


Abstract
Available genomic data for pathogens has created new opportunities for drug discovery and development to fight them, including new resistant and multiresistant strains. In particular structural data must be integrated with both, gene information and experimental results. In this sense, there is a lack of an online resource that allows genome wide-based data consolidation from diverse sources together with thorough bioinformatic analysis that allows easy filtering and scoring for fast target selection for drug discovery. Here, we present Target-Pathogen database (http://target.sbg.qb.fcen.uba.ar/patho), designed and developed as an online resource that allows the integration and weighting of protein information such as: function, metabolic role, off-targeting, structural properties including druggability, essentiality and omic experiments, to facilitate the identification and prioritization of candidate drug targets in pathogens. We include in the database 10 genomes of some of the most relevant microorganisms for human health (Mycobacterium tuberculosis, Mycobacterium leprae, Klebsiella pneumoniae, Plasmodium vivax, Toxoplasma gondii, Leishmania major, Wolbachia bancrofti, Trypanosoma brucei, Shigella dysenteriae and Schistosoma Smanosoni) and show its applicability. New genomes can be uploaded upon request.
INTRODUCTION
The successful use of antibiotics has been facing challenges because microbial pathogens are developing various forms of resistance in the last decades (1). Despite this critical situation, new drug development projects have been inadequate for reasons ranging from bad selection of targets to reduced antimicrobial drug discovery efforts by pharmaceutical companies (2,3). Currently, it is accepted that identification and validation of appropriate targets are critical steps for designing new drugs. In this sense, next generation sequencing is increasingly aiding the evaluation of gene function, essentiality and suitability for drug development. Nevertheless there is a lack of online resources that allows genome wide based data consolidation from diverse sources in order to define a list of potential targets. Here, we present Target-Pathogen database (target.sbg.qb.fcen.uba.ar/patho), an online resource that allows genome wide based target ranking and identification. By integrating and weighting the data, focusing on essentiality, metabolic role and structural druggability prediction of proteins, Target-Pathogen facilitates the identification and prioritization of candidate targets suitable for new drug development projects.
MATERIALS AND METHODS
All data present in Target-Pathogen is based either on the in silico calculation of selected properties for each protein or on the integration and meta-analysis of publicly available data. All open reading frames (ORFs) derived from each complete genome sequencing were downloaded from the UniProt database (4). All ORFs were analysed with the HMMER (5) program and assigned to PFAM (6) families or domains. The pipeline-engine which we call Target-Pathogen is schematically shown in Figure 1.
Figure 1.

General sketch of Target-Pathogen pipeline. Structural druggability and metabolic analyses are integrated with available experimental data and in silico analysis data. After all data are integrated in Target-Pathogen, a user designed scoring function is used to weight different features in order to obtain a ranked list of candidate drug targets.
Generation of structural homology-based models
Experimental structures were searched in the PDB. Structures of entire proteins or PFAM domains present in the PDB with 95% sequence identity were assigned to the query.
For all remaining ORFs we attempted to build homology-based models using MODELLER. For or all the structures (crystals and models) we then compute several structural properties like: (i) DrugScore (DS), (ii) Active site residues (7) and (iii) PFAM family relevant residues. For a complete set of properties check supplementary information. Further description of this methodology can be found in Supplementary Material.
Structural assessment of druggability
Druggability is a concept that describes the ability of a given protein to bind a drug-like compound, which in turn modulates its function in a craved way (8,9). Druggable proteins should have a well-defined pocket with suitable physicochemical attributes to allow drug binding-sites prediction (9,10). Luque et al., developed a fast method for druggability prediction based on fpocket (11,12). Using fpocket and druggability prediction as the starting point we have previously built a tuberculosis whole-genome protein druggability database (13).
