


Abstract
Regular monitoring of drug regulatory agency web sites and similar resources for information on new drug approvals and changes to legal status of marketed drugs is impractical. It requires navigation through several resources to find complete information about a drug as none of the publicly accessible drug databases provide all features essential to complement in silico drug discovery. Here, we propose SuperDRUG2 (http://cheminfo.charite.de/superdrug2) as a comprehensive knowledge-base of approved and marketed drugs. We provide the largest collection of drugs (containing 4587 active pharmaceutical ingredients) which include small molecules, biological products and other drugs. The database is intended to serve as a one-stop resource providing data on: chemical structures, regulatory details, indications, drug targets, side-effects, physicochemical properties, pharmacokinetics and drug–drug interactions. We provide a 3D-superposition feature that facilitates estimation of the fit of a drug in the active site of a target with a known ligand bound to it. Apart from multiple other search options, we introduced pharmacokinetics simulation as a unique feature that allows users to visualise the ‘plasma concentration versus time’ profile for a given dose of drug with few other adjustable parameters to simulate the kinetics in a healthy individual and poor or extensive metabolisers.
INTRODUCTION
Bioinformatics and cheminformatics are research fields in which huge amounts of data are being generated each day at a rapid pace. This vast amount of data is distributed across several online databases that are either publicly accessible or often accessible only via subscription. This decentralized distribution of data restrains linking of the current wealth of information with the enormous amount of data that has been accumulating over decades. We witnessed a significant progress in the last 10–15 years through several remarkable contributions that attempted to bridge this ‘information/informatics gap’. Comprehensive small molecule databases such as DrugBank (1), KEGG (2) and ChEBI (3) have been established as expert curated resources. On the other hand, PubChem (4), ChEMBL (5) and Binding DB (6) serve as major resources for bioactivity. Therapeutic Target Database (TTD) (7) and Comparative Toxicogenomics Database (CTD) (8) focus on known or explored therapeutic targets of drugs and literature references that report chemical-gene/protein interactions. A recent addition to the league of publicly accessible drug databases is DrugCentral (9) which serves as an online drug compendium with a special focus on active pharmaceutical ingredients that are approved by FDA and other drug regulatory agencies. Further, resources like Protein Data Bank (PDB) (10) and Cambridge Structural Database (CSD) (11) archive the experimentally determined three dimensional (3D) structures of biological macromolecules and low molecular weight structures. Despite constant enrichment of data at each of these platforms, there has always been a need for a resource that could connect several layers of information on drugs in the context of in silico research. Especially, no dedicated resources exist for 3D structures of drugs, with rare exceptions such as e-Drug3D database (12). In this context, we previously came up with SuperDrug database containing a total of 2396 experimentally determined and computed 3D structures for active ingredients present in the WHO’s essential marketed drugs (13). Although some of the aforementioned resources focus on the pharmacological aspects of drugs to variable extents, none provide comprehensive pharmacokinetic data which facilitates simulation of pharmacokinetics of approved drugs.
Here we present SuperDRUG2, an update of our previous conformational drug database, currently containing information for 4587 active pharmaceutical ingredients that are present in pharmaceutical products. We aim to integrate data that is widely distributed across multiple resources and serve as a one-stop source. The database features multiple search options that facilitate two-dimensional (2D) and 3D similarity calculation, identification of potential drug–drug interactions in complex drug regimens among several other features. A special focus of the database lies in simulation of the ‘plasma-concentration versus time’ curves using pharmacokinetic data extracted from various sources such as drug labels and scientific literature. We introduce for the first time a 3D-superposition feature that superimposes drugs of interest with those ligands already known to bind with protein targets in experimentally determined 3D structures.
MATERIALS AND METHODS
Approved and marketed drugs
Several online public resources including the most recent pharmaceutical product collections from the U.S. Food and Drug Administration (US FDA), the European Medicines Agency (EMA), Health Canada, the Korea's FDA (KFDA), and China's FDA (CFDA) were searched for active ingredients used in pharmaceutical products (see Section 1 in supplementary information (S2) for detailed list of resources and methods). For convenience, we will use the term ‘drug’ instead of ‘active ingredient’ which is widely accepted by chemists and biologists in the field of drug discovery. Currently, the database comprises a total of 4587 drugs grouped into two categories: small molecules (3,982 drugs) and biological/other drugs (605 drugs). Both 2D and 3D structures were standardized in ChemAxon software (https://www.chemaxon.com) for all small molecules entries. The standardization procedure is detailed in one of our former database papers (14). The 3D conformations were also generated using the same software. The 2D depictions on the web site are generated using RDKit toolkit (http://www.rdkit.org) whereas the interactive 3D structure visualisation is enabled via 3Dmol.js library (15).
