

Abstract
Background
Secretory Carrier-Associated Membrane Proteins (SCAMPs) are highly conserved 32–38 kDa proteins that are involved in membrane trafficking. A systems approach was taken to elucidate function of SCAMPs in wood formation of Populus trees. Phenotypic and multi-omics analyses were performed in woody tissues of transgenic Populus trees carrying an RNAi construct for Populus tremula x tremuloides SCAMP3 (PttSCAMP3; Potri.019G104000).
Results
The woody tissues of the transgenic trees displayed increased amounts of both polysaccharides and lignin oligomers, indicating increased deposition of both the carbohydrate and lignin components of the secondary cell walls. This coincided with a tendency towards increased wood density as well as significantly increased thickness of the suberized cork in the transgenic lines. Multivariate OnPLS (orthogonal projections to latent structures) modeling of five different omics datasets (the transcriptome, proteome, GC-MS metabolome, LC-MS metabolome and pyrolysis-GC/MS metabolome) collected from the secondary xylem tissues of the stem revealed systemic variation in the different variables in the transgenic lines, including changes that correlated with the changes in the secondary cell wall composition. The OnPLS model also identified a rather large number of proteins that were more abundant in the transgenic lines than in the wild type. Several of these were related to secretion and/or endocytosis as well as both primary and secondary cell wall biosynthesis.
Conclusions
Populus SCAMP proteins were shown to influence accumulation of secondary cell wall components, including polysaccharides and phenolic compounds, in the woody tissues of Populus tree stems. Our multi-omics analyses combined with the OnPLS modelling suggest that this function is mediated by changes in membrane trafficking to fine-tune the abundance of cell wall precursors and/or proteins involved in cell wall biosynthesis and transport. The data provides a multi-level source of information for future studies on the function of the SCAMP proteins in plant stem tissues.
Electronic supplementary material
The online version of this article (10.1186/s12864-017-4411-1) contains supplementary material, which is available to authorized users.
Keywords: Secretory Carrier-Associated Membrane Protein (SCAMP), Populus, Wood chemistry, Wood density, Biomass, Bioprocessing, Cork, Multi-omics
Background
Forest trees are an important source of renewable products such as biofuels and bioenergy. They are highly efficient in incorporating atmospheric carbon into the tree trunk, and increased forest stocks have recently been identified by the Intergovernmental Panel on Climate Change as, possibly, the most efficient way to combat further increases in atmospheric CO2 [1]. Forest stocks can be increased by increasing carbon flow into the secondary cell walls in the woody tissues of the stem. Several different approaches have been taken to modify biosynthesis of the individual secondary cell wall components for the purpose of increased biomass production. One of them is reducing lignin content by genetic engineering, in natural variants or in forest tree hybrids, which has in some cases been shown to increase the growth of forest trees [2–4], but in other cases to impair tree growth and also the water transport capacity of the trees [5–8]. Only a few reports exist on targeted modification or natural variants of genes that promote cellulose or hemicellulose biosynthesis. Overexpression of a sucrose synthase induced an increase in cellulose biosynthesis and wood density without interfering with growth of hybrid poplar trees [9]. Overexpression of the hemicellulose catabolic xyloglucanase also increased cellulose content and density of the wood in white poplar [10], but reduced tree growth in long-term cultivation [11]. Due to the imminent risk for growth penalty when modifying secondary cell wall biosynthesis, it is important to find additional genetic engineering strategies to improve biomass production of forest trees in a manner that does not have adverse effects on the growth of the trees.
Multi-omics approaches, including simultaneous profiling of the transcriptome, proteome and the metabolome, can be helpful when trying to improve complex processes such as growth and biomass production [12, 13]. Such multi-omics approaches have so far not been extensively utilized in forest trees [14] but have proven instrumental in other contexts, for example in creating strategies to increase carotenoid biosynthesis in maize kernels [15] or secondary metabolite accumulation in grape berries [16]. In addition to being quite expensive, a multidisciplinary approach can be challenging due to difficulties encountered when interpreting the multilayered datasets. A good solution for the latter problem is to employ multivariate methods such as OnPLS (orthogonal projections to latent structures) [17, 18] which can handle noisy, multicollinear datasets with many more variables than samples and identify significant variation in several datasets at the same time [14].
Here we took a multidisciplinary approach, combined with the most recent development of the OnPLS method, to analyze data from a set of transgenic Populus trees that had been identified among a large number of transgenic trees as having increased biomass production rates. The transgenic lines carried an RNAi construct for the PttSCAMP3 (Populus tremula x tremuloides Secretory Carrier-Associated Membrane Protein3) gene. The SCAMPs are highly conserved 32–38 kDa proteins that are localized in the endomembranes and the plasmamembrane and that, in animals, seem to be mainly involved in exocytosis in specialized secretory cells but also endocytosis and multivesicular endosome biogenesis [19]. In plants the function is unclear, although a role in lily pollen tube growth has been suggested [20]. Arabidopsis mutants in the SCAMP genes have not exhibited any obvious phenotypes [19]. Our analysis identified changes in wood chemistry, wood density and enzyme-catalyzed cell wall digestibility of the PttSCAMP3 RNAi lines. The underlying mechanisms were elucidated by analyses of the transcriptomes, proteomes and metabolomes combined with the OnPLS modelling. These results revealed not only a critical function for the SCAMP-dependent pathway in wood chemistry but also provided a systems-level interpretation of biological responses and pathways controlled by the SCAMP proteins in the woody tissues of Populus.
Methods
Transformations, growth conditions and tissue sampling
An RNAi construct for PttSCAMP3 was created by amplifying a fragment from a cDNA clone “EST G066P24” corresponding to Potri.019G104000 (SCAMP3) with the forward primer GGGGACCACTTTGTACAAGAAAGCTGGGTCTGGAGGCTATGTTATGTGGTATCG and reverse primer GGGGACAAGTTTGTACAAAAAAGCAGGCTGACACTGAGGAGTGATTCAACGC, followed by recombination into pDONOR201 and further into pK7GWIWG2(I), resulting in a hairpin structure of two inverted PttSCAMP3 fragments under the control of the Cauliflower Mosaic Virus 35S promoter. The resulting vector was transformed into hybrid aspen (Populus tremula x tremuloides) clone T89 according to [21]. A large number of transgenic lines were regenerated, of which three were selected for detailed analyses.
Material from wild type (WT) and transgenic trees was amplified in vitro, and 33 wild type trees and five trees for each of the transgenic lines were grown in the greenhouse in K-soil (Hasselfors Light peat with sand and clay, Hasselfors Garden AB, Örebro, Sweden) with an 18 h day length, day/night temperature of 20/15 °C and relative humidity of 50–70%. The trees were grown in a random order, rotated on a weekly basis, and fertilized once a week after 3 weeks of growth with Horto NPK 7–1-5 Rika-S (Weibulls, Åby, Sweden). After 2 months of growth, the stem height and diameter of the stem at the base of each tree was measured. Next day trees were harvested and the bottom part of the stem, excluding the lowermost 10 cm, was collected for the various analyses. A seven-cm piece from the stem (10–17 cm from the base of the stem) was collected for the analyses of the metabolome, proteome and transcriptome, flash frozen with liquid nitrogen and stored at −80 °C. A three-cm piece (33–36 cm from the base) was collected for anatomical inspection and placed in FAA (5% formaldehyde, 5% acetic acid, 50% ethanol) and stored at 4 °C. A further ten-cm piece (36–46 cm from the base) was cut and used for density measurements, pyrolysis-gas chromatography/mass spectrometry (Py-GC/MS), monosaccharide analyses and analytical enzymatic saccharification, and stored at −20 °C. Stem dry weight was measured (together with a repetition of density measurements) from a separate experiment where whole stems were dried and weighed.
Analyses of the transcriptome, proteome and metabolome
Seven WT trees and five trees for each of the transgenic lines were selected for transcriptome, proteome and metabolome analyses. The wild type trees were selected on the basis of a PCA score plot to cover the variation in growth across the whole population of the wild type trees. The seven-cm piece that was collected from the stem was peeled, and the living part of the xylem was scraped away with a scalpel and flash frozen in liquid nitrogen as described in [22]. All samples were ground to a fine powder in a mortar cooled with liquid nitrogen and stored at -80 °C.
