

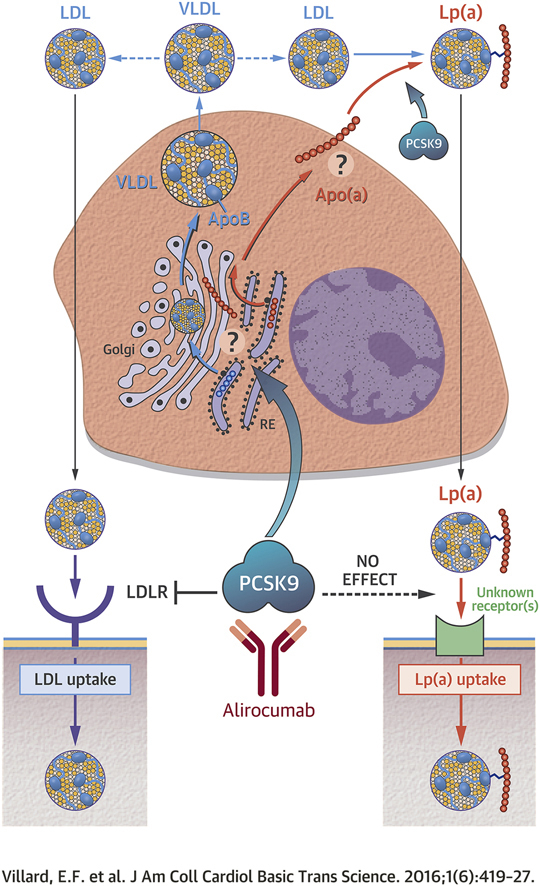
Visual Abstract
Key Words: familial hypercholesterolemia, LDL receptor, lipoprotein(a), PCSK9, primary human hepatocytes
Abbreviations and Acronyms: apo, apolipoprotein; BODIPY, boron dipyrromethene; FH, familial hypercholesterolemia; He, heterozygote; Ho, homozygote; LC-MS/MS, liquid chromatography-tandem mass spectrometry; LDL-C, low-density lipoprotein cholesterol; LDLR, low-density lipoprotein receptor; Lp(a), lipoprotein(a); mAbs, monoclonal antibodies; PCSK9, proprotein convertase subtilisin kexin type 9
Highlights
-
•
Unlike LDL uptake, Lp(a) uptake is not altered by PCSK9 or PCSK9 inhibition in primary human hepatocytes and in primary dermal fibroblasts isolated from familial hypercholesterolemic and non–familial hypercholesterolemic patients.
-
•
Lp(a) uptake is occurring in the absence of a functional LDL receptor and is not affected by LDL receptor blockade with monoclonal antibodies.
-
•
Lp(a) cellular binding and whole particle uptake are not altered by PCSK9.
-
•
The secretion of Lp(a) from primary human hepatocytes is enhanced by PCSK9, an effect that is blunted by PCSK9 inhibition with alirocumab.
Summary
To elucidate how the proprotein convertase subtilisin kexin type 9 (PCSK9) inhibitor alirocumab modulates lipoprotein(a) [Lp(a)] plasma levels, the authors performed a series of Lp(a) uptake studies in primary human hepatocytes and dermal fibroblasts and measured Lp(a) secretion from human hepatocytes. They found that Lp(a) cellular uptake occurred in a low-density lipoprotein receptor–independent manner. Neither PCSK9 nor alirocumab altered Lp(a) internalization. By contrast, the secretion of apolipoprotein (a) from human hepatocytes was sharply increased by PCSK9, an effect that was reversed by alirocumab. They propose that PCSK9 does not significantly modulate Lp(a) catabolism, but rather enhances the secretion of Lp(a) from liver cells.
Proprotein convertase subtilisin kexin type 9 (PCSK9) inhibition with monoclonal antibodies (mAbs), either as monotherapy or in combination with statins, recently emerged as a promising strategy to lower circulating low-density lipoprotein cholesterol (LDL-C) in patients with dyslipidemia and cardiovascular disease risk 1, 2, 3. PCSK9 is a natural circulating inhibitor of the low-density lipoprotein receptor (LDLR). It binds to the LDLR, and after endocytosis, targets the LDLR that would otherwise recycle back to the cell surface for lysosomal degradation (4). Both statins and anti-PCSK9 mAbs act by increasing the abundance of LDLR at the surface of hepatocytes, promoting an accelerated clearance of circulating low-density lipoprotein (LDL) particles, thus lowering LDL-C levels 4, 5. But, unlike statins (6), anti-PCSK9 mAbs also promote an unexplained 25% to 30% reduction in circulating lipoprotein (a) [Lp(a)] levels 1, 2, 7.