Further, physicochemical properties and chemical structure identifiers were generated using the RDKit nodes in KNIME (https://www.knime.com). In order to ensure connectivity with well-known drug databases, every drug entry was annotated with links to external resources including the WHO’s index of ATC codes (https://www.whocc.no/atc_ddd_index). Drug labels were extensively text-mined for regulatory details (of approval), therapeutic indications and the recommended doses. In addition, we also flagged some entries as withdrawn drugs. These drugs were previously known to cause adverse effects and eventually withdrawn in one or more countries and sometimes world-wide (14), (16). It must be noted that sometimes only a particular pharmaceutical product or a specific dose or dosage form of the drug is withdrawn which does not necessarily indicate that the drug does not exist in any currently approved/marketed pharmaceutical products.
Drug targets
We extracted target information from DrugBank (v. 5) (1), TTD (7) and ChEMBL (v. 22) (5). Confirmed drug-target interactions were found at the first two resources while ChEMBL provides experimental activity data. Information from ChEMBL was pre-processed using filter criteria suggested by Bajorath et al. (17) to retain only high confidence activity data (detailed procedure is described under Section 2 of supplementary information (S2)). Overall, the database comprises >20 000 confirmed drug-target interactions covering more than 2300 drugs interacting with 3000 distinct targets. In order to understand the interactions in the context of side-effects, we used a list of side-effect targets on the Novartis Safety Panel proposed by Lounkine et al. (18) and annotated our drug-target relations into two categories: safety and non-safety. Identification of previously undetected targets for known drugs can provide valuable insights and leads in drug repurposing endeavours. Our previously published target prediction server, SuperPred (19) was used to collect >17 000 drug-target interactions (more than 2500 drugs). Further, protein structures and their co-crystallized ligands were extracted from PDB (10) and mapped to the targets in our database, resulting in a total of 23 260 structures that are used for 3D-superposition.
2D and 3D similarity
The 2D structures of small molecules are converted to MDL MACCS key based fingerprints to facilitate chemical similarity search. Tanimoto coefficient is used as the standard 2D similarity metric. Additionally, we implemented the Ullmann's algorithm for subgraph isomerism using the open source Chemistry Development toolkit (20) for substructure similarity search. Up to 200 conformations per drug were calculated in order to perform pairwise 3D structure comparisons. Atoms were assigned by minimal distance and superimposed by using the Kabsch algorithm (21). In a co-ordinate system comprising normalized set of atoms, the centre of masses of both conformers are calculated and superimposed. A root-mean-square-deviation (RMSD) score is derived for each comparison which signifies the extent of similarity between the two structures. A detailed methodology on how 3D similarity is calculated can be found in our previous work (22).
Side effects
The current version of database includes >100 000 side effect relations for nearly 950 approved drugs that not only cover the adverse events recorded during the clinical trials prior to drug approval but also those identified during the post-marketing surveillance. The side effect data was collected from SIDER resource (v. 4.1) (23). We also extracted the frequency information for side effects for each drug and labelled the relations according to the SIDER frequency scale. A total of 4964 distinct side effects identified by MEDRA concept identifiers are currently linked from our resource to the SIDER database.