The analysis of the transcriptome
Total RNA was extracted using a RNeasy mini kit (Qiagen) supplemented with the RNAse-free DNAse set (Qiagen) and RNeasy MinElute cleanup kit (Qiagen). The protocol was based on the standard in-house protocol and on the manufacturer’s instructions. RNA integrity was assessed by gel electrophoresis on agarose gel (staining with gel-red) and using a Bioanalyser 2100 (Agilent Technologies, Waldbronn, Germany). RNA sequencing (Illumina, 100 bp paired-end reads) was performed at the Beijing Genome Institute (China), and the analysis was carried out according to their standard procedure. Raw data were pre-processed and aligned using the RNA-Seq pipeline described in [23]. In short, reads were filtered for ribosomal RNA, trimmed and aligned to version 3 of the Populus trichocarpa reference genome [24–26] with STAR [27]. The number of reads aligning to annotated gene models was determined using HTSeq [28]. Read counts were normalized with a variance stabilizing transformation (VST) implemented in the R-package DESeq2 [29]. These gene expression values were used in further downstream analyses.
Quantitative PCR (qPCR) analysis was run for RNA samples from three replicate trees per genotype after a DNAse treatment with DNA-free TM kit (Ambion), cDNA synthesis by iScript cDNA synthesis kit (Bio-Rad) and qPCR with LightCycler® 480 II (Roche) to analyse expression of PttSCAMP3 using primers GGAGGCTATGTTATGTGGTATCGC and CAGAGCACTATCTGTCCTCATTGC. A cyclophilin gene (Potri.004G168800) [30] was chosen as a reference gene using GeNorm software as described earlier [31], and amplified with primers GGCTAATTTTGCCGATGAGA and ACGTCCATCCCTTCAACAAC.
The analysis of the proteome
Total proteins were extracted from 20 mg of frozen stem tissue powder as described earlier [32]. The samples were run on a Synapt™ G2 HDMS mass spectrometer (Waters, UK) equipped with a nanoflow electrospray ionization interface according to [33]. Protein identification and peptide quantification was described earlier [14].
The analysis of the metabolome with GC-MS and LC-MS
For gas chromatography-mass spectrometry (GC-MS), metabolites were extracted and their profiles analyzed using an Agilent 6890 GC coupled to a Pegasus III time of flight MS, as described in [34]. The generated files were processed and the metabolites identified as described in [35].
For ultra high performance liquid chromatography-mass spectrometry (UHPLC-MS) analysis, one mL of extraction buffer (20/20/60 v/v chloroform:water:methanol) including the internal standards Reserpine (Sigma), Sulfadimethoxine (Fluka), Leucine-Enkephalin (Fluka) and Val-Tyr-Val (Bachem) was added to 9–12 mg of the plant material. The sample was shaken with a tungsten bead in a mixer mill at 30 Hz for 3 min, the bead was removed and the sample was centrifuged at +4 °C, 14,000 rpm, for 10 min. Then, 200 μL of supernatant were transferred to a micro vial and the solvents were evaporated. Before analysis, the sample was re-suspended in 10 + 10 μL methanol and water (with 0.1% formic acid). The chromatographic separation was performed on an Agilent 1290 Infinity UHPLC-system (Agilent Technologies, Waldbronn, Germany). Two μL of re-suspended aliquots of extracted plant sample were injected onto a 2.1 × 100 mm, 1.7 μm Kinetex C18 column (Phenomenex, Torrace, USA) held at 40 °C. The gradient elution buffers were A (H2O, 0.1% formic acid) and B (acetonitrile, 0.1% formic acid), and the flow-rate was 0.5 mL min−1. The compounds were eluted with a linear gradient consisting of 1–20% B over 0–4 min, 20–40% B over 4–6 min, 40–95% B over 6–9 min, the composition was held at 95% B for 4.5 min, and returned to 1% B at 14.50 min, the composition was kept at 1% B for a further 4.5 min before the next injection. The diode array detector was set to scan the interval 190–640 nm with a step length of 2 nm and a slit width of 2 nm. The compounds were detected with an Agilent 6540 Q-TOF mass spectrometer equipped with a jet stream electrospray ion source operating in negative ion mode. The settings were kept identical between the modes, with the exception of the capillary voltage. A reference interface was connected for accurate mass measurements; the reference ions purine (4 μM) and HP-0921 (Hexakis(1H, 1H, 3H–tetrafluoropropoxy phosphazine) (1 μM), both purchased from Agilent Technologies (Santa Clara, CA, USA), were infused directly into the MS at a flow rate of 0.05 mL min−1 for internal calibration, and their monitored ions were m/z 119.03632 and m/z 966.000725 for negative mode, respectively. The gas temperature was set to 300 °C, the drying gas flow to 8 L min−1 and the nebulizer pressure to 40 psig. The sheath gas temp was set to 350 °C and the sheath gas flow to 11 L min−1. The capillary voltage was set to 4000 V. The nozzle voltage was 0 V. The fragmentor voltage was 100 V, the skimmer 45 V and the OCT 1 RF Vpp 750 V. The collision energy was set to 0 V. The m/z range was 70–1700, and data were collected in centroid mode with an acquisition rate of 4 scans s−1 (1974 transients/spectrum). Mass Feature Extraction (MFE) from the data acquired was performed using the MassHunter™ Qualitative Analysis software package, version B05.00 (Agilent Technologies Inc., Santa Clara, CA, USA). Extracted features were aligned and matched between samples using Mass Profiler Professional™ 12.5 (Agilent Technologies Inc., Santa Clara, CA, USA).
The metabolite annotation was done by manual interpretation of the fragments with high mass accuracy or by searches in an in house database. For critical samples, extracts from transgenic and wild-type plants were re-analyzed by Liquid Chromatography Quadrupole Time-of-Flight Mass Spectrometry (LC-Qtof) targeted MS/MS approach using the same chromatographic and mass spectrometry conditions as described above, with collision energy set up from 10 to 40 V. The metabolomic extracts were also re-analyzed by a lipidomic approach [36] to improve annotations of metabolites with long retention times.
Wood chemical analyses
The ten-cm stem piece (36–46 cm from the base) that was collected for the chemical analyses was freeze-dried for about 48–72 h, cut into small pieces (1 cm long × 1 mm diameter) and ground into a rough powder using a centrifugal mill (Retsch ZM 200, Haan, Germany). For analytical enzymatic saccharification and monosaccharide analysis by acid hydrolysis, the rough powder was sieved to a particle size between 0.1 and 0.5 mm using an analytical sieve shaker AS 200 (Retsch). For pyrolysis-GC/MS, the rough powder was further ground into a fine powder using a ball mill (Retsch MM400) for 150 s at 30 Hz, as described previously [37].
Pyrolysis-gas chromatography/mass spectrometry (Py-GC/MS)
About 50 μg of fine wood powder, weighed using a micro balance (XP6U, Mettler Toledo, USA) was analyzed by a pyrolyzer (PY-2020iD and AS-1020E, Frontier Lab, Japan) connected to a GC-MS (7890A/5975C; Agilent Technologies AB, Sweden), as described previously [38]. All 33 wild type trees and 5 replicate trees from the RNAi lines were analyzed.
Monosaccharide analysis by acid hydrolysis
Sieved rough wood powder from each of the transgenic lines (with three technical replicates, each of which containing equal amounts of wood powder pooled from five biological replicates of the transgenic trees and from five pools of wild type trees) was used to determine total monosaccharide content after acid hydrolysis. Dry wood powder (100 mg, after moisture analysis using Mettler Toledo HG63, Switzerland) was hydrolyzed with sulfuric acid (3 ml, 72% (w/w)] for 1 h at 30 °C). The reaction mixture was then diluted to 4% sulfuric acid using deionized water and autoclaved for 1 h at 120 °C. After centrifugation (14,000 g for 20 min), the supernatant was collected and analyzed for monosaccharide sugars using high-performance anion-exchange chromatography (HPAEC), as described previously [37].
Analytical enzymatic saccharification
Sieved wood powder (50 mg) from each sample (each transgenic line containing five biological replicates and the wild type containing five pools of biological replicates, each pool consisting of equal amounts of wood from 4 to 6 wild type trees) was subjected to enzymatic hydrolysis, with and without prior thermochemical pretreatment. The thermochemical pretreatment was performed as previously described [37] by impregnation with 1% (w/w) sulfuric acid and treatment at 165 °C for 10 min using an Initiator single-mode microwave instrument (Biotage, Uppsala, Sweden). The pretreated wood was divided into a liquid phase, referred to as pretreatment liquid, and a solid phase consisting mainly of cellulose and lignin, which, after washing, was used as a substrate for cellulolytic enzymes. Analytical enzymatic saccharification of non-pretreated and pretreated wood was described earlier [37]. Briefly, milled and sieved wood or the solid phase after the pretreatment was digested enzymatically for 72 h at 45 °C by addition of commercially available liquid preparations of Celluclast 1.5 L (Sigma-Aldrich) and Novozyme 188 (Sigma-Aldrich). Samples for rapid glucose analysis using a glucometer [37] were withdrawn after 2 h for determination of the glucose production rate (GPR). The monosaccharide contents of samples taken at the end of the reaction, after 72 h, were analyzed using High-performance anion-exchange chromatography (HPAEC) for determination of the sugar yields, as previously described [37]. The monosaccharide contents of the pretreatment liquid were also analyzed using HPAEC.