Structural druggability of each potential target was assessed by using the fpocket program (11) and DrugScore (DS) index (11,12). Based on a preliminary analysis of DS distribution for all pockets that host a drug-like compound in the PDB (11–12,14), pockets are classified in four categories: (i) non druggable (ND; DS ≤ 0.2), (ii) poorly druggable (PD; 0.2 < DS ≤ 0.5), (iii) druggable (D; 0.5 < DS ≤ 0.7) and (iv) highly druggable (HD; DS > 0.7).
Off-target criteria
All proteins in the database were subjected to NCBI-BLASTp (E-value <10−7) against human proteome (ncbi assembly accession GCF_000001405.36) to identify non-host homologous targets. The metadata score (human_offtarget) reflects this value with the scale 1—maximum alignment identity. For pathways off target criteria, a bacterial protein was considered a human homologue when sequence similarity was >50% with coverage of more than 50% of the pathogen query protein and with an E-value less than 10−4 using the BLOSUM62 matrix.
Essentiality
All proteomes were submitted to the Database of Essential Genes (DEG) (15,16) for homology analyses (15,17). The BLASTp cut-off values used were: E-value = 1e−05, identity ≥ 80% (18). If homologous genes are found, it is possible that the queried genes are also essential, since functions encoded by essential genes are broadly conserved in microorganisms (19). Essential genes can also be identified by specific experiments and can be added to the Target-Pathogen as metadata. In this sense, we have included in Target-Pathogen data of Mycobacterium tuberculosis essential genes identified by Transposon Site Hybridization (13,14).
Metabolic network analysis
Genome-scale metabolic network (MN) of microorganisms has been widely used to aid in expediting drug discovery in the past decade. MN reconstruction allows identifying choke points reactions (20,21). It is assumed that the blocking of such reactions may either lead to the accumulation of a potentially toxic metabolite in the cell or the lack of essential compound. For these reason genes associated to these reactions are supposed to be relevant from the drug discovery point of view. Graph representations of MNs also allow obtaining important topological measures as betweenness centrality (22). High values of node betweenness centrality from the metabolic perspective, reflects the participation of a reaction as intermediary in many other transformations, and its blockage would generate disequilibrium in many different pathways.
MNs were built by using Pathway Tools v. 19.0 (23), using as input pathogen Genbank files downloaded from NCBI (http://www.ncbi.nlm.nih.gov/). The reconstructed MN was exported in systems biology markup language (SBML) format for downstream analyses. After MN reconstruction, we generated a reaction graph, where nodes represent reactions (i.e usually enzymes) and there is an edge between two nodes if the product of one reaction is used as substrate on the reaction that follows. Cytoscape v. 2.8.3 was used for data visualization and further MN analyses (23,24). Choke-point analysis was conducted in order to identify potential drug targets from the metabolic perspective. Using Cytoscape we also calculated the betweenness centrality (22) of every node.
RESULTS
Browsing the available genomic information in Target-Pathogen
A large amount of information for genes and proteins within the available genomes is actually present in Target-Pathogen. The genome browser can be queried using the web interface at http://target.sbg.qb.fcen.uba.ar/patho. Here, you can start by selecting one of the available genomes. There are three easy ways to explore them. First, the genome can be navigated in a fast, scrolling and zooming way using JBrowse (25). Another interface offers a main search menu with several options to retrieve the Gene records (i.e Keyword, Gene or pathways). Finally, genomes can also be easily explored by EC number (26) or the different categories of Gene Ontology (GO) (27) using Krona (28).
Once group of genes is selected the resulting records are listed. In this page one could click on a desired protein product, and the specific protein data page will be shown. By selecting one model or crystal in the Structure-Tab, the user will be directed to the structures visualization module that allows to (i) select a pocket for graphical display, (ii) display heteroatoms, assigned PFAM and CSA relevant residues and (iii) display drug binding residues; allowing the user to analyse pocket relevance. The displayed protein is available to download as a VMD file (29). Let’s assume, that we are interesting in the pcaA gene of Mycobacterium tuberculosis, thus we simply type ‘pcaA’ in the ‘Gene’ field, to retrieve this record. In the present example, our protein of interest has been crystallized (PDB ID = 1l1e). To access the record, click on the 1l1e structure. A view of this structure is shown in Figure 2. Interestingly, alpha spheres of the druggable pocket ‘number 1’ overlap with the crystallized drug binding site.