Pharmacokinetic parameters
The data on pharmacokinetics of drugs is scarce in many publicly available resources. However, having such data is essential to simulate the kinetic profile of a drug under varying physiological conditions to improve personalized therapy. We extracted half-life, volume of distribution, protein binding, bioavailability, and time to peak among various other parameters that correspond to the ADME phases. The majority of pharmacokinetic data for humans is extracted from scientific literature while databases such as DrugBank and dedicated drug information portal Drugs.com (https://www.drugs.com) provided partial information for some drugs. Other sources include drug labels and product monographs. More than 50% of all drugs with pharmacokinetic data were annotated with therapeutic minimum and maximum plasma levels extracted from literature (24).
drug–drug interactions
We extracted the drug–drug interaction data mainly from DrugBank and additionally extracted information from package inserts, labels of pharmaceutical products and scientific literature through semi-automated text-mining. The interactions are classified into risk categories (1: monitor therapy; 2: consider replacement; 3: avoid combination) which are widely used at other public and commercial resources for drug–drug interactions. Further, we annotated some drugs as potentially inappropriate medications based on the ‘Beers criteria’ (25) proposed by the American Geriatrics Society, originally published in 2012 and last updated in 2015. The medications covered in this list are considered to be associated with poor outcomes in older adults and are recommended to be avoided for all individuals in this group, except those in palliative and hospital care. A German variant of the Beers list, known as the ‘PRISCUS list’ (26) was also used to annotate drugs that are potentially unsuitable for the elderly.
Web application, system requirements and data availability
All the data in SuperDRUG2 is stored in a relational MySQL database and the web site is set up as Java web application on a virtual Linux (Ubuntu 14.04 LTS) server, accessible at http://cheminfo.charite.de/superdrug2. JavaScript is key to almost all search options we offer. Therefore, we strongly recommend using modern web browsers such as Safari, Google Chrome or Firefox (with JavaScript enabled). The contents of the database are made available via customized download links on the web site.
DATABASE SEARCH OPTIONS
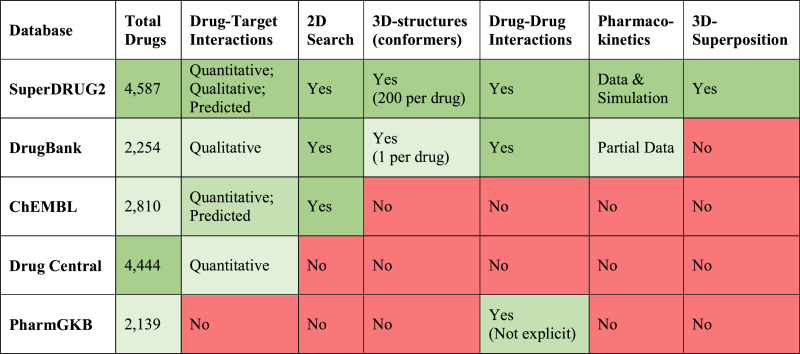
The integrated data in SuperDRUG2 can be accessed via multiple interactive features described below and are schematically represented in Figure 1. A detailed comparison of the contents, coverage and the uniqueness of our database with existing drug databases is presented in Table 1. Although the resources compared with are not necessarily exclusive drug databases, the details presented in Table 1 are expected to justify the novelty of our database as a one stop-resource. A list of web links to the list of pharmaceutical products approved for use in several countries world-wide is provided in the supplementary information sheet S1. The national drug lists can also be accessed through a map visualization on the web site.
Figure 1.

A schematic representation of the data and search options in SuperDRUG2.
Table 1. A detailed comparison of SuperDRUG2 database with four other existing drug databases in terms of their content, content type and coverage. The coverage information of the listed resources is based on our access on 26/05/2017.
|
Drug search
A simple way to search for drug records is to use the ‘Name Search’ option under the Drug Search page. In case an exact name or synonym match does not yield any result, the search query is used to look up the chemical structure at PubChem and five most similar drugs from the database are displayed and ranked by the similarity towards the input molecule. A molecule sketching tool provided in the ‘Structure Search’ section facilitates structure-based search. Three different search types (exact match, similarity search and substructure search) are provided. Users have the flexibility to choose a similarity threshold and the maximum number of results. A detailed drug record contains multiple sections that provide: basic details such as synonyms, indications, ATC codes and marketing status; 2D and 3D molecular structures; regulatory details; drug targets; side-effects, pharmacokinetic data; physicochemical properties, links to external databases via specific identifiers; and marketed drug products.