Data integration and statistical analysis by OnPLS
Data acquired from five platforms (transcriptomics, proteomics, GC-MS, LC-MS and Py-GC/MS) were integrated by OnPLS. The data sets from the three different transgenic lines were combined into one dataset and preprocessed as described earlier [39]. In short, the datasets from the transgenic lines were normalized relative to WT by subtracting the average WT value from the value of each data point and dividing by the standard deviation (SD) of the wild type. Model significance was determined using the so-called leave-one-out cross-validation [40], and implemented here using p(CORR) value as a measure of the significance of the variation [41]. An arbitrary cutoff value |p(CORR)| > 0.5 was applied here to identify statistically significant variation between the transgenic lines and the wild type similar to the approaches taken by Tulipani et al. [42] and Llorach et al. [43–45].
Results
Description of the SCAMP gene family in Populus
The SCAMP genes encode highly conserved proteins which normally form small gene families. Populus genus has eight SCAMP gene family members. Phylogenetic analysis was performed to investigate Populus trichocarpa SCAMP (PtrSCAMP) sequence similarity with two angiosperm species, Arabidopsis thaliana and Amborella trichopoda, the bryophyte Physcomitrella patens and the lycophyte Selaginella moellendorffii. The analysis revealed two clusters containing the SCAMP sequences from the three angiosperm species (Fig. 1a), similar to what was described previously [19]. A third cluster contained sequences from Physcomitrella patens and Selaginella moellendorffii. Two Populus trichocarpa sequences (PtrSCAMP7 and PtrSCAMP8) were separate from these three clusters, suggesting they might be functionally divergent. It has previously been reported that most plant SCAMP proteins have a cytoplasmic N-terminal with NPF motifs, four transmembrane domains, and a cytoplasmic C-terminal containing the tyrosine sorting motif YXXF [19, 46]. The domain structure of the Populus SCAMP gene family was analysed here, showing that PtrSCAMP1–4, PtrSCAMP6, and PtrSCAMP8 have cytoplasmic tails and four transmembrane domains each, PtrSCAMP1 and PtrSCAMP3–6 have the C-terminal YXXF motif and PtrSCAMP1–6 have two N-terminal NPF motifs (Fig. 1b).
Fig. 1.
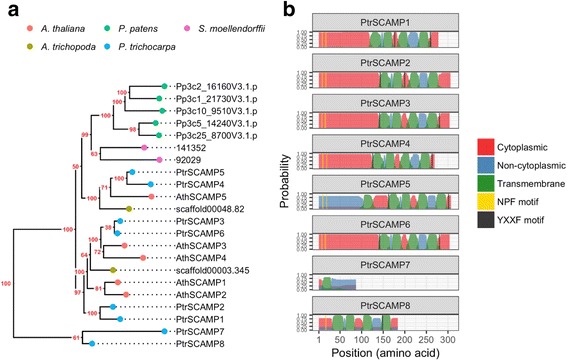
Phylogenetic analysis of SCAMP gene families and transmembrane topology analysis of P. trichocarpa SCAMP proteins. a The gene families were retrieved from the Joint Genome Institute (JGI) (http://genome.jgi.doe.gov/). The SCAMP genes are evm_27.model.AmTr_v1.0_scaffold00003.345 and evm_27.model.AmTr_v1.0_scaffold00048.82 in Amborella trichopoda, AT1G61250 (AthSCAMP1), AT1G11180 (AthSCAMP2) AT2G20840 (AthSCAMP3), AT1G03550 (AthSCAMP4) and AT1G32050 (AthSCAMP5) in Arabidopsis thaliana, Phpat.001G082700 (Pp3c1_21730V3.1.p), Phpat.002G068200 (Pp3c2_16160V3.1.p), Phpat.025G032600 (Pp3c25_8700V3.1.p), Phpat.010G037000 (Pp3c10_9510V3.1.p) and Phpat.005G053500 (Pp3c5_14240V3.1.p) in Physcomitrella patens, Potri.004G036600 (PtrSCAMP1), Potri.011G045100 (PtrSCAMP2), Potri.019G104000 (PtrSCAMP3), Potri.001G134100 (PtrSCAMP4), Potri.003G099300 (PtrSCAMP5), Potri.013G144700 (PtrSCAMP6), Potri.011G045200 (PtrSCAMP7) and Potri.004G036700 (PtrSCAMP8) in Populus trichocarpa, and 141,352 and 92,029 in Selaginella moellendorffii. The evolutionary history was inferred using the WAG substitution model [58]. Evolutionary analyses were conducted in R (https://www.R-project.org/) using the phangorn package (v2.2.0) and visualised using the ggtree extension for ggplot2. Numbers are bootstrap support values based on 1000 runs. b Transmembrane topology prediction was performed using Phobius and visualised using ggplot2. The NPF motif and the tyrosine sorting motif YXXF were identified using the regular expressions NPF and YXXF, respectively, in R (https://www.R-project.org/)
Next, we analyzed expression of the Populus SCAMP gene family members in the woody tissues of the stem using the AspWood gene expression database (http://aspwood.popgenie.org) which contains high-resolution RNA sequencing data from the different tissue types of the aspen (Populus tremula) stem [47]. The analysis revealed that all Populus tremula SCAMPs (PtSCAMP) except for PtSCAMP8 are expressed in the stem (Fig. 2). They all have quite similar expression patterns, with a peak of expression right at the beginning of the maturation zone which is the location for the initiation of secondary cell wall formation. Notably, PtSCAMP3 shows a peak of expression in the xylem a little earlier than the others. PtSCAMP7, which was divergent from the others in the phylogenetic analysis, exhibited similar, although somewhat lower, expression than the other Populus SCAMP genes. On the basis of this, we can conclude that there are seven SCAMP genes in Populus that are expressed in the woody tissues in a manner suggestive of roles during xylem expansion and/or initiation of xylem maturation.
Fig. 2.
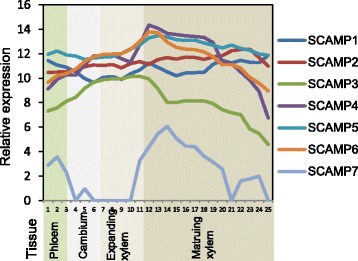
Expression profile of the Populus SCAMP gene family members in Populus stem. The data were retrieved from the AspWood database (http://aspwood.popgenie.org) where relative expression (relative to the number of RNA-Seq reads in the sample and VST normalized) is shown for aspen stem samples which consist primarily of phloem, cambium, expanding xylem and maturing xylem. Data is shown for tree 1. Similar results were obtained for three additional replicate trees in the AspWood database
Transgenic lines modified in the expression of Populus SCAMP3 were analyzed in this study, and we therefore used the AspWood database to find genes that were co-expressed with PtSCAMP3 in the aspen woody tissues. Interestingly, several nucleotide-diphospho-sugar transferases, as well as genes related to cell wall biosynthesis, were among the most co-expressed genes (Additional file 1).
Suppression of two SCAMP genes in transgenic Populus trees results in increased accumulation of secondary cell wall components in the stem
Functional analyses were performed in transgenic Populus tremula x tremuloides (Ptt) trees carrying an RNAi construct for PttSCAMP3. RNA sequencing of three transgenic lines showed a 2–69% decrease in the expression of PttSCAMP3. PttSCAMP3 is paralogous with PttSCAMP6, and the RNAi construct resulted in 9–75% decrease in the expression of PttSCAMP6 as well, while the expression of the other PttSCAMP genes were only slightly changed in the different lines (Fig. 3a). The RNAi lines therefore reflect the combined function of PttSCAMP3 and PttSCAMP6 in line three and the function of PttSCAMP6 in line 1. Verification of the RNAseq results by qPCR revealed suppression of PttSCAMP3 also in line 2 (Fig. 3b).
Fig. 3.