Figure 2.

Protein structures visualization in Target-Pathogen. The different visualization tabs that are available when searching for proteins in Target-Pathogen are shown. The table above shows the alignment to the corresponding crystal structure or template model. Other tabs present structure related data, including the interactive pocket visualization module. The visualization module allows (i) to select which pocket to show (ticking the corresponding pocket Select field), (ii) display present HETATMS (37), assigned CSA (38) or PFAM relevant residues, (iii) display the protein in different styles. In the druggable pocket of the example shown below, we depict polar alpha spheres of pocket ‘1’ in black while its apolar α spheres in white. The HETATMS found in the crystal structure are shown as balls and sticks in different colours.
Selection of targets for pathogens
Traditional target prioritization approaches, such as searching the literature and trying to mentally integrate diverse criteria, can quickly become overwhelming. Alternatively, Target-Pathogen can help you to computationally apply a set of filters to obtain a short list of proteins that fulfil user predefined criteria such as protein function, metabolic role, off-targeting, structural druggability, essentiality and omic experiments. To perform this task you have to choose a specific organism and click ‘Prioritize targets’. You will be directed to a three tabs page. The ‘Filter tab’, allows researchers to select targets based on whether or not they fulfil a set of user defined criteria. As an example, we show how to obtain a list of proteins with attractive characteristics for drug targeting in Leishmania major (30). Just, by applying a set of of filters we obtained a 381 proteins that are essential (has a hit in DEG), highly druggable (DS > 0.7), have residues inside the druggable pocket reported in the Catalytic Site Atlas database (CSA) and don’t have close homologues in the human genome (off-target > 0.6). Interestingly, more than a quarter (105) of these proteins were annotated with kinase activity (GO Activity GO:0016301), previously reported as promising candidates for the development of new drugs against this protozoan (31–33). If we filter the proteins annotated with the GO Biological Process term ‘intracellular signal transduction’, the database returns a short list of proteins (27) that fulfil all these criteria. Most of these protein are kinases (26) and among them MKK was previously reported as a drug target in Leishmania (32). By using simple filters we have been able to select a short list of relevant proteins to develop new drugs against L. major with available structure and information about the druggable pockets (Figure 3).
Figure 3.

Number of proteins in Leishmania major genome with desirable properties for drug targets.
Querying Target-Pathogen to rank and prioritize drug targets
Target-Pathogen database not only allow researchers to query and filter proteins, users also have the option of assigning a numerical weight value to different protein properties to build a user defined scoring function (SF). Moreover, users can combine different filters with the SF to obtain a particular list of ranked genes according to a user designed criteria. We have previously proposed a set of nitrosative stress sensible targets in order to develop a new drug to combat latent tuberculosis using in silico detailed evaluation of the Mtb druggable proteome (14). Target-Pathogen can now help us to make a similar analysis. We firstly select only those essential proteins which have druggable pockets and lack of a close homologue in humans (See tutorial in Supplementary Material). Target-Pathogen will return a set of 762 druggable, essential and without close homologues in the human host (off-target > 0.6) targets. To further rank this group, we performed a SF including an analysis of available data concerning their expression level under infection mimicking conditions and the metabolic context of each protein, according to the following equation:
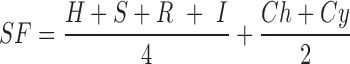 |
The first term integrates available expression data under different conditions mimicking infection. The selected conditions, which group different reports, comprise Hypoxia (H), Starvation (S), RNOS stress (R) and infection in mice (I). The second term focuses in metabolic context of the proteins. In this way Ch and Cy determines if the reaction associated to the protein is a chokepoint or central in the bacteria metabolism. Each variable takes the value of 1 if the protein comply the criteria and 0 if not. A high value of SF would mean that the protein fulfil most of the criteria that defines a promising drug target. Supplementary Figure S4 shows the five proteins which have the higher values. The top five proteins are essential, druggable and do not present homology with any human protein. They are also important from the metabolic point of view and are overexpressed in different assays that mimic the conditions encountered by the bacilli inside the granuloma. Among them are Rv2245 and Rv1285. The former codes a protein involved in the mycolate biosynthesis, a pathway targeted by first-line tuberculosis drugs such as isoniazid and ethambutol (34). Rv1285 is a protein implicated in the sulfate activation pathway and has been previously proposed as a drug target against tuberculosis (35). In this example, we have shown how filtering and ranking together allows to highlight relevant targets. Finally, users can upload their own data in order to include it for calculating new scoring functions or as a filter for a personalized ranking (For further instructions see Supplementary Material).