3D superposition
The feature of 3D superposition could be used in two ways. The first option is to look up for two small molecule drugs using the name search fields. Once a user selects the drugs, a 3D superposition of the two structures is calculated and an interactive 3D visualisation of the superimposed structures is displayed along with an RMSD score that indicates the structural similarity. The second option is to superimpose a drug from the database with a ligand that is known to bind to a protein in a PDB complex. To start using the feature, the user has to first search for a protein target of interest. PDB structures associated with this target are displayed along with the chain identifiers and ligands. After choosing a combination of PDB structure and ligand, the user is allowed to search for a small molecule drug of interest in the database. An interactive 3D visualisation of the overlapped molecules is provided in the context of the binding site of the ligand. This would be an interesting feature to understand the fit of the drug into the binding pocket of the target protein of interest. Figure 2 shows an exemplary 3D superposition result in which niraparib, a well-known poly ADP ribose polymerase (PARP) inhibitor is superimposed with a small molecule inhibitor (PDB ligand ID: 1SX) in the 3D structure of tankyrase 1 (PDB ID: 4KRS), an important regulator of the Wnt/β-catenin signalling. Dual inhibitors of PARP1/2 and tankyrase 1 are known to inhibit growth of DNA repair deficient tumours (27). Understanding the role of known PARP1/2 inhibitors such as niraparib and olaparib in the inhibition of tankyrase 1 could be useful in exploring opportunities to repurpose these drugs for other cancer types.
Figure 2.

3D visualisation of the result of the superposition of niraparib and PDB ligand 1KS in the crystal structure (4KRS) of Tankyrase 1. Both molecules (niraparib: white colour; 1SX: red color) are well superposed in the 1SX binding region of chain A.
Pharmacokinetics simulation
To the best of our knowledge, SuperDRUG2 is the first academic resource to provide simulation of pharmacokinetics of approved drugs as an easily accessible feature. The users can simply search for a drug by its name to see if a simulation is available within our database. The concentration vs. time curve for a recommended dose of the drug is displayed, assuming that it is administered once per day. A therapeutic window is displayed whenever the experimentally determined therapeutic minimum and maximum concentrations are found. The users are provided with interactive sliders to adjust the dose, intake interval and the time period of simulation. Furthermore, approximate changes in drug plasma levels for poor and ultra-rapid metabolisers can be visualised relative to the plasma levels for a healthy adult. Optionally, users can provide a dose of interest to observe changes in plasma level. A use case for dose adaptation based on the pharmacokinetic simulation feature is presented in the next section. It should be noted that this feature is not aimed at providing recommendations or alternatives to dosing schemes to healthcare practitioners in clinical practice but may provide hints for possible problems and solutions. A brief description of the pharmacokinetic model behind the simulation is provided under Section 4 of supplementary information (S2).
Drug–drug interactions
Our drug–drug interaction checker takes a list of medications and provides a list of possible drug–drug interactions associated with the co-administration of these drugs. The users are alerted through a ‘traffic light signal’ adaption displaying one three risk levels whenever a potential drug–drug interaction is found. In addition, to provide the context of metabolic effects on a drug combination, the users are linked to our TRANSFORMER resource (28) which provides detailed report on the effects of a drug on metabolizing enzymes. Further, in order to provide special recommendations to the elderly patient group, we mark those drugs in the input list that are present in the PRISCUS and Beer's list of potentially inappropriate medications. If a drug is known to be present in the PRICSUS list, all possible alternative drugs and dose levels are provided as recommendations.
USE CASE
The following use case illustrates the utility of pharmacokinetics simulation feature of SuperDRUG2 to provide early recommendations for dose adaption. We use the antihypertensive drug losartan as an example. The minimum and maximum recommended doses per day are 25mg and 50mg, respectively. For hypertensive patients with left ventricular hypertrophy or type 2 diabetic nephropathy, a maximum of 100mg per day is recommended. Losartan undergoes hepatic metabolism via cytochrome enzymes 2C9 and 3A4 to form an active metabolite which is 10–40 times more potent. Previous studies indicate that decreased levels of losartan metabolites are observed in carriers of CYP2C9*2 and/or CYP2C9*3 alleles (29) due to the lowered rate of oxidation of losartan (29) into its metabolite and a higher plasma AUC losartan/AUC metabolite ratio (30).