There is strong epidemiological evidence that Lp(a) is a highly atherogenic lipoprotein (8), because elevated Lp(a) levels are independently and significantly associated with cardiovascular disease 6, 9, 10. Lp(a) consists of a unique protein homologous to plasminogen, apolipoprotein (apo) (a) [apo(a)], that is covalently tethered to the apolipoprotein B100 [apoB100] moiety of an LDL particle by a unique disulfide bond (11). Apo(a) is a high-molecular-weight glycoprotein (∼300 to 800 kDa), expressed exclusively by the liver, that contains from 3 to more than 40 identical Kringle IV2 domains. A strong inverse relationship exists between the apo(a) isoform size and Lp(a) plasma concentration in humans (12). Circulating Lp(a) concentrations appear to be primarily controlled by synthesis rather than catabolism (12). The molecular and cellular pathways governing apo(a)/Lp(a) hepatic production and Lp(a) cellular uptake and degradation are not well understood (13). The potential role of the LDLR in Lp(a) clearance remains controversial 8, 14.
There is currently a clear need to understand how, unlike statins, anti-PCSK9 mAbs reduce the circulating levels of Lp(a) in patients. This could result from an enhanced clearance and/or a reduced production of Lp(a). To answer this question, we have investigated the role of PCSK9 and of the LDLR in mediating Lp(a) cellular uptake. We also investigated the effects of PCSK9 and of the anti-PCSK9 monoclonal antibody alirocumab on Lp(a) secretion from primary human hepatocytes. We report that Lp(a) production from hepatocytes is enhanced by PCSK9 and blunted by alirocumab, whereas neither PCSK9 nor the LDLR seem to significantly modulate Lp(a) catabolism.
Methods
Fibroblasts
Primary normal human dermal fibroblasts were purchased from PromoCell (Heidelberg, Germany), and LDLR defective and negative dermal fibroblasts were obtained either from the Corriel Cell Repository (Camden, New Jersey) or isolated from forearm biopsies of heterozygous or homozygous familial hypercholesterolemic (FH) patients at Groote Schuur Hospital (Cape Town, South Africa), after obtaining written informed consent 5, 15. Dr. Jean-Pierre Rabès (University of Versailles St-Quentin, France) genotype-checked each of the HDF lines to ascertain its LDLR mutation status. Fibroblasts were grown in DMEM (Thermo Fisher Scientific, Saint-Aubin, France) containing 20% fetal calf serum.
Hepatocytes
Human primary hepatocytes isolated from 2 donors (BioreclamationIVT, Baltimore, Maryland) were thawed in GRO-CP culture medium. After 5 h, GRO-CP was replaced with GRO-HI culture medium (BioreclamationIVT). Hepatocytes were seeded in 6 collagen-I–coated well plates with increasing concentrations (0, 1.2, and 3.1 μg/ml) of recombinant PCSK9 (Cyclex, Nagano, Japan) in the absence or presence of saturating concentrations of alirocumab (8 μg/ml) for 72 h, by replacing one-half of the culture medium every day with fresh GRO-HI medium, supplemented with or without PCSK9 and/or alirocumab.
apoB100 and apo(a) secretion from hepatocytes
The culture medium was collected, spun to get rid of cell debris, and assessed for apoB100 concentration with the EA 7001-1 sandwich enzyme-linked immunosorbent assay (ELISA) kit (Assaypro, St Charles, Missouri), and for apo(a) concentration by ELISA with the EL3001-1 sandwich ELISA kit (Assaypro) as well as by liquid chromatography-tandem mass spectrometry (LC-MS/MS). Density KBr gradient ultracentrifugation was performed on 72-h–conditioned hepatocyte media to separately quantify lipidated Lp(a) (density >1.21 g/ml) from lipid-free apo(a) (density <1.21 g/ml) using apo(a) and apoB ELISA kits. Apo(a) quantifications were performed by LC-MS/MS (16); detailed methods of this assay can be found in the Supplemental Appendix.
LDLR expression
Hepatocytes were washed twice with phosphate-buffered saline (PBS), and cellular proteins were extracted in 100-μl cell lysis buffer 9803 (Cell Signaling Technology, Saint Quentin en Yvelines, France) supplemented with complete mini-protease inhibitor cocktail (Sigma-Aldrich, St. Louis, Missouri). LDLR immunoblot analyses were performed with a rabbit antihuman-LDLR polyclonal antibody PAB8804 (Abnova, Walnut, California) on a Sally Sue Simple Western device, using the 66-440 kDa separation master kit PS-MK12 (Protein Simple, San Jose, California), and normalized to transferrin receptor expression assessed using the AF2474 primary antibody (R&D Systems, Lille, France).