Choosing promising pathways for drug development
Bioinformatic reconstruction of a pathogens MNs has now become important to explore novel molecular targets.Target-Pathogen allows users to select and study proteins in the context of their metabolic pathways. Moreover, it allows users to rank and prioritize entire pathways as good candidates for novel therapies with a defined criterion. One fundamental advantage of studying the metabolic context of putative targets is that results are expected to allow the design of possible combined therapies (targeting more than one target from the same metabolic pathway).
For example, we could be interested in determining attractive pathways for new drug development against polymyxin B-resistant Klebsiella pneumoniae. In this case, we should be interested in a scoring function that takes account metabolic features such as proportion of chokepoints, centrality and completeness of the pathway (i.e total number of reactions of a pathway associated with a gene/total number of reactions present in the pathway). Other features to consider are essentiality, sequence conservation and homology with human proteins of the enzymes involved in the pathways. Also we may be interested to incorporate in our scoring function a term related to overexpression during polymyxin B (PB) exposure. Ultimately, we could define a pathway as druggable if at least one of the proteins involved is druggable and rule out completely non-druggable pathways. For more detail see tutorial in Supplementary Material.
The mode of action of polymyxin B generally involves interaction with lipopolysaccharides (LPS) located in the outer membrane of Gram-negative bacteria, competing with the calcium and magnetic ions that stabilize LPS, allowing for drug uptake to the cell interior (36). In this sense, it is interesting to note that four of the five best-ranked pathways (obtained as a result of a scoring function that is the linear sum of all the criteria mentioned above) are related to the synthesis of membrane components known to be essential in bacterial survival which make them attractive as drug targets. Moreover, most of the genes associated to these pathways are overexpressed in polymyxin B resistance induction (37).
DISCUSSION
In this work, we present Target-Pathogen, a database that allows query and prioritization of drug targets in pathogens. It is an easy-to-use web-based platform which allows genome wide based data consolidation from diverse sources in a user-friendly manner, with graphical interface for structural visualization and manipulation.
There are few existing databases that are designed for target selection in a set of relevant pathogens but most of these databases focuses on a single protein analysis or focus on specific protein characteristics. For example, Drug Target Database (38) is a useful resource to select potential targets using a reverse docking program. The Therapeutic Targets Database (39) provides a large volume of data of already known therapeutic targets. Another database that includes data of known targets is TargetDB/TargetTrack (40), but only focuses on structural information. TDR targets (41) is an interesting resource focused on tropical neglected diseases.
With structurome and drugome information Target-Pathogen is a unique resource to analyse whole genomes of relevant pathogens. Using Target-Pathogen, researchers can quickly prioritize genes of interest in a fast and intuitive manner, running simple queries (such as looking for proteins with high druggability score or associated with choke-points reactions), filtering by different data, assigning numerical weights for different properties and combining these results to produce a ranked list of targets. In this sense, an aspect that distinguishes Target-Pathogen is that it permits users to upload their own data. Once the data are uploaded, they can be included to obtain a personalized ranking of candidate drug targets in pathogens. Another key feature where Target-Pathogen stands alone is its ability to rank not only proteins but entire pathways. This characteristic allows prioritizing promising pathways to develop new drugs in order to synergistically attack several proteins of the same pathway. We have now selected ten relevant genomes but the database can be easily updated with other relevant pathogens as the bioinformatic pipelines have been automatized. In this sense, we will extend Target-Pathogen to more organisms than the current version, that’s why we also encourage users to request new genomes of interest and join our team by mailing to target@biargentina.com.ar.