In Figure 3A, one can see that the plasma levels of losartan even at a maximum dose for special indications of 100 mg do not remain within the therapeutic window in order to provide a longer duration of action. Therefore, a twice daily administration of 50–70 mg might improve the coverage of the therapeutic window (see Figure 3B). Consistently, a recent study also reported that twice daily administration of the same daily dose of losartan is more effective in comparison to once daily administration of a single dose (31). Additional use cases can be found in Section 5 of supplementary information (S2).
Figure 3.

Plasma concentration versus time curves generated using the pharmacokinetics simulation feature for losartan in two different cases: (A) dose = 100 mg/day and (B) dose = 70 mg twice daily.
FUTURE DIRECTIONS
We will regularly update the database with new entries to ensure excellent coverage and data quality standards. Especially, the pharmacokinetic data needed for simulation of plasma levels of drug will be further enriched to provide simulations for as many drugs as possible. We also plan to improve the list of drugs that have side effects by adding information from large collections such as the FDA’s adverse event reporting system. Multiple other ways to browse the contents of the database will be eventually added to improve the user experience.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR online.
FUNDING
Berlin-Brandenburg research platform BB3R, Federal Ministry of Education and Research (BMBF), Germany [031A262C]; DKTK. Funding for open access charge: Charité - University Medicine Berlin.
Conflict of interest statement. None declared.
REFERENCES
- 1. Wishart D.S., Knox C., Guo A.C., Shrivastava S., Hassanali M., Stothard P., Chang Z., Woolsey J.. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006; 34:D668–D672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kanehisa M., Goto S., Sato Y., Furumichi M., Tanabe M.. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012; 40:D109–D114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hastings J., Owen G., Dekker A., Ennis M., Kale N., Muthukrishnan V., Turner S., Swainston N., Mendes P., Steinbeck C.. ChEBI in 2016: Improved services and an expanding collection of metabolites. Nucleic Acids Res. 2016; 44:D1214–D1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kim S., Thiessen P.A., Bolton E.E., Chen J., Fu G., Gindulyte A., Han L., He J., He S., Shoemaker B.A. et al. PubChem substance and compound databases. Nucleic Acids Res. 2016; 44:D1202–D1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bento A.P., Gaulton A., Hersey A., Bellis L.J., Chambers J., Davies M., Kruger F.A., Light Y., Mak L., McGlinchey S. et al. The ChEMBL bioactivity database: an update. Nucleic Acids Res. 2014; 42:D1083–D1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gilson M.K., Liu T., Baitaluk M., Nicola G., Hwang L., Chong J.. BindingDB in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2016; 44:D1045–D1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhu F., Shi Z., Qin C., Tao L., Liu X., Xu F., Zhang L., Song Y., Liu X., Zhang J. et al. Therapeutic target database update 2012: a resource for facilitating target-oriented drug discovery. Nucleic Acids Res. 2012; 40:D1128–D1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Davis A.P., Grondin C.J., Johnson R.J., Sciaky D., King B.L., McMorran R., Wiegers J., Wiegers T.C., Mattingly C.J.. The Comparative Toxicogenomics Database: update 2017. Nucleic Acids Res. 2017; 45:D972–D978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ursu O., Holmes J., Knockel J., Bologa C.G., Yang J.J., Mathias S.L., Nelson S.J., Oprea T.I.. DrugCentral: online drug compendium. Nucleic Acids Res. 2017; 45:D932–D939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E.. The Protein Data Bank. Nucleic Acids Res. 2000; 28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Allen F.H. The Cambridge Structural Database: a quarter of a million crystal structures and rising. Acta Crystallogr. B. 2002; 58:380–388. [DOI] [PubMed] [Google Scholar]
- 12. Pihan E., Colliandre L., Guichou J.F., Douguet D.. e-Drug3D: 3D structure collections dedicated to drug repurposing and fragment-based drug design. Bioinformatics. 2012; 28:1540–1541. [DOI] [PubMed] [Google Scholar]