Lp(a) isolation, purification, and labeling
Human plasma from 6 anonymous donors with Lp(a) levels above 30 mg/dl was purchased from BioreclamationIVT. We determined by LC-MS/MS their mean number of Kringle IV2 domains to be 9.8, 9.9, 11.6, 12.1, 17.5, and 19.1. Lp(a) was isolated by sequential ultracentrifugation (1.050 < d <1.125 g/ml) at 40,000 g. Lp(a) fraction was dialyzed against PBS (137 mmol/l NaCl, 2.7 mmol/l KCl 8 mmol/l Na2HPO4, and 2 mmol/l KH2PO4) and purified by fast performance liquid chromatography on a lysine sepharose 4B column (GE Healthcare, Velizy-Villacoublay, France). Lp(a) was subsequently dialyzed against PBS, fluorescently labeled with boron dipyrromethene (BODIPY) (Thermo Fisher Scientific), and purified by gel filtration on Sephadex G-25 columns (NAP-5, Sephadex G25 0.5 ml, GE Healthcare). The absence of free label was checked by high-performance liquid chromatography on BioSec5 columns (Agilent Technologies, Santa Clara, California). The structural integrity of Lp(a) was verified by electrophoresis migration on buffered agarose hydrogels (pH 8.5) using the Hydrasis electrophoresis system (Sebia, Norcross, Georgia) and Sudan Black staining (Supplemental Figure 1). Lp(a), either reduced with 50 mmol/l dithiothreitol or not reduced, was subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis on 4% to 12% Tris-Glycine gels (Thermo Fisher Scientific); transferred onto nitrocellulose membrane; and immunoblotted with a rabbit monoclonal anti-human apo(a) antibody, clone EPR6474, and an antirabbit immunoglobulin G horseradish peroxidase secondary antibody (OriGene, Rockville, Maryland). Immunoreactive bands were visualized with Amersham ECL western blotting detection reagent (GE Healthcare).
Fluorescent LDL and Lp(a) uptake in fibroblasts
Control and FH fibroblasts were seeded in flat-bottomed, 96-well plates (35,000 cells/well) in DMEM containing 0.5% fetal calf serum for 24 h at 37°C and were subsequently supplemented with 10 μg/ml mevastatin (Sigma-Aldrich) for 24 h. The medium was replaced with fresh medium containing 10 μg/ml mevastatin with or without recombinant gain-of-function PCSK9 (600 ng/ml) (Cyclex) with or without saturating concentrations of alirocumab (19.2 μg/ml) for 5 h at 37°C 5, 15. LDL-BODIPY (Life Technologies, Saint Aubin, France) or Lp(a)-BODIPY (10 μg/ml final concentration) were added to the culture medium for the final 3 h of the incubation time. In a subset of experiments, an excess of unlabeled Lp(a) or LDL (200 μg/ml) was added 5 min before the addition of fluorescent lipoproteins. Fibroblasts were washed gently with PBS, lifted with Accutase (Sigma-Aldrich), and resuspended in ice-cold PBS containing 1% bovine serum albumin (BSA). Cells were then washed twice in ice-cold PBS with 1% BSA and once in ice-cold PBS before fixation with PBS containing 0.5% paraformaldehyde (Sigma-Aldrich). Finally, cells were resuspended in PBS supplemented with 0.2% trypan blue to quench cell-surface bound fluorescent LDL or Lp(a) prior to flow cytometry analysis (17). Cells were analyzed using an LSRII cytometer (Beckton Dickinson, Le Pont de Claix, France) and the FlowJo software (Tree Star, Ashland, Oregon) of the Cellular and Tissular Imaging Core Facility of Nantes University (MicroPICell). The mean fluorescence intensity of cells incubated without fluorescent lipoproteins was subtracted from the mean fluorescence intensity of cells incubated with fluorescent lipoproteins to determine the specific mean fluorescence intensity level of lipoprotein uptake, expressed in arbitrary units 5, 15. In a subset of experiments, control and LDLR defective homozygote (Ho) FH fibroblasts were seeded in poly-L-lysine–coated 8-well Millicell EZ slides (Merck-Millipore, Fontenay Sous Bois, France) (15,000 cells/well). Cells were treated as described in the previous text. LDL-BODIPY or Lp(a)-BODIPY (20 μg/ml) was added to the medium for the final 4 h of the incubation. Cells were washed twice in ice-cold PBS with 1% BSA, rinsed twice in ice-cold PBS, fixed in PBS containing 4% paraformaldehyde for 15 min at room temperature, and then rinsed twice in PBS. Slides were mounted with cover slides in Prolong antifade reagent containing DAPI (Life Technologies) and visualized on a confocal A1 N-SIM microscope (Nikon, Melville, New York).