The goal of Target-Pathogen is not to replace wet strategies for target identification. Rather, our goal is to become a useful resource for researchers working in the field of target identification and/or drug discovery to translate biological questions in a computational tractable way by exploring, filtering and weighting the vast quantity of genome-scale data sets. Target-Pathogen will be continuously updated as part of a national initiative to develop tools for pathogens.
AVAILABILITY
JBrowse is a fast, embeddable genome browser built completely with JavaScript and HTML5, with optional run-once data formatting tools written in Perl.
(https://github.com/GMOD/jbrowse/tree/master)
GLmol is a molecular viewer written in Javascript and WebGL.
(https://github.com/biochem-fan/GLmol)
Krona allows hierarchical data to be explored with zooming, multi-layered pie charts.
(https://github.com/marbl/Krona/wiki)
Biojs-vis-sequence displays a protein/nucleotide sequence.
(http://biojs.io/d/biojs-vis-sequence)
Modular BioJS component for a multiple sequence alignment.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Consejo Nacional de Investigaciones Científicas y Técnicas [CONICET PIP 11220130100469CO to A.G.T.]; Agencia Nacional de Promoción Científica y Tecnológica [ANPCyT, PICT-2015–1863 to D.F.D.]; CONICET Postdoctoral Fellowships (to L.A.D., E.L., G.B., L.R.); CONICET Memberships (to D.F.D., A.G.T., M.A.M., E.J.S.). Funding for open access charge: Agencia Nacional de Promoción Científica y Tecnológica [PICT-2015-1863]; Ministerio de Ciencia; Tecnología e Innovación Productiva.
Conflict of interest statement. None declared.
REFERENCES
- 1. Martens E., Demain A.L.. The antibiotic resistance crisis, with a focus on the United States. J. Antibiot. 2017; 70:520–526. [DOI] [PubMed] [Google Scholar]
- 2. Fernandes P. The global challenge of new classes of antibacterial agents: an industry perspective. Curr. Opin. Pharmacol. 2015; 24:7–11. [DOI] [PubMed] [Google Scholar]
- 3. Wenzel R.P. The antibiotic pipeline—challenges, costs, and values. N. Engl. J. Med. 2004; 351:523–526. [DOI] [PubMed] [Google Scholar]
- 4. Pundir S., Magrane M., Martin M.J., O’Donovan C. The UniProt Consortium . Searching and navigating UniProt Databases. Curr. Protoc. Bioinform. 2015; 50, doi:10.1002/0471250953.bi0127s50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wheeler T.J., Eddy S.R.. nhmmer: DNA homology search with profile HMMs. Bioinformatics. 2013; 29:2487–2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Finn R.D., Coggill P., Eberhardt R.Y., Eddy S.R., Mistry J., Mitchell A.L., Potter S.C., Punta M., Qureshi M., Sangrador-Vegas A. et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 2016; 44:D279–D285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Furnham N., Holliday G.L., de Beer T.A.P., Jacobsen J.O.B., Pearson W.R., Thornton J.M.. The Catalytic Site Atlas 2.0: cataloging catalytic sites and residues identified in enzymes. Nucleic Acids Res. 2013; 42:D485–D489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hopkins A.L., Groom C.R.. The druggable genome. Nat. Rev. Drug Discov. 2002; 1:727–730. [DOI] [PubMed] [Google Scholar]
- 9. Cheng A.C., Coleman R.G., Smyth K.T., Cao Q., Soulard P., Caffrey D.R., Salzberg A.C., Huang E.S.. Structure-based maximal affinity model predicts small-molecule druggability. Nat. Biotechnol. 2007; 25:71–75. [DOI] [PubMed] [Google Scholar]