- 13. Goede A., Dunkel M., Mester N., Frommel C., Preissner R.. SuperDrug: a conformational drug database. Bioinformatics. 2005; 21:1751–1753. [DOI] [PubMed] [Google Scholar]
- 14. Siramshetty V.B., Nickel J., Omieczynski C., Gohlke B.O., Drwal M.N., Preissner R.. WITHDRAWN–a resource for withdrawn and discontinued drugs. Nucleic Acids Res. 2016; 44:D1080–D1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rego N., Koes D.. 3Dmol.js: molecular visualization with WebGL. Bioinformatics. 2015; 31:1322–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Onakpoya I.J., Heneghan C.J., Aronson J.K.. Post-marketing withdrawal of 462 medicinal products because of adverse drug reactions: a systematic review of the world literature. BMC medicine. 2016; 14:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hu Y., Bajorath J.. Influence of search parameters and criteria on compound selection, promiscuity, and pan assay interference characteristics. J. Chem. Inf. Model. 2014; 54:3056–3066. [DOI] [PubMed] [Google Scholar]
- 18. Lounkine E., Keiser M.J., Whitebread S., Mikhailov D., Hamon J., Jenkins J.L., Lavan P., Weber E., Doak A.K., Cote S. et al. Large-scale prediction and testing of drug activity on side-effect targets. Nature. 2012; 486:361–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nickel J., Gohlke B.O., Erehman J., Banerjee P., Rong W.W., Goede A., Dunkel M., Preissner R.. SuperPred: update on drug classification and target prediction. Nucleic Acids Res. 2014; 42:W26–W31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Steinbeck C., Han Y., Kuhn S., Horlacher O., Luttmann E., Willighagen E.. The Chemistry Development Kit (CDK): an open-source Java library for Chemo- and Bioinformatics. J. Chem. Inf. Comput. Sci. 2003; 43:493–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kabsch W. A solution for the best rotation to relate two sets of vectors. Acta Cryst. 1976; A32:922–923. [Google Scholar]
- 22. Gohlke B.O., Overkamp T., Richter A., Richter A., Daniel P.T., Gillissen B., Preissner R.. 2D and 3D similarity landscape analysis identifies PARP as a novel off-target for the drug Vatalanib. BMC Bioinformatics. 2015; 16:308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kuhn M., Letunic I., Jensen L.J., Bork P.. The SIDER database of drugs and side effects. Nucleic Acids Res. 2016; 44:D1075–D1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schulz M., Iwersen-Bergmann S., Andresen H., Schmoldt A.. Therapeutic and toxic blood concentrations of nearly 1,000 drugs and other xenobiotics. Crit. Care. 2012; 16:R136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. By the American Geriatrics Society 2015 Beers Criteria Update Expert Panel American Geriatrics Society 2015 updated beers criteria for potentially inappropriate medication use in older adults. J. Am. Geriatr. Soc. 2015; 63:2227–2246. [DOI] [PubMed] [Google Scholar]
- 26. Holt S., Schmiedl S., Thurmann P.A.. Potentially inappropriate medications in the elderly: the PRISCUS list. Deutsches Arzteblatt Int. 2010; 107:543–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. McGonigle S., Chen Z., Wu J., Chang P., Kolber-Simonds D., Ackermann K., Twine N.C., Shie J.L., Miu J.T., Huang K.C. et al. E7449: A dual inhibitor of PARP1/2 and tankyrase1/2 inhibits growth of DNA repair deficient tumors and antagonizes Wnt signaling. Oncotarget. 2015; 6:41307–41323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hoffmann M.F., Preissner S.C., Nickel J., Dunkel M., Preissner R., Preissner S.. The Transformer database: biotransformation of xenobiotics. Nucleic Acids Res. 2014; 42:D1113–D1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yasar U., Forslund-Bergengren C., Tybring G., Dorado P., Llerena A., Sjoqvist F., Eliasson E., Dahl M.L.. Pharmacokinetics of losartan and its metabolite E-3174 in relation to the CYP2C9 genotype. Clin. Pharmacol. Ther. 2002; 71:89–98. [DOI] [PubMed] [Google Scholar]
- 30. Yasar U., Tybring G., Hidestrand M., Oscarson M., Ingelman-Sundberg M., Dahl M.L., Eliasson E.. Role of CYP2C9 polymorphism in losartan oxidation. Drug Metab. Disposition. 2001; 29:1051–1056. [PubMed] [Google Scholar]
- 31. Szauder I., Csajagi E., Major Z., Pavlik G., Ujhelyi G.. Treatment of hypertension: favourable effect of the twice-daily compared to the once-daily (evening) administration of perindopril and losartan. Kidney Blood Pressure Res. 2015; 40:374–385. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.