, 81472617, 81502430]; Shanghai Sailing Program [17YF1402600]. Funding for open access charge: National Natural Science Foundation of China.
Conflict of interest statement. None declared.
REFERENCES
- 1. Colombo M., Raposo G., Thery C.. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014; 30:255–289. [DOI] [PubMed] [Google Scholar]
- 2. Thery C., Zitvogel L., Amigorena S.. Exosomes: composition, biogenesis and function. Nat. Rev. Immunol. 2002; 2:569–579. [DOI] [PubMed] [Google Scholar]
- 3. Goldvaser H., Gutkin A., Beery E., Edel Y., Nordenberg J., Wolach O., Rabizadeh E., Uziel O., Lahav M.. Characterisation of blood-derived exosomal hTERT mRNA secretion in cancer patients: a potential pan-cancer marker. Br. J. Cancer. 2017; 117:353–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Del Re M., Biasco E., Crucitta S., Derosa L., Rofi E., Orlandini C., Miccoli M., Galli L., Falcone A., Jenster G.W. et al. The detection of androgen receptor splice variant 7 in plasma-derived exosomal RNA strongly predicts resistance to hormonal therapy in metastatic prostate cancer patients. Eur. Urol. 2017; 71:680–687. [DOI] [PubMed] [Google Scholar]
- 5. Aoki J., Ohashi K., Mitsuhashi M., Murakami T., Oakes M., Kobayashi T., Doki N., Kakihana K., Sakamaki H.. Posttransplantation bone marrow assessment by quantifying hematopoietic cell-derived mRNAs in plasma exosomes/microvesicles. Clin. Chem. 2014; 60:675–682. [DOI] [PubMed] [Google Scholar]
- 6. Dong L., Lin W., Qi P., Xu M.D., Wu X., Ni S., Huang D., Weng W.W., Tan C., Sheng W. et al. Circulating long RNAs in serum extracellular vesicles: their characterization and potential application as biomarkers for diagnosis of colorectal cancer. Cancer Epidemiol. Biomarkers Prev. 2016; 25:1158–1166. [DOI] [PubMed] [Google Scholar]
- 7. Kim K.M., Abdelmohsen K., Mustapic M., Kapogiannis D., Gorospe M.. RNA in extracellular vesicles. Wiley Interdiscip. Rev. RNA. 2017; 8, doi:10.1002/wrna.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang J., Zhou Y., Lu J., Sun Y., Xiao H., Liu M., Tian L.. Combined detection of serum exosomal miR-21 and HOTAIR as diagnostic and prognostic biomarkers for laryngeal squamous cell carcinoma. Med. Oncol. 2014; 31:148. [DOI] [PubMed] [Google Scholar]
- 9. Song J., Kim D., Han J., Kim Y., Lee M., Jin E.J.. PBMC and exosome-derived Hotair is a critical regulator and potent marker for rheumatoid arthritis. Clin. Exp. Med. 2015; 15:121–126. [DOI] [PubMed] [Google Scholar]
- 10. Li Y., Zheng Q., Bao C., Li S., Guo W., Zhao J., Chen D., Gu J., He X., Huang S.. Circular RNA is enriched and stable in exosomes: a promising biomarker for cancer diagnosis. Cell Res. 2015; 25:981–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zheng Q., Bao C., Guo W., Li S., Chen J., Chen B., Luo Y., Lyu D., Li Y., Shi G. et al. Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat. Commun. 2016; 7:11215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nabet B.Y., Qiu Y., Shabason J.E., Wu T.J., Yoon T., Kim B.C., Benci J.L., DeMichele A.M., Tchou J., Marcotrigiano J. et al. Exosome RNA unshielding couples stromal activation to pattern recognition receptor signaling in cancer. Cell. 2017; 170:352–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Consortium G.T. The genotype-tissue expression (GTEx) project. Nat. Genet. 2013; 45:580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Barrett T., Wilhite S.E., Ledoux P., Evangelista C., Kim I.F., Tomashevsky M., Marshall K.A., Phillippy K.H., Sherman P.M., Holko M. et al. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. 2013; 41:D991–D995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Leinonen R., Sugawara H., Shumway M. International Nucleotide Sequence Database, C. . The sequence read archive. Nucleic Acids Res. 2011; 39:D19–D21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Glazar P., Papavasileiou P., Rajewsky N.. circBase: a database for circular RNAs. RNA. 2014; 20:1666–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bolger A.M., Lohse M., Usadel B.. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014; 30:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim D., Langmead B., Salzberg S.L.. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 2015; 12:357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liao Y., Smyth G.K., Shi W.. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014; 30:923–930. [DOI] [PubMed] [Google Scholar]
- 20. Li B., Dewey C.N.. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011; 12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Harrow J., Frankish A., Gonzalez J.M., Tapanari E., Diekhans M., Kokocinski F., Aken B.L., Barrell D., Zadissa A., Searle S. et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012; 22:1760–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. You X., Conrad T.O.. Acfs: accurate circRNA identification and quantification from RNA-Seq data. Sci. Rep. 2016; 6:38820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Memczak S., Jens M., Elefsinioti A., Torti F., Krueger J., Rybak A., Maier L., Mackowiak S.D., Gregersen L.H., Munschauer M. et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013; 495:333–338. [DOI] [PubMed] [Google Scholar]
- 24. Gerstberger S., Hafner M., Tuschl T.. A census of human RNA-binding proteins. Nat. Rev. Genet. 2014; 15:829–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lotvall J., Hill A.F., Hochberg F., Buzas E.I., Di Vizio D., Gardiner C., Gho Y.S., Kurochkin I.V., Mathivanan S., Quesenberry P. et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles. J. Extracell. Vesicles. 2014; 3:26913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Salzman J., Chen R.E., Olsen M.N., Wang P.L., Brown P.O.. Cell-type specific features of circular RNA expression. PLoS Genet. 2013; 9:e1003777. [DOI] [PMC free article] [PubMed] [Google Scholar]