- 10. Xie L., Bourne P.E.. A robust and efficient algorithm for the shape description of protein structures and its application in predicting ligand binding sites. BMC Bioinformatics. 2007; 8(Suppl. 4):S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schmidtke P., Le Guilloux V., Maupetit J., Tufféry P.. fpocket: online tools for protein ensemble pocket detection and tracking. Nucleic Acids Res. 2010; 38:W582–W589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schmidtke P., Barril X.. Understanding and predicting druggability. A high-throughput method for detection of drug binding sites. J. Med. Chem. 2010; 53:5858–5867. [DOI] [PubMed] [Google Scholar]
- 13. Radusky L., Defelipe L.A., Lanzarotti E., Luque J., Barril X., Marti M.A., Turjanski A.G.. TuberQ: a Mycobacterium tuberculosis protein druggability database. Database. 2014; 2014:bau035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Defelipe L.A., Do Porto D.F., Pereira Ramos P.I., Nicolás M.F., Sosa E., Radusky L., Lanzarotti E., Turjanski A.G., Marti M.A.. A whole genome bioinformatic approach to determine potential latent phase specific targets in Mycobacterium tuberculosis. Tuberculosis. 2016; 97:181–192. [DOI] [PubMed] [Google Scholar]
- 15. Zhang R. DEG: a database of essential genes. Nucleic Acids Res. 2004; 32:D271–D272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Luo H., Lin Y., Gao F., Zhang C.-T., Zhang R.. DEG 10, an update of the database of essential genes that includes both protein-coding genes and noncoding genomic elements. Nucleic Acids Res. 2014; 42:D574–D580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Barh D., Gupta K., Jain N., Khatri G., León-Sicairos N., Canizalez-Roman A., Tiwari S., Verma A., Rahangdale S., Hassan S.S. et al. Conserved host–pathogen PPIs: globally conserved inter-species bacterial PPIs based conserved host-pathogen interactome derived novel target in C. pseudotuberculosis, C. diphtheriae, M. tuberculosis, C. ulcerans, Y. pestis, and E. coli targeted by Piper betel compounds. Integr. Biol. 2013; 5:495–509. [DOI] [PubMed] [Google Scholar]
- 18. Barh D., Jain N., Tiwari S., Parida B.P., D’Afonseca V., Li L., Ali A., Santos A.R., Guimarães L.C., de Castro Soares S. et al. A novel comparative genomics analysis for common drug and vaccine targets in Corynebacterium pseudotuberculosis and other CMN group of human pathogens. Chem. Biol. Drug Des. 2011; 78:73–84. [DOI] [PubMed] [Google Scholar]
- 19. Kobayashi K., Ehrlich S.D., Albertini A., Amati G., Andersen K.K., Arnaud M., Asai K., Ashikaga S., Aymerich S., Bessieres P. et al. Essential Bacillus subtilis genes. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:4678–4683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Albert R. Scale-free networks in cell biology. J. Cell Sci. 2005; 118:4947–4957. [DOI] [PubMed] [Google Scholar]
- 21. Yeh I., Hanekamp T., Tsoka S., Karp P.D., Altman R.B.. Computational analysis of Plasmodium falciparum metabolism: organizing genomic information to facilitate drug discovery. Genome Res. 2004; 14:917–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lacroix V., Cottret L., Thébault P., Sagot M.-F.. An introduction to metabolic networks and their structural analysis. IEEE/ACM Trans. Comput. Biol. Bioinform. 2008; 5:594–617. [DOI] [PubMed] [Google Scholar]
- 23. Karp P.D., Latendresse M., Paley S.M., Krummenacker M., Ong Q.D., Billington R., Kothari A., Weaver D., Lee T., Subhraveti P. et al. Pathway Tools version 19.0 update: software for pathway/genome informatics and systems biology. Brief. Bioinform. 2016; 17:877–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Russell J., Cohn R.. Cytoscape Book on Demand Limited. 2012; California. [Google Scholar]