LDL and Lp(a) cellular uptake in hepatocytes
Human primary hepatocytes were seeded in flat-bottom 96-well collagen-I–coated plates (50,000 cells/well) in GRO-CP culture medium for 5 h at 37°C and then grown overnight in serum-free GRO-HI medium. Hepatocytes were then treated with or without recombinant wild-type PCSK9 (3.1 μg/ml) for 24 h. LDL and Lp(a) uptake experiments were conducted for 5 h, as described in the previous text for primary fibroblasts, in the presence or absence of recombinant wild-type PCSK9 (0, 1.2, and 3.1 μg/ml), with or without an LDLR-blocking antibody (500 μg/ml) or immunoglobulin G isotype control (clones 472,413 and 11,711, respectively, R&D Systems) and/or saturating concentrations (8 μg/ml) of alirocumab. Supernatants were harvested for Lp(a) integrity assessment by western blot under reducing and nonreducing conditions, as described in the previous text, and cells were washed twice with PBS before fluorescence measurement on a Tecan spectrofluorometer (Tecan, Männedorf, Switzerland). In a subset of experiments, hepatocytes were washed intensively (i.e., 3× with PBS with 0.8% BSA, twice with PBS with 0.8% BSA supplemented with 0.2 mol/l epsilon aminocaproic acid, and twice with 0.2 mol/l acetic acid/0.5 mol/l NaCl solution [pH 2.5]), as previously described (14), before fluorescence measurements. Cellular proteins were collected in cell lysis buffer 9803 (Cell Signaling Technology) supplemented with cOmplete Mini protease inhibitor cocktail and intracellular apo(a) contents assessed by ELISA, as in the previous text.
Statistical analyses
Analyses were performed using SAS version 9.2 for Windows 7 (SAS Institute, Cary, North Carolina). Data are presented as mean ± SEM. Comparisons between groups were made using 1- and 2-way analysis of variance with Newman-Keuls post hoc tests. All p values <0.05 are considered statistically significant.
Results
The LDLR does not promote Lp(a) cellular uptake ex vivo
The role of the LDLR on Lp(a) cellular uptake was first investigated in primary fibroblasts isolated from 1 wild-type control, 1 heterozygous (He) FH, 7 LDLR-defective HoFH, and 4 LDLR-negative HoFH patients (Supplemental Table 1) treated with mevastatin with or without PCSK9 in the presence or absence of alirocumab. We verified that LDL cellular uptake correlated with LDLR cell surface expression levels 5, 15. Maximal fluorescent LDL internalization was observed in control fibroblasts, followed by HeFH fibroblasts, and then receptor-defective and receptor-negative HoFH fibroblasts (Figure 1A). PCSK9 reduced whereas alirocumab restored LDL uptake in all fibroblast lines, except for those receptor-negative HoFH fibroblasts totally lacking LDLR function (Figures 1A and 1B). Fluorescent LDL uptake was reduced by the addition of 20-fold excess unlabeled LDL to the culture medium (Figure 1C). By contrast, Lp(a) uptake was similar in all fibroblast lines, irrespective of the LDLR mutation status or LDLR expression level. Neither PCSK9 nor alirocumab significantly altered the ability of any tested fibroblast line to internalize fluorescent Lp(a) (Figures 1D and 1E). Fluorescent Lp(a) uptake was reduced similarly by the addition of 20-fold excess unlabeled Lp(a) into the culture medium in control and FH fibroblasts (Figure 1F). These data demonstrate that Lp(a) cellular uptake is not dependent on the presence of a functional LDLR.
Figure 1.

Lp(a) Uptake Is Not Modulated by LDLR Expression in Human Fibroblasts
Flow cytometric assessment of fluorescent low-density lipoprotein (LDL) (A) and fluorescent lipoprotein(a) [Lp(a)] (D) uptake in dermal skin fibroblast isolated from 1 non–familial hypercholesterolemia (FH) (control), 1 heterozygous (He) FH, 7 receptor-defective homozygous (Ho) FH, and 4 receptor-negative HoFH patients. Cells were treated with mevastatin with or without recombinant gain-of-function proprotein convertase subtilisin kexin type 9 (PCSK9) (600 ng/ml) and/or alirocumab (19.2 μg/ml). Representative confocal microscopy images (Z stacks) of fluorescent LDL (red hot)(B) and fluorescent Lp(a) (yellow hot) (E) cellular uptake in control and HoFH fibroblasts are displayed. Nuclei are shown in blue. Relative uptake of fluorescent LDL (C) and fluorescent Lp(a) (F) in the presence of 20-fold excess unlabeled LDL and Lp(a), respectively, in fibroblasts from 1 non-FH control and 3 HoFH patients. ΔMFI is the mean fluorescence intensity difference between cells incubated with and cells incubated without fluorescent lipoproteins (i.e., autofluorescence). ΔMFI is in arbitrary units. *p < 0.001, **p < 0.03 vs. non-FH control without PCSK9 and without LDL excess, #p < 0.005 vs. non-FH control without PCSK9 and without alirocumab, using 2-way analysis of variance on rank-transformed values with post-hoc Newman-Keuls test. Histograms represent the mean ± SEM of a minimum of 3 independent experiments (except for HeFH, n = 1).