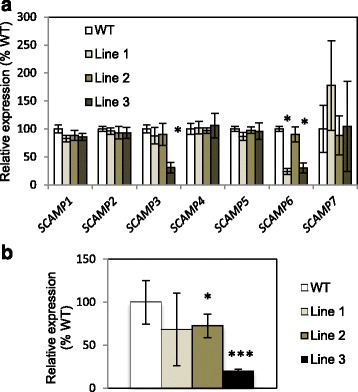
Expression of the PttSCAMP genes in the wild type and the three PttSCAMP3 RNAi lines. a Relative expression (as a percentage of the WT) is shown for differentiating xylem samples scraped from the base of two-month-old trees. The expression is based on gene expression values in the RNA sequencing datasets, where read counts were normalized for the RNAseq library size. Asterisks indicate significant difference from the wild type at P(Benjamini-Hochberg adjusted) < 0.05 according to the R-package DESeq2. Vertical bars indicate ± SD. n = 5. b Relative expression of PttSCAMP3 (as a percentage of the WT) by qPCR analysis. Asterisks indicate significant difference from the wild type at P < 0.05 (*) or P < 0.001 (***) according to Welch corrected t-test. Vertical bars indicate ± SD. Three biological replicates were analyzed in three technical replicates each
Detailed phenotypic analysis of 2-month-old, greenhouse-grown trees revealed an increase in the density of the wood in the RNAi lines compared to the wild type, although this was statistically significant only for line 3 (Fig. 4a). These young RNAi trees displayed slight differences in the total volume of the stem (Fig. 4b), which together with the changes in the density resulted in slight, but not statistically significant increase in the dry weight of the stem in lines 1 and 3 (Fig. 4c). Interestingly, trees that were grown for 6 months in the greenhouse developed a brown, striated bark (Fig. 4d) in contrast to the green and smooth bark of the wild-type trees of the same age (Fig. 4e). Anatomical inspection of the bark revealed that also the thickness of the suberized cork was significantly increased in all the three transgenic lines compared to the wild type (Fig. 4f-h).
Fig. 4.
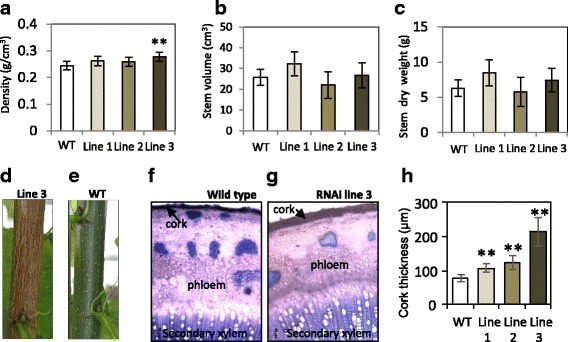
Phenotypic analysis of the wild type and the PttSCAMP3 RNAi lines. Wood density at the base of the stem (a), stem volume (b) and stem dry weight (c) in two-months-old greenhouse grown trees. Representative images of the stems of six-month-old wild type (d) and PttSCAMP3 RNAi line 3 (e) trees. f-g Light microscopy images of toluidine blue stained transverse sections taken from the base of the stems of two-months-old greenhouse grown wild type (f) and PttSCAMP3 RNAi line 3 (g) trees. H. Thickness of the cork in the wild type and the PttSCAMP3 RNAi lines at the base of the stems of two-months-old greenhouse grown trees. Asterisks indicate significant differences from the wild type at P < 0.01 (**) according to a Welch-corrected t-test. The volume of the stem is estimated with the formula volume = π · radius2 · height / 3. Vertical bars indicate ± SD. n = 5
The expression pattern and changes in wood density prompted us to investigate the effect of PttSCAMP3 RNAi expression on cell wall chemistry. A high-throughput analysis by pyrolysis gas chromatography/mass spectrometry (Py-GC/MS) did not reveal any significant differences in the relative content of carbohydrates and lignin even though a slight tendency towards higher lignin content was present especially in line 3 (Fig. 5). As Py-GC/MS reveals only the relative content of the cell wall components, alternative methods were used to identify possible differences in the absolute amounts of the cell wall components. An LC-MS metabolomic analysis revealed that the abundance of small phenolic compounds which were earlier identified as oligolignols [48] increased in abundance in the transgenic lines compared to the wild type (Fig. 6). The only compound that had a lower abundance in the transgenic lines was 5-O-caffeoyl shikimic acid which has been reported to inhibit activity of the lignin biosynthetic 4-coumaric acid:coenzyme A ligase (4CL) [49]. Detailed analysis of the carbohydrate composition by acid hydrolysis followed by HPAEC analysis also revealed increased abundance of monosaccharides derived from the major secondary cell wall carbohydrates glucan and xylan and from the minor carbohydrates arabinan and galactan in the RNAi lines compared to the wild type (Fig. 7). Taken together, the results support enhanced accumulation of both the carbohydrate and lignin components of the secondary cell walls in the woody tissues of the PttSCAMP3 RNAi lines 1 and 3.
Fig. 5.
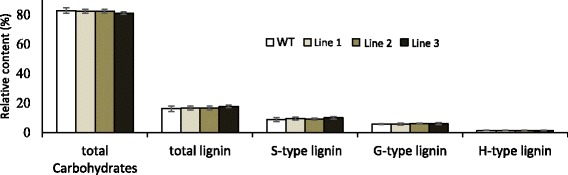
Pyrolysis gas chromatography/mass spectrometry (Py-GC/MS) analysis of the secondary xylem tissues. The relative content of carbohydrates and lignin is shown for mature xylem samples taken from the base of wild type and the PttSCAMP3 RNAi trees. The relative content is calculated as the sum of the peak areas for the pyrolysis products derived from either the carbohydrate or lignin polymers, and are shown as a percentage of the total peak area from the GC-MS analysis. The composition of lignin is further shown as the relative content of the pyrolysis products derived from the S, G and H type lignin. n = 5
Fig. 6.
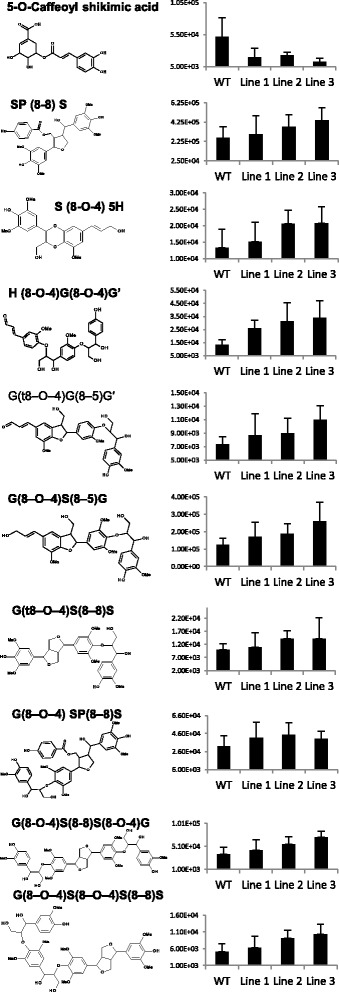
Small phenolic compounds accumulate in the secondary xylem of the PttSCAMP3 RNAi lines. The graphs depict the abundance (peak area/mg fresh weight) and chemical structure of putative lignin-related oligomers in the PttSCAMP3 RNAi lines and the wild type in the LC-MS metabolome analysis. Only metabolites having a |p(CORR)| ≥ 0.6 in the OnPLS analysis were included. Full data set for the LC-MS metabolome analysis can be found in Additional file 5. G, guaiacyl unit; S, syringyl unit; SP, unit derived from sinapyl p-hydroxybenzoate; H, p-hydroxyphenyl unit. n = 5
Fig. 7.