- 25. Skinner M.E., Uzilov A.V., Stein L.D., Mungall C.J., Holmes I.H.. JBrowse: a next-generation genome browser. Genome Res. 2009; 19:1630–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bairoch A. The ENZYME database in 2000. Nucleic Acids Res. 2000; 28:304–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gene Ontology Consortium and Gene Ontology Consortium The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004; 32:D258–D261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ondov B.D., Bergman N.H., Phillippy A.M.. Interactive metagenomic visualization in a web browser. BMC Bioinformatics. 2011; 12:385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Humphrey W., Dalke A., Schulten K.. VMD: visual molecular dynamics. J. Mol. Graph. 1996; 14:33–38. [DOI] [PubMed] [Google Scholar]
- 30. Tiwari N., Gedda M.R., Tiwari V.K., Singh S.P., Singh R.K.. Limitations of current therapeutic options, possible drug targets and scope of natural products in control of leishmaniasis. Mini Rev. Med. Chem. 2017; 17:182–189. [DOI] [PubMed] [Google Scholar]
- 31. Chawla B., Madhubala R.. Drug targets in Leishmania. J. Parasit. Dis. 2010; 34:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Naula C., Parsons M., Mottram J.C.. Protein kinases as drug targets in trypanosomes and Leishmania. Biochim. Biophys. Acta. 2005; 1754:151–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Brumlik M.J., Pandeswara S., Ludwig S.M., Murthy K., Curiel T.J.. Parasite mitogen-activated protein kinases as drug discovery targets to treat human protozoan pathogens. J. Signal Transduct. 2011; 2011:971968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Barry C.E., Crick D.C., McNeil M.R.. Targeting the formation of the cell wall core of M. tuberculosis. Infect. Disord. Drug Targets. 2007; 7:182–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Raman K., Yeturu K., Chandra N.. targetTB: a target identification pipeline for Mycobacterium tuberculosis through an interactome, reactome and genome-scale structural analysis. BMC Syst. Biol. 2008; 2:109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zavascki A.P., Goldani L.Z., Li J., Nation R.L.. Polymyxin B for the treatment of multidrug-resistant pathogens: a critical review. J. Antimicrob. Chemother. 2007; 60:1206–1215. [DOI] [PubMed] [Google Scholar]
- 37. Ramos P.I.P., Custódio M.G.F., Quispe Saji G.D.R., Cardoso T., da Silva G.L., Braun G., Martins W.M.B.S., Girardello R., de Vasconcelos A.T.R., Fernández E. et al. The polymyxin B-induced transcriptomic response of a clinical, multidrug-resistant Klebsiella pneumoniae involves multiple regulatory elements and intracellular targets. BMC Genomics. 2016; 17:737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gao Z., Li H., Zhang H., Liu X., Kang L., Luo X., Zhu W., Chen K., Wang X., Jiang H.. PDTD: a web-accessible protein database for drug target identification. BMC Bioinformatics. 2008; 9:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhu F., Han B., Kumar P., Liu X., Ma X., Wei X., Huang L., Guo Y., Han L., Zheng C. et al. Update of TTD: Therapeutic Target Database. Nucleic Acids Res. 2010; 38:D787–D791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen L., Oughtred R., Berman H.M., Westbrook J.. TargetDB: a target registration database for structural genomics projects. Bioinformatics. 2004; 20:2860–2862. [DOI] [PubMed] [Google Scholar]
- 41. Magariños M.P., Carmona S.J., Crowther G.J., Ralph S.A., Roos D.S., Shanmugam D., Van Voorhis W.C., Agüero F.. TDR Targets: a chemogenomics resource for neglected diseases. Nucleic Acids Res. 2012; 40:D1118–D1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.