PCSK9 inhibits LDL, but not Lp(a), cellular uptake by human primary hepatocytes
To elucidate whether PCSK9 alters Lp(a) hepatic clearance, primary human hepatocytes isolated from 2 donors were treated with increasing concentrations of PCSK9 in the absence or presence of saturating concentrations of alirocumab. Cellular LDLR expression assessed by Western blot was reduced in a dose-dependent manner in hepatocytes treated with increasing doses of PCSK9, an effect that was abolished by alirocumab (Figure 2). LDL cellular uptake by primary human hepatocytes correlated with LDLR expression levels and was reduced in cells treated with PCSK9. This effect of PCSK9 on LDL cellular uptake was reversed by alirocumab (Figure 3A). We verified the functionality of the LDLR in primary human hepatocytes by adding an antibody targeting the extracellular domain of the LDLR to the culture medium. Blocking the LDLR with this antibody reduced the cellular uptake of fluorescent LDL (Figure 3A).
Figure 2.

PCSK9 Reduces LDLR Expression in Human Primary Hepatocytes: An Effect Blocked by Alirocumab
Low-density lipoprotein receptor (LDLR) expression was measured in hepatocytes, treated for 72 h with increasing concentrations of recombinant PCSK9 with or without saturating concentrations (8 μg/ml) of alirocumab, by simple western blot. LDLR levels were normalized to transferrin receptor expression. Histograms represent the mean ± SEM of 3 independent experiments. *p < 0.01 vs. control (no PCSK9); **p < 0.05 vs. same dose of PCSK9 without alirocumab, using 1-way analysis of variance on rank-transformed values with post-hoc Newman-Keuls test. A representative simple Western image is displayed. Abbreviations as in Figure 1.
Figure 3.

Inhibiting LDLR Expression With PCSK9 or LDLR Function With a Blocking Antibody Does Not Affect Lp(a) Uptake by Human Primary Hepatocytes
Cells were treated with recombinant PCSK9 (± alirocumab) or an anti-LDLR blocking antibody (Ab). An isotype control antibody was used as control (Ctrl). Fluorescent LDL (A) and fluorescent Lp(a) (B) cellular uptake was measured. *p < 0.001 vs. control (black bars). **p < 0.001 vs. PCSK9 treatment (white bars). ***p < 0.001 vs. anti-LDLR blocking antibody (light gray bars), using 1-way analysis of variance analysis on rank-transformed values with post-hoc Newman-Keuls test. Histograms represent the mean ± SEM of 7 independent experiments from 2 hepatocyte donors. Abbreviations as in Figures 1 and 2.
By contrast, PCSK9, alirocumab, and the antibody targeting the extracellular domain of the LDLR did not significantly alter the cellular uptake of fluorescent Lp(a) in human primary hepatocytes (Figure 3B). The inability of PCSK9 to modulate Lp(a) cellular uptake was observed across a broad range of Lp(a) concentrations (Supplemental Figure 2). To demonstrate that primary hepatocytes effectively promote cellular endocytosis of both LDL and Lp(a), cells were washed with a highly stringent buffer to eliminate any LDL and Lp(a) particles that were bound to the cell surface. After stringent washing, PCSK9 reduced fluorescent LDL endocytosis but had no effect on fluorescent Lp(a) endocytosis (Supplemental Figure 3). We confirmed that native Lp(a) (i.e., unlabeled) and fluorescently labeled Lp(a) were equally internalized by primary hepatocytes by measuring intracellular apo(a) contents. Furthermore, we demonstrated that PCSK9 had no effect on native Lp(a) uptake by primary hepatocytes (Supplemental Figure 4). The integrity of Lp(a) particles, in particular the covalent tethering of apo(a) to apoB100 during the cellular incubation process, was verified by immunoblot analysis under denaturing and nondenaturing conditions (Supplemental Figure 5). Taken together, these data demonstrate that PCSK9 does not modulate Lp(a) cellular uptake in human hepatocytes.
PCSK9 enhances apo(a)/Lp(a) cellular secretion by human primary hepatocytes
To elucidate the potential role of PCSK9 in modulating Lp(a) secretion, we used primary human hepatocytes isolated from 2 donors and grown in the absence of serum lipoproteins. Cells were incubated with increasing concentrations of PCSK9 in the absence or presence of saturating concentrations of alirocumab. The amounts of human apoB100 and apo(a) in the culture medium were quantified by ELISA and/or LC-MS/MS. Both lipid-free and lipidated apoB and apo(a) were detected in the culture medium of human primary hepatocytes, indicating that these cells produce and secrete apoB-containing lipoproteins (LpB) as well as Lp(a). The addition of PCSK9 increased the concentration of apoB in the culture medium by 2-fold and apo(a) in the culture medium by up to 3-fold; both effects were reversed by alirocumab (Figures 4A and 4B). The effects of PCSK9 were similar in hepatocytes isolated from both donors.
Figure 4.

PCSK9 Treatment Increases Apo(a) Secretion From Primary Human Hepatocytes: An Effect Blocked by Alirocumab
Cells were treated with increasing concentrations of recombinant PCSK9 with or without alirocumab for 72 h. ApoB100 (A) and apo(a) (B) were quantified in the culture medium by enzyme-linked immunosorbent assay and liquid chromatography-tandem mass spectrometry, respectively. *p < 0.05 vs. control (no PCSK9). **p < 0.05 vs. same dose of PCSK9 without alirocumab, using 1-way analysis of variance on rank-transformed values with post-hoc Newman-Keuls test. Histograms represent the mean ± SEM of 3 independent experiments from 2 hepatocyte donors. Abbreviations as in Figure 1.