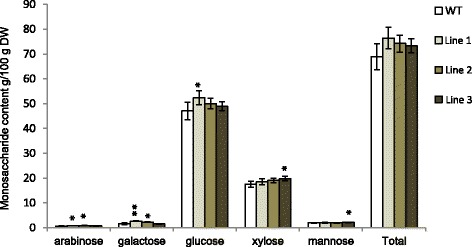
Monosaccharide yields in hydrolysates of the woody tissues of PttSCAMP3 RNAi lines and wild type. Monosaccharide composition in wild type (WT) and PttSCAMP3 RNAi lines detected after acid hydrolysis (72% w/w sulfuric acid). The hydrolysis releases arabinose, galactose, glucose, xylose and mannose from the secondary cell wall arabinan, galactan, glucan, xylan and mannan, respectively. Values are given as a percentage of g monosaccharide (in anhydrous form) per g dry weight of wood ± SD. Values are mean of three technical replicates, each of which containing equal amounts of wood powder pooled from five biological replicates for the transgenic lines and from five replicate pools of wild type trees. The “total” columns indicate the sum of the individual monosaccharide abundances. Asterisks indicate significant differences from the wild type at P < 0.05 (*) and P < 0.01 (**) according to Welch-corrected t-test
Suppression of the PttSCAMP genes influences the bioprocessing properties of the wood
An increasingly important trait of forest trees is the susceptibility of the lignocellulosic raw material to enzymatic hydrolysis. To evaluate the effect of PttSCAMP3 RNAi on this trait, an analytical scale pretreatment and enzymatic hydrolysis experiment was conducted for the wild type and the three different transgenic lines, and sugar yields were measured in woody material with and without an acid pretreatment. Interestingly, the lines behaved differently in these analyses. While line 1 showed a tendency towards increased glucose production rate (GPR; after 2 h of enzymatic hydrolysis) and significant increases in the yield of glucose both with and without the acid pretreatment, line 3 showed quite the opposite tendency towards decreased GPR and decreased yields of glucose and xylose that are the main sugars in the woody polymers (Fig. 8). The difference in the saccharification potential of the two lines might be due to the difference in the carbohydrate to lignin ratio of these lines; both lines showed increases in level of the carbohydrates (Fig. 7) that is expected to increase sugar yields after enzymatic hydrolysis, but this effect might be counteracted in line 3 by the increase in the relative content of lignin (Fig. 5) that is known to have an adverse effect on the saccharification potential.
Fig. 8.
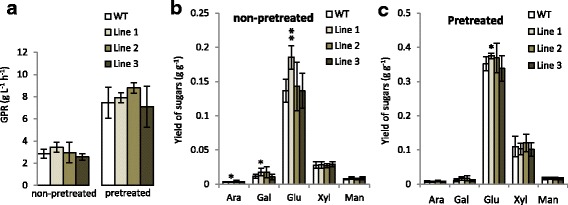
Susceptibility of the PttSCAMP3 RNAi lines and wild type to enzymatic hydrolysis. a Glucose production rates (GPR) of wild type (WT) and PttSCAMP3 RNAi lines after 2 h of enzymatic hydrolysis. The values represent means of GPR (g L−1 h−1 ± SD) in samples without (non-pretreated) and with an acid pretreatment (pretreated). b Sugar yields after enzymatic hydrolysis of non-pretreated woody tissues. The values represent amounts of the different monosaccharides (g monosaccharide per g dry weight) ± SD. c Sugar yields after enzymatic hydrolysis in pretreated woody tissues. The values represent combined sugar yields (g monosaccharide per g dry weight) ± SD from the pretreatment liquid and the enzymatic hydrolysate. Ara, arabinose; Gal, galactose, Glu, glucose; Xyl, xylose; Man, mannose. Asterisks indicate significant differences from the wild type at P < 0.05 (*) and P < 0.01 (**) according to a Welch-corrected t-test. n = 5
Integration of the multi-omics data by OnPLS modelling
To understand the mechanisms underlying the phenotypic changes in the PttSCAMP3 RNAi lines, multi-omics analysis including transcriptomic, metabolomic and proteomic analysis of the transgenic lines and the wild type was performed in the living secondary xylem tissues from the same stem samples where the dead, mature tissues had been collected for the analyses of the wood chemistry and saccharification. To identify the most significant changes in relation to the phenotypic changes, OnPLS (orthogonal projections to latent structures) analysis was performed for data combined for the three transgenic lines from the five different omics platforms (transcriptomic, proteomic, GC-MS metabolomic, LC-MS metabolomic and Py-GC/MS metabolomic) and compared to the wild type. OnPLS [17, 18] is an extension of O2PLS [50, 51], and suitable for simultaneous analysis of more than two blocks of data or, as in our case, data from more than two platforms. It separates each data block into three parts: one where the data variation is globally joint (shared between all blocks or platforms in this case), one where the variation is locally joint (shared between some, but not all blocks/platforms) and one where the variation is unique to one block/platform. The analysis was introgressed into an OnPLS model, and the overview of the model was visualized by principal component analysis (Additional file 2A). The PCA analysis of the model revealed clear separation between the wild type and the transgenic PttSCAMP3 lines. The majority of the modeled variation was either globally joint or locally joint; the sum of the global and location variation was 61% for the transcriptome, 70% for the proteome, 66% for the LC-MS metabolome, 73% for the GC-MS metabolome and 57% for the Py-GC/MS metabolome. A linear analysis of the five datablocks revealed the first component of each dataset as the main contributor accounting for the separation between the wild type and the transgenic lines (Additional file 2B). Statistical analysis was therefore performed on the basis of the first component. Large number of the variables were significantly different (|p(CORR)| > 0.5) between the transgenic lines and the wild type (Table 1; Additional files 3, 4, 5, 6 and 7). For both the proteome and the metabolome, the majority of the statistically significant changes resulted from increased rather than decreased abundance of the variables in the RNAi lines compared to the wild type (Table 1).
Table 1.
Significantly different variables between the transgenic lines and the wild type in the different datasets on the basis of the OnPLS multivariate analysis
| Dataset | Total number of variables | Number of the variables downregulated in transgenic lines compared to the wild type | Number of the variables upregulated in the transgenic lines compared to the wild type |
|---|---|---|---|
| Transcriptome | 27,929 | 2639 | 2951 |
| Proteome | 1208 | 9 | 95 |
| Metabolome (LC-MS) | 1141 | 54 | 317 |
| Metabolome (GC-MS) | 214 | 12 | 22 |
| Metabolome (Py-GC/MS) | 109 | 17 | 38 |
The comparisons are done on the basis of the first component of the OnPLS analysis. |p(CORR)| > 0.5 was applied here as an arbitrary cutoff value to identify statistically significant variation between the transgenic lines and the wild type
Multi-omics data provides clues to the function of the PttSCAMP proteins
Consistent with the expected function of the SCAMP proteins in membrane trafficking, a number of proteins that are known to be involved in secretion and/or endocytosis, such as secretion-associated RABA GTPase family protein Potri.016G000400 and a golgi snare protein Potri.014G066800, were significantly upregulated in the transgenic lines compared to the wild type (Table 2). Vice versa, three other RAB GTPase homologs (Potri.008G061300, Potri.003G081800, Potri.003G004100) were among the most downregulated proteins in the transgenic lines (Table 3).
Table 2.