Discussion
In this work, we investigated the mechanisms by which anti-PCSK9 mAbs reduce circulating Lp(a) levels in humans. We showed that Lp(a) cellular uptake was not modulated by the LDLR or by PCSK9 ex vivo. Furthermore, we demonstrated that PCSK9 enhanced Lp(a) production from primary hepatocytes, an effect that was blunted by alirocumab.
The physiological role of the LDLR in mediating Lp(a) plasma clearance is controversial. We used a well-characterized collection of dermal fibroblasts (from non-FH, HeFH, and HoFH patients) (5), to show that Lp(a) cellular uptake occurs to a similar extent whether the expression and function of the LDLR is maximal, abrogated genetically, or reduced by PCSK9. The functionality of the LDLR was confirmed by performing LDL uptake experiments in parallel (5). In line with our results, Reblin et al. (18) showed that the specific uptake of I125-Lp(a) (but not that of I125-LDL) was similar in wild-type and LDLR-knockout mouse embryonic fibroblasts. Furthermore, Niemeier et al. (19) found that Lp(a) was a very weak ligand for the LDLR in BIAcore studies. Our results are also in agreement with clinical studies showing that PCSK9 inhibition (with evolocumab) promoted significant reductions in both circulating LDL-C (−24%) and Lp(a) (−19%) in 6 receptor-defective HoFH patients and reduced Lp(a) (−18%), but not LDL-C (+15%) levels in 2 receptor-negative HoFH patients (20). Finally, our results are concordant with the previously published observations that up-regulating LDLR expression even with the most potent statins does not reduce Lp(a) in humans (6), and that Lp(a) plasma clearance occurs at similar catabolic rates in non-FH, HeFH, and HoFH patients 12, 21.
In this study, we also performed Lp(a) and LDL uptake experiments in primary hepatocytes obtained from 2 donors. In line with our findings in fibroblasts, we showed that PCSK9 reduces LDLR expression and function (i.e., LDL uptake) without altering Lp(a) cellular uptake. Blocking LDLR function with an antibody yielded similar results. In contrast to our findings, a recent study found that PCSK9 reduced the internalization of Lp(a) in HepG2 cells (14). We are uncertain how to explain the discrepancies between the 2 studies. One possible explanation would be that we used primary human hepatocytes, whereas Romagnuolo et al. (14) used a hepatoma cell line. However, Romagnuolo et al. (14) found that PCSK9 reduced the uptake of Lp(a) in human dermal fibroblasts expressing the LDLR. Because Romagnuolo et al. (14) observed that recombinant apo(a) uptake, unlike Lp(a) particle uptake, was not altered by PCSK9 and/or LDLR expression in dermal fibroblasts, we confirmed in the present study that apo(a) had not detached from Lp(a) particles during our incubation process (Supplemental Figure 5). A less likely explanation for the discrepancies between the 2 studies is that there were differences in apo(a) isoforms, as we used Lp(a) with apo(a) isoforms containing 9 to 19 Kringle IV2 domains, whereas Romagnuolo et al. (14) used apo(a) isoforms containing 7 to 8 Kringle IV2 domains (14). A third possible explanation for the discrepancies between the 2 studies is that we used PCSK9 concentrations (600 ng/ml of gain-of-function PCSK9 and 1.2 to 3.1 μg/ml of wild-type PCSK9) closer to the physiological range than those used by Romagnuolo et al. (14) (10 to 20 μg/ml).
Because we did not observe any effect of PCSK9 on Lp(a) cellular uptake by human hepatocytes, we tested whether PCSK9 might modulate Lp(a) secretion from these cells. We showed a PCSK9 dose-dependent increase in both apoB and apo(a) (free and lipidated) in the culture medium, an effect that was blunted by alirocumab. Because PCSK9 reduces LDLR expression, the variations in apoB levels observed in the medium of primary hepatocytes treated with PCSK9 and/or alirocumab cannot be attributed to an increased production of LpB, but must rather be a consequence of reduced clearance of newly secreted LpB, as the LDLR simultaneously promotes apoB uptake. By contrast, we found that PCSK9 and alirocumab did not modulate Lp(a) uptake in primary hepatocytes. The increase in apo(a) levels measured in the medium of human hepatocytes treated with PCSK9 therefore indicates that PCSK9 enhances the production/secretion of Lp(a) by those cells, an effect that is abolished by alirocumab. The molecular mechanisms by which PCSK9 enhances apo(a) secretion and Lp(a) assembly (i.e., disulfide bond formation between apo(a) and apoB100 on LDL particles), which likely occurs at the surface of hepatocytes (7), certainly merits further investigation. In that respect, another class of lipid-lowering drugs, the CETP inhibitors, which do not act on the LDLR, but rather on the lipid content of LDL particles, also significantly reduces Lp(a) levels. It is farfetched to speculate that PCSK9, which is known to associate with LDL particles in the plasma 22, 23, might enhance the conformation/ability of LDL particles to serve as substrates for Lp(a) formation.
Conclusions
Therefore, we propose that physiological concentrations of PCSK9 do not significantly modulate Lp(a) catabolism, but rather enhance the generation of Lp(a), and that anti-PCSK9 mAbs reduce Lp(a) levels primarily by altering this pathway. This hypothesis could be evaluated by performing a series of Lp(a) turnover studies in patients before and after treatment with monoclonal antibodies targeting PCSK9.
Perspectives.
COMPETENCY IN MEDICAL KNOWLEDGE: Statins reduce LDL-C levels by increasing the expression of the LDL receptor. Monoclonal antibodies targeting PCSK9, a new class of lipid-lowering drugs, also reduce LDL-C levels by decreasing the degradation of the LDL receptor. We now report a cellular mechanism indicating that PCSK9 enhances the hepatic production of Lp(a), a highly atherogenic lipoprotein subclass. These results underline why unlike statins, monoclonal antibodies targeting PCSK9 also reduce circulating Lp(a) in dyslipidemic patients.
TRANSLATIONAL OUTLOOK: Our results pave the way for in vivo turnover studies of major Lp(a) apolipoprotein components [apo(a) and apoB100] in patients with elevated Lp(a) plasma levels. The exact molecular and cellular mechanisms governing the secretion and assembly of Lp(a) and its modulation by PCSK9 remain to be fully elucidated.
Footnotes
This work funded by an Investigator Initiated Study Concept Research Grant from Sanofi R&D and Regeneron, and by the Agence Nationale de la Recherche Programme Recherche Hospitalière en Santé 2016 Projet CHOPIN. Drs. Villard, Blankenstein, Tran, Poirier, Le Bail, Illiano, Janiak, Muslin, and Guillot are full-time employees of Sanofi. Dr. Krempf has received consulting fees and lecture honoraria from AstraZeneca, Amgen, Merck Sharp & Dohme, and Sanofi. Dr. Blom has served on the advisory boards of Amgen, Aegerion, and Sanofi; and has received lecture honoraria from Amgen, Pfizer, AstraZeneca, and Aegerion. Dr. Lambert has received honoraria and research funding from Amgen, Sanofi-Regeneron, and Pfizer. All other authors have reported that they have no relationships relevant to the contents of this paper to disclose. Drs. Villard and Thedrez contributed equally to this work.
Contributor Information
Etienne Guillot, Email: etienne.guillot@sanofi.com.
Gilles Lambert, Email: gilles.lambert@univ-reunion.fr.
Appendix
References
- 1.Sabatine M.S., Giugliano R.P., Wiviott S.D. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372:1500–1509. doi: 10.1056/NEJMoa1500858. [DOI] [PubMed] [Google Scholar]
- 2.Robinson J.G., Farnier M., Krempf M. Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372:1489–1499. doi: 10.1056/NEJMoa1501031. [DOI] [PubMed] [Google Scholar]
- 3.Ballantyne C.M., Neutel J., Cropp A. Results of bococizumab, a monoclonal antibody against proprotein convertase subtilisin/kexin type 9, from a randomized, placebo-controlled, dose-ranging study in statin-treated subjects with hypercholesterolemia. Am J Cardiol. 2015;115:1212–1221. doi: 10.1016/j.amjcard.2015.02.006. [DOI] [PubMed] [Google Scholar]
- 4.Marais A.D., Kim J.B., Wasserman S.M., Lambert G. PCSK9 inhibition in LDL cholesterol reduction: genetics and therapeutic implications of very low plasma lipoprotein levels. Pharmacol Ther. 2015;145:58–66. doi: 10.1016/j.pharmthera.2014.07.004. [DOI] [PubMed] [Google Scholar]
- 5.Lambert G., Chatelais M., Petrides F. Normalization of low-density lipoprotein receptor expression in receptor defective homozygous familial hypercholesterolemia by inhibition of PCSK9 with alirocumab. J Am Coll Cardiol. 2014;64:2299–2300. doi: 10.1016/j.jacc.2014.07.995. [DOI] [PubMed] [Google Scholar]
- 6.Khera A.V., Everett B.M., Caulfield M.P. Lipoprotein(a) concentrations, rosuvastatin therapy, and residual vascular risk: an analysis from the JUPITER trial (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin) Circulation. 2014;129:635–642. doi: 10.1161/CIRCULATIONAHA.113.004406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raal F.J., Giugliano R.P., Sabatine M.S. Reduction in lipoprotein(a) with PCSK9 monoclonal antibody evolocumab (AMG 145): a pooled analysis of more than 1,300 patients in 4 phase II trials. J Am Coll Cardiol. 2014;63:1278–1288. doi: 10.1016/j.jacc.2014.01.006. [DOI] [PubMed] [Google Scholar]