The most upregulated variables in the transgenic PttSCAMP3 RNAi lines from the OnPLS analysis
| Transcript | Protein | Metabolite (LC-MS) | Metabolite (GC-MS) |
||||
|---|---|---|---|---|---|---|---|
| Potri.010G017600 (receptor kinase 3) |
−0.9 | Potri.002G251600 (Actin binding Calponin homology (CH) domain-containing protein) | −0.8 | 324.0377@0.60600036 | −0.9 | Glycerol 1-palmitate | −0.8 |
| Potri.008G125700 (RING-H2 finger A2A) |
−0.9 | Potri.006G168900 (SET domain-containing protein) | −0.8 | Suberic acid | −0.9 | Erythrose | −0.6 |
| Potri.006G230700 (homeobox protein ATH1) | −0.9 | Potri.015G108300 | −0.8 | 396.1603@9.253999 | −0.9 | Myo-inositol-1-phosphate | −0.6 |
| Potri.008G098300 | −0.9 | Potri.014G066800 (golgi snare 12) | −0.8 | Monogalactosyldiacyl-glycerol (36:6) | −0.8 | Gluconic acid-6-phosphate | −0.5 |
| Potri.018G093100 (homolog of X-ray repair cross complementing 3 | −0.9 | Potri.018G060600 | −0.8 | 888.5163@9.920005 | −0.8 | Oxalic acid | −0.5 |
| Potri.011G081900 | −0.9 | Potri.019G053700 (isopentenyl-diphosphate delta-isomerase) | −0.8 | Monogalactosyldiacyl-glycerol (36:6) | −0.8 | Linoleic acid | −0.5 |
| Potri.018G106700 (DNAse I-like superfamily protein) | −0.9 | Potri.010G081600 (similar to auxin down-regulated protein ARG10) | −0.8 | Monogalactosyldiacyl-glycerol (36:6) | −0.8 | alpha-Linolenic acid | −0.5 |
| Potri.009G166100 | −0.9 | Potri.012G074900 (B12D protein) |
−0.8 | Phosphatidic acid (36:6) | −0.8 | ||
| Potri.015G071100 | −0.9 | Potri.001G309200 (BolA-like family protein) |
−0.8 | 458.0594@0.5979997 | −0.8 | ||
| Potri.003G165800 | −0.9 | Potri.016G000400 (RAB GTPase homolog A2B) |
−0.8 | 380.1651@9.253999 | −0.8 | ||
| Potri.001G058600 (Squamosa promoter-binding protein-like) | −0.8 | Potri.010G038400 (pyruvate dehydrogenase complex E1 α subunit) | −0.8 | Hydroxycaproic acid | −0.8 | ||
| Potri.009G056200 (NB-ARC disease resistance protein) | −0.8 | Potri.006G255800 (Myosin heavy chain-related protein) |
−0.7 | octadecadienoic acid | −0.8 | ||
| Potri.002G191900 (gibberellin 2-oxidase 6) | −0.8 | Potri.006G107100 (aspartate aminotransferase 1) |
−0.7 | 283.0899@1.0639995 | −0.8 | ||
| Potri.003G063900 (ELF4-like 4) | −0.8 | Potri.010G127500 (protein phosphatase 2A subunit A2) |
−0.7 | digalactosyldiacylglycerol (34:3) | −0.8 | ||
| Potri.003G100100 (Homeodomain-like protein) | −0.8 | Potri.007G013400 (peptidylprolyl cis/trans isomerase) | −0.7 | 516.0613@0.6520003 | −0.8 | ||
| Potri.016G110300 (Josephin family protein) | −0.8 | Potri.008G217700 (cullin 1) |
−0.7 | digalactosyldiacylglycerol (34:3) | −0.8 | ||
| Potri.016G026600 (alpha-L-arabinofuranosidase 1) | −0.8 | Potri.003G098400 (Proteasome component (PCI) domain protein) | −0.7 | 1118.559@9.75 | −0.8 | ||
| Potri.002G065900 (5-FORMYLTETRAHYDRO-FOLATE CYCLO-LIGASE-RELATED) | −0.8 | Potri.015G042600 (TCP-1/cpn60 chaperonin family protein) | −0.7 | 920.3991@11.602999 | −0.8 | ||
| Potri.002G024300 (SAUR-like auxin-responsive protein family) | −0.8 | Potri.018G091100 (Transducin/WD40 repeat-like superfamily protein) | −0.7 | Digalactosyldiacyl-glycerol (36:6) | −0.8 | ||
| Potri.016G140200 (Leucine-rich repeat protein kinase) | −0.8 | Potri.007G091000(Lipase/lipooxygenase, PLAT/LH2 family protein) | −0.7 | 934.5073@9.919004 | −0.8 | ||
| Potri.014G129800 (complex 1 family protein/LVR family protein) | −0.8 | Potri.012G069000 (METHYL-TRANSFERASE PMT2-RELATED) | −0.7 | 490.2831@10.1900015 | −0.8 | ||
| Potri.003G111000 | −0.8 | Potri.016G087900 (adenylosuccinate synthase) | −0.7 | 5′-AMP | −0.8 | ||
| Potri.T128200 (Disease resistance protein family) | −0.8 | Potri.001G206400 (Polymerase/ histidinol phosphatase-like) |
−0.7 | Phosphatidic acid (36:6) | −0.8 | ||
| Potri.T034300 | −0.8 | Potri.002G105100 (Clathrin adaptor complexes medium subunit family protein) | −0.7 | 830.4298@11.596002 | −0.8 | ||
| Potri.005G148400 (AP2-LIKE ETHYLENE-RESPONSIVE TRANSCRIPTION FACTOR ANT | −0.8 | Potri.T002400 (Disease resistance protein (TIR-NBS-LRR class) family) | −0.7 | 812.4986@9.919004 | −0.8 | ||
| Potri.010G194400 (Yos1-like protein) | −0.8 | Potri.001G021900 | −0.7 | 737.4975@10.446007 | −0.8 | ||
| Potri.017G059900 (ralf-like 32) | −0.8 | Potri.006G138600 (chaperonin 20) | −0.7 | 124.0619@0.7949998 | −0.8 | ||
| Potri.017G027800 (RHOMBOID-like protein 13) | −0.8 | Potri.014G090100 (exocyst complex component sec10) | −0.7 | 453.9293@9.467004 | −0.8 | ||
| Potri.016G073500 | −0.8 | Potri.004G140900 (cytochrome P450, family 707, subfamily A, polypeptide 4) | −0.7 | Monogalactosyldiacyl-glycerol (36:6) | −0.8 | ||
| Potri.001G281600 (Lateral organ boundaries domain protein) | −0.8 | Potri.013G128600 (ribosomal protein L5 B) | −0.7 | 689.1812@0.5330003 | −0.8 | ||
The variables are listed in descending order of statistical significance (p(CORR) from the OnPLS analysis. The p(CORR) value is depicted next to each variable. The transcript and protein annotations are according to JGI V3.0. LC-MS metabolites without annotations could not be identified and are listed as mass@retention time. Galactolipids can be present as adduct forms. Full datasets are listed in Additional files 3,4,5 and 6
Table 3.
The most downregulated variables in the transgenic PttSCAMP3 RNAi lines from the OnPLS analysis
| Transcript | Protein | Metabolite (LC-MS) | Metabolite (GC-MS) |
||||
|---|---|---|---|---|---|---|---|
| Potri.002G066100 (Alba DNA/RNA-binding protein) | 0.9 | Potri.006G079700 (PREFOLDIN 1) | 0.7 | 260.0366@7.6459966 | 0.8 | Fructose-6-Phosphate | 0.7 |
| Potri.002G026600 (Regulator of chromosome condensation) | 0.9 | Potri.008G061300 (RAB GTPase homolog A2B) | 0.6 | 170.0909@6.5109987 | 0.8 | Succinic acid | 0.6 |
| Potri.008G014700 (calcium dependent protein kinase 1) | 0.9 | Potri.001G304700 (Ribosomal protein S5 domain 2-like superfamily protein) | 0.6 | ‘UDP-galactose | 0.8 | Threonic acid | 0.6 |
| Potri.003G098200 (extra-large GTP-binding protein 3) | 0.9 | Potri.005G108100 (aconitase 1) | 0.6 | 2-cis,4-trans-xanthoxin | 0.7 | Adenosine | 0.5 |
| Potri.018G044300 | 0.9 | Potri.016G032500 (Single-stranded nucleic acid binding R3H protein) | 0.6 | 312.2202@7.9389973 | 0.7 | Malic acid | 0.5 |
| Potri.001G306700 (Protein of unknown function (DUF1278)) | 0.8 | Potri.008G060000 (eukaryotic translation initiation factor 3G1) | 0.6 | iso-erythritol | 0.7 | Caffeic acid | 0.5 |
| Potri.001G428100 (NB-ARC disease resistance protein) | 0.8 | Potri.004G070000 (UDP-glucosyl transferase 88A1) | 0.5 | 263.2075@8.526 | 0.7 | ||
| Potri.007G002400 (cytochrome P450, family 716, subfamily A,) | 0.8 | Potri.001G400900 (similar to cytidylyltransferase family) | 0.5 | 86.041@0.7590002 | 0.7 | ||
| Potri.006G093800 (Inositol phosphorylceramide synthase 2) | 0.8 | Potri.001G464500 (germin-like protein 2) | 0.5 | 280.1638@7.9479985 | 0.7 | ||
| Potri.005G181800 (Protein kinase superfamily protein) | 0.8 | Potri.019G050500 (Protein kinase superfamily protein) | 0.5 | 438.3488@9.589997 | 0.7 | ||
| Potri.001G435700 (endoribo-nuclease L-PSP protein) | 0.8 | Potri.003G168500 | 0.5 | 157.0447@2.569 | 0.7 | ||
| Potri.015G079500 (scramblase-related) | 0.8 | Potri.013G062500 | 0.5 | 5-O-Caffeoylshikimic acid | 0.7 | ||
| Potri.011G100900 (ARF-GAP domain 13) | 0.8 | Potri.015G090900 (26S proteasome, regulatory subunit Rpn7) | 0.5 | 392.0447@0.6760002 | 0.6 | ||
| Potri.006G187500 (Calcineurin-like metallo-phosphoesterase) | 0.8 | Potri.009G120500 (regulatory particle triple-A ATPase 4A) | 0.5 | 98.0651@4.285999 | 0.6 | ||
| Potri.014G148800 (DNA topoisomerase, type IA, core) | 0.8 | Potri.010G069900 (Ribosomal protein L14) | 0.5 | 404.0859@3.440001 | 0.6 | ||
| Potri.016G018700 | 0.8 | Potri.011G110900 (general regulatory factor 9) | 0.5 | 157.0442@2.1259997 | 0.6 | ||
| Potri.006G085400 (aminoacyl-tRNA and biotin synthetase) | 0.8 | Potri.006G073200 (Ribosomal protein L30/L7 family protein) | 0.5 | 243.1821@6.7790008 | 0.6 | ||
| Potri.001G420400 (SMAD/FHA domain-containing protein) | 0.8 | Potri.012G062600 (ribulose-bisphosphate carboxylase) | 0.4 | 310.1765@7.268003 | 0.6 | ||
| Potri.017G082800 | 0.8 | Potri.002G082101 | 0.4 | 103.0391@0.7649997 | 0.6 | ||
| Potri.011G169200 | 0.8 | Potri.002G057300 (Pleckstrin homology (PH) domain-containing protein) | 0.4 | 422.2356@9.213004 | 0.6 | ||
| Potri.003G098100 (GHMP kinase family protein) | 0.8 | Potri.002G182500 (CARBON CATABOLITE REPRESSOR PROTEIN 4) | 0.4 | 537.1221@1.2070005 | 0.6 | ||
| Potri.002G006700 (MLO family protein) | 0.8 | Potri.008G012400 (FASCICLIN-like arabinogalactan protein 17 precursor) | 0.4 | 537.1187@1.2060003 | 0.6 | ||
| Potri.008G159700 (Pyruvate kinase family protein) | 0.8 | Potri.018G078200 (ankyrin repeat family protein) | 0.4 | Succinic acid | 0.6 | ||
| Potri.004G106200 (phosphoglycerate mutase) | 0.8 | Potri.002G045700 (tryptophan synthase alpha chain) | 0.4 | 148.0429@0.859 | 0.6 | ||
| Potri.011G167000 (amino acid permease 7) | 0.8 | Potri.006G275000 (H(+)-ATPase 5) | 0.4 | 464.1305@4.291 | 0.6 | ||
| Potri.001G004600 (tubulin α-3) | 0.8 | Potri.003G081800 (RAS 5) | 0.4 | 464.1145@4.291 | 0.6 | ||
| Potri.002G171800 (cytochrome P450, family 703, subfamily A) | 0.8 | Potri.001G194000 (Ribosomal L28e protein family) | 0.4 | 7-Hydroxyflavone | 0.6 | ||
| Potri.005G028900 (Rab escort prot) | 0.8 | Potri.003G004100 (RAB GTPase 11C) | 0.4 | 305.9969@0.57099974 | 0.6 | ||
| Potri.T060400 (NB-ARC disease resistance protein) | 0.8 | Potri.T106200 (Peroxidase superfamily protein) | 0.4 | 252.0948@6.714998 | 0.6 | ||
| Potri.001G132900 (ENHANCED DISEASE RESISTANCE 2) | 0.8 | Potri.007G014300 (Histone superfamily protein) | 0.4 | 419.9017@0.41500008 | 0.6 | ||
The variables are listed in descending order of statistical significance (p(CORR) from the OnPLS analysis. The p(CORR) value is depicted next to each variable. The transcript and protein annotations are according to JGI V3.0. LC-MS metabolites without annotations could not be identified and are listed as mass@retention time. Full datasets are listed in Additional files 3,4,5 and 6
One of the most striking changes in the PttSCAMP3 RNAi lines concerned increased abundance of lipids, such as linolenic acid, linoleic acid and glycerol-1-palmitate (Table 2; Additional file 6). Also several galactolipids such as monogalactosyldiacylglycerol (MGDG) and diagalactosyldiacylglycerol (DGDG) as well as their precursor phosphatidic acid (PA) were increased in abundance (Additional file 8). Since galactolipids are known to be localized in the chloroplast membranes, their localization in the differentiating xylem elements is most probably in the parenchymatic ray cells that are the only chloroplast-containing cells of the xylem.
Multi-omic analysis of the cell wall biosynthetic pathways
The multi-omic analyses revealed numerous differences in carbon metabolism and cell wall biosynthesis of the PttSCAMP3 RNAi lines. A detailed analysis revealed increased abundance of sucrose, fructose and glucose in the transgenic lines compared to the wild type (Additional file 6; Fig. 9). In addition, enzymes catalyzing cell wall monomer biosynthesis were frequently more abundant in the transgenic lines. Both a sucrose synthase (SuSy3) and a cytosolic invertase which produce nucleotide sugars and neutral fructose and glucose for biosynthesis of various cell wall components, were more abundant in the transgenic lines compared to the wild type (Additional file 4; Fig. 9). Also enzymes that produce monomers for secondary cell wall hemicellulose (xylan) biosynthesis (UDP-glucose 6-dehydrogenase and UDP-xylose synthase) were more abundant on a protein level, but mostly suppressed on the transcript level in the transgenic lines compared to the wild type (Fig. 9). Also enzymes that are responsible for producing precursors for pectin in the primary cell walls were mostly more abundant in the transgenic lines compared to the wild type (Fig. 9). Overall, the enzymes corresponding to cell wall biosynthesis were more abundant on the protein level but suppressed on the transcriptional level.
Fig. 9.
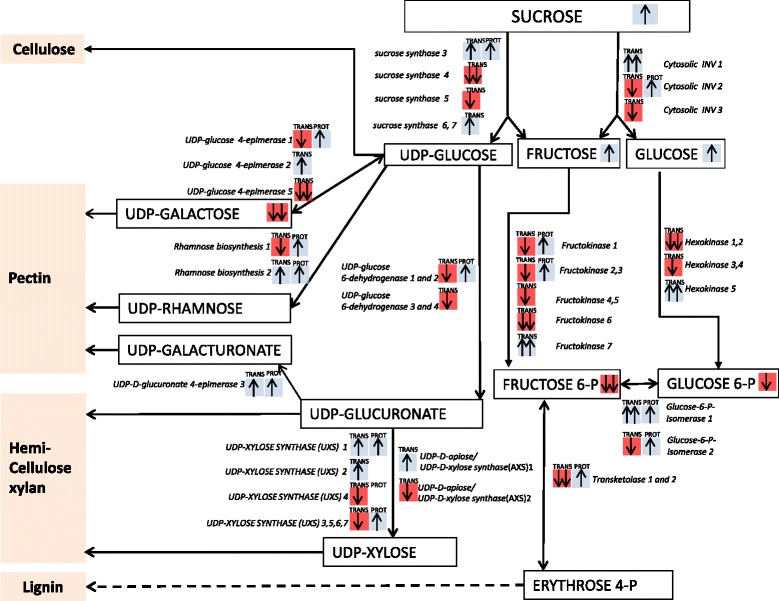
Analysis of the cell wall biosynthetic pathway on the transcript, protein and metabolite levels. The abundance of the secondary cell wall-related metabolites, transcripts (Trans) and proteins (Prot) are shown in the PttSCAMP3 RNAi lines compared to the wild type. ↑ in the blue boxes indicates upregulation in PttSCAMP3 RNAi lines; ↑↑ in the blue boxes indicates significant (|p(CORR)| >0.5) upregulation in PttSCAMP3 RNAi lines; ↓ in the red boxes indicates downregulation in the PttSCAMP3 RNAi lines; ↓↓ in the red boxes indicates significant (|p(CORR)| > 0.5) downregulation in PttSCAMP3 RNAi lines. Metabolites are shown in bold in boxes with the arrow/color codes as indicated above. The genes and proteins correspond to the following Populus trichocarpa gene models according to JGI V3.0: Fructokinase 1 (Potri.017G126300); Fructokinase 2 (Potri.007G129700); Fructokinase 3 (Potri.017G029000); Fructokinase 4 (Potri.012G132700); Fructokinase 5 (Potri.004G089300); Fructokinase 6 (Potri.015G134900); Fructokinase 7 (Potri.019G063600); Hexokinase 1 (Potri.001G190400); Hexokinase 2 (Potri.005G238600); Hexokinase 3 (Potri.009G050000); Hexokinase 4 (Potri.018G088300); Hexokinase 5 (Potri.001G254800); Cytosolic INV 1 (Potri.014G188100); Cytosolic INV 2 (Potri.013G110800); Cytosolic INV3 (Potri.019G082000); sucrose synthase 3 (Potri.002G202300); sucrose synthase 4 (Potri.006G136700); sucrose synthase 5 (Potri.018G063500); sucrose synthase 6 (Potri.004G081300); sucrose synthase 7 (Potri.012G037200); glucose-6-phosphate isomerase 1 (Potri.008G118900); glucose-6-phosphate isomerase 2 (Potri.002G104000); UDP-D-apiose/UDP-D-xylose synthase 1 (Potri.009G150600); UDP-D-apiose/UDP-D-xylose synthase 2 (Potri.004G189900); UDP-glucose 6-dehydrogenase 1 (Potri.017G092000); UDP-glucose 6-dehydrogenase 2 (Potri.004G118600); UDP-glucose 6-dehydrogenase 3 (Potri.010G159800); UDP-glucose 6-dehydrogenase 4 (Potri.008G094300); UDP-glucose 4-epimerase 1 (Potri.003G123700); UDP-glucose 4-epimerase 2 (Potri.003G140900); UDP-glucose 4-epimerase 5 (Potri.001G090700); UDP-D-xylose synthase (UXS) 1 (Potri.006G214000); UDP-D-xylose synthase (UXS) 2 (Potri.014G129200); UDP-D-xylose synthase (UXS) 3 (Potri.010G207200); UDP-D-xylose synthase (UXS) 4 (Potri.002G204400); UDP-D-xylose synthase (UXS) 5 (Potri.001G237200); UDP-D-xylose synthase (UXS) 6 (Potri.008G053100); UDP-D-xylose synthase (UXS) 7 (Potri.016G080500); Transketolase 1 (Potri.002G146300); Transketolase 2 (Potri.014G068200); UDP-D-glucuronate 4-epimerase (Potri.002G146500); Rhamnose biosynthesis 1 (Potri.006G272700); Rhamnose biosynthesis 2 (Potri.001G383500). Observe that the numbering of the genes is arbitrary to allow identification of the different members of the gene family
Discussion
The plant SCAMP proteins are believed to function in various membrane trafficking pathways on the basis of structural conservation with the animal systems as well the plant localization studies, but no functional evidence has been reported earlier most probably due to functional redundancy of the gene family [19]. Our study in Populus trees identified a role for plant SCAMPs in wood formation. The PttSCAMP3 RNAi lines exhibited increased deposition of both the carbohydrate and the phenolic components of the woody tissues of the stem, suggesting function of the studied SCAMP genes in suppressing deposition of the secondary cell wall components. In spite of the changes in cell wall polymers no growth penalties were observed in the transgenic lines, which demonstrates the potential of SCAMP3 suppression in improving biomass properties in forest trees.
The different RNAi lines differed somewhat in their phenotypes. Line 1 showed increased content of the glucan polysaccharide (Fig. 7), which correlated with increased glucose yields after enzymatic saccharification of this line (Fig. 8). Line 3 showed slightly increased lignin content and increased accumulation of xylan which is the main secondary cell wall hemicellulose (Figs. 6 and 7). Both xylan and lignin are known to act negatively on enzymatic hydrolysis of the secondary cell walls, possibly then explaining the absence of increased sugar yields in the saccharification assays of the woody tissues in line 3. These phenotypic differences could depend on the effect of the RNAi construct in suppressing only PttSCAMP6 in line 1 and both PttSCAMP3 and PttSCAMP6 in line 3 (Fig. 3a), and hence slightly different roles of these two Populus SCAMP proteins in membrane trafficking. Differential action of PttSCAMP3 and PttSCAMP6 is supported by the slightly different expression patterns in the differentiating xylem tissues of the stem (Fig. 2). The two earlier characterized plant SCAMP proteins, OsSCAMP1 in rice and NtSCAMP2, were also shown to have partially differential localization along the secretory pathway and hence most probably at least partially different functions. While OsSCAMP1 was localized to the plasmamembrane, trans-golgi network and cell plate [52], NtSCAMP2 was in addition found on the secretory vesicle cluster [46].
Overaccumulation of the cell wall components can be achieved by increased secretion of these compounds and/or increased trafficking of proteins, such as cell wall monomer biosynthetic enzymes or transporters, that are critical for cell wall deposition. Increased secretion would explain increased accumulation of xylan that is synthesized in the golgi and secreted to the cell wall, whereas increased trafficking of cellulose biosynthetic CesA enzymes would in turn enhance accumulation of cellulose. Overaccumulation of the lignin precursors is more difficult to explain since the transport mechanisms of lignin monomers are not fully understood even though ABC transporters are believed to be involved [53–55]. Four ABC transporter proteins were identified in the PttSCAMP3 transgenic lines (Potri.002G036400, Potri.010G003000, Potri.014G113200 and Potri.015G023800). Two of these (Potri.010G003000 and Potri.015G023800) were more abundant in the transgenic lines compared to the wild type. According to the proposed function of the Arabidopsis homolog, Potri.010G003000 is involved in auxin efflux. The function of Potri.015G023800 is not known and it is possible that this ABC transporter could be involved in transport of monomers for lignin.
Another mechanism to control accumulation of secreted compounds and proteins is to affect the secretory machinery itself. The tobacco NtSCAMP2 was localized to the secretory vesicle clusters which are responsible for mass secretion to the cell wall [46], and it is possible that the Populus SCAMP proteins control some function of such vesicles. It was interesting that PttSCAMP3 was highly co-expressed with nucleotide-diphospho-sugar transferases (Additional file 1). Accordingly, a nucleotide-diphospho-sugar transferase (Potri.001G400900) was significantly suppressed in abundance in the transgenic RNAi lines (Table 3), which supports the role of the PttSCAMPs in control of this kind of proteins. Yet another indication in this direction is provided by DeBolt et al. [56] who showed that a mutation in a dinucleotide sugar transferase UGT80B1 decreased suberin accumulation in Arabidopsis, and proposed that UGT80B1 glycosylates sterols that control trafficking of lipid precursors for instance for suberin biosynthesis. It is therefore possible that the PttSCAMPs are crucial for the function of some dinucleotide sugar transferases that, like UGT80B1, affect membrane trafficking and hence secretion of cell wall components and/or biosynthetic enzymes. According to this scenario, the PttSCAMPs would function as a safeguard that normally suppress secretion of cell wall precursors. Changes in the PttSCAMP-mediated trafficking would then allow rapid modification in the level of the cell wall polymers whenever necessary.
Conclusions
In the current study, we demonstrated a function for Populus SCAMP proteins in deposition of cell wall components in woody tissues of Populus trees. Even though only small differences were evident in comparisons using traditional statistical methods, the OnPLS model provided clear separation between the transgenic lines and the wild type. This demonstrates the strength of OnPLS modeling in handling simultaneously very different kinds of datasets and the intrinsic property of the model to readily identify small but consistent variation between the different datasets. The modelling also allowed identification of possible mechanisms underlying the phenotypic changes in the PttSCAMP3 transgenic trees and hence putative functions for the Populus SCAMP genes. These datasets provide a solid basis for understanding and further exploration of this poorly understood gene family in plants.
Additional files
Coexpression analysis of PttSCAMP3 in AspWood. Closest neighbours of PttSCAMP3 were identified in AspWood database (http://aspwood.popgenie.org/aspwood-v3.0/) using the default settings. (XLSX 21 kb)
The multivariate modelling of the data. A. PCA scores overview of the OnPLS model for the wild type (WT) and transgenic PttSCAMP3 RNAi lines. PCA is based on all variation (global, local and unique variation) in the OnPLS model. B. Contribution of the PCA loadings for the different datasets to the overall variation between the transgenic lines and the wild type in the OnPLS model. The datasets are the following: LCMS, LC-MS metabolomic dataset; Trans; transcriptomic dataset; Prot, proteomic dataset; GC, GC-MS metabolomic dataset; MSPy, Pyrolysis-gas chromatography/mass spectrometry dataset. The numbers 1–7 refer to the number of PCA component for each particular dataset. (PPTX 126 kb)
RNAseq analysis of wild type and PttSCAMP3 RNAi plants. (XLSX 1841 kb)
Proteomic analysis of wild type and PttSCAMP3 RNAi plants. 4.1. List of all peptides identified in the proteomic study. Statistical (OnPLS) analysis was done at the peptide level to compare the data from the wild type and the combined data from the three transgenic lines. The statistical analysis on the protein level is based on the p(CORR) values of the cognate peptides. 4.2. List of all proteins identified in the proteomic study on the basis of the cognate unique peptides. The p(CORR) value is a median for the p(CORR) values of the unique peptides corresponding to each of the proteins, and depict statistical significance between the wild type and the transgenic lines. (XLSX 698 kb)
LC-MS metabolomic analysis of wild type and PttSCAMP3 RNAi plants. (XLSX 64 kb)
GC-MS metabolomic analysis of wild type and PttSCAMP3 RNAi plants. (XLSX 19 kb)
Py-GC/MS analysis of wild type and PttSCAMP