- 8.Lamon-Fava S., Diffenderfer M.R., Marcovina S.M. Lipoprotein(a) metabolism. Curr Opin Lipidol. 2014;25:189–193. doi: 10.1097/MOL.0000000000000070. [DOI] [PubMed] [Google Scholar]
- 9.Alonso R., Andres E., Mata N. Lipoprotein(a) levels in hypercholesterolemia: an important predictor of cardiovascular disease independent of the type of LDL receptor mutation. J Am Coll Cardiol. 2014;63:1982–1989. doi: 10.1016/j.jacc.2014.01.063. [DOI] [PubMed] [Google Scholar]
- 10.Erqou S., Kaptoge S., Perry P.L., for the Emerging Risk Factors Collaboration Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA. 2009;302:412–423. doi: 10.1001/jama.2009.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kronenberg F., Utermann G. Lipoprotein(a): resurrected by genetics. J Intern Med. 2013;273:6–30. doi: 10.1111/j.1365-2796.2012.02592.x. [DOI] [PubMed] [Google Scholar]
- 12.Rader D.J., Cain W., Ikewaki K. The inverse association of plasma lipoprotein(a) concentrations with apolipoprotein(a) isoform size is not due to differences in Lp(a) catabolism but to differences in production rate. J Clin Invest. 1994;93:2758–2763. doi: 10.1172/JCI117292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsimikas S., Hall J.L. Lipoprotein(a) as a potential causal genetic risk factor of cardiovascular disease: a rationale for increased efforts to understand its pathophysiology and develop targeted therapies. J Am Coll Cardiol. 2012;60:716–721. doi: 10.1016/j.jacc.2012.04.038. [DOI] [PubMed] [Google Scholar]
- 14.Romagnuolo R., Scipione C.A., Boffa M.B., Marcovina S.M., Seidah N.G., Koschinsky M.L. Lipoprotein(a) catabolism is regulated by proprotein convertase subtilisin/kexin type 9 through the low density lipoprotein receptor. J Biol Chem. 2015;290:11649–11662. doi: 10.1074/jbc.M114.611988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lambert G., Petrides F., Chatelais M. Elevated plasma PCSK9 level is equally detrimental for patients with nonfamilial hypercholesterolemia and heterozygous familial hypercholesterolemia, irrespective of low-density lipoprotein receptor defects. J Am Coll Cardiol. 2014;63:2365–2373. doi: 10.1016/j.jacc.2014.02.538. [DOI] [PubMed] [Google Scholar]
- 16.Croyal M., Ouguerram K., Passard M. Effects of extended-release nicotinic acid on apolipoprotein (a) kinetics in hypertriglyceridemic patients. Arterioscler Thromb Vasc Biol. 2015;35:2042–2047. doi: 10.1161/ATVBAHA.115.305835. [DOI] [PubMed] [Google Scholar]
- 17.Etxebarria A., Benito-Vicente A., Palacios L. Functional characterization and classification of frequent low-density lipoprotein receptor variants. Hum Mutat. 2015;36:129–141. doi: 10.1002/humu.22721. [DOI] [PubMed] [Google Scholar]
- 18.Reblin T., Niemeier A., Meyer N. Cellular uptake of lipoprotein[a] by mouse embryonic fibroblasts via the LDL receptor and the LDL receptor-related protein. J Lipid Res. 1997;38:2103–2110. [PubMed] [Google Scholar]
- 19.Niemeier A., Willnow T., Dieplinger H. Identification of megalin/gp330 as a receptor for lipoprotein(a) in vitro. Arterioscler Thromb Vasc Biol. 1999;19:552–561. doi: 10.1161/01.atv.19.3.552. [DOI] [PubMed] [Google Scholar]
- 20.Stein E.A., Honarpour N., Wasserman S.M., Xu F., Scott R., Raal F.J. Effect of the proprotein convertase subtilisin/kexin 9 monoclonal antibody, AMG 145, in homozygous familial hypercholesterolemia. Circulation. 2013;128:2113–2120. doi: 10.1161/CIRCULATIONAHA.113.004678. [DOI] [PubMed] [Google Scholar]
- 21.Rader D.J., Mann W.A., Cain W. The low density lipoprotein receptor is not required for normal catabolism of Lp(a) in humans. J Clin Invest. 1995;95:1403–1408. doi: 10.1172/JCI117794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tavori H., Rashid S., Fazio S. On the function and homeostasis of PCSK9: reciprocal interaction with LDLR and additional lipid effects. Atherosclerosis. 2015;238:264–270. doi: 10.1016/j.atherosclerosis.2014.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tavori H., Giunzioni I., Linton M.F., Fazio S. Loss of plasma proprotein convertase subtilisin/kexin 9 (PCSK9) after lipoprotein apheresis. Circ Res. 2013;113:1290–1295. doi: 10.1161/CIRCRESAHA.113.302655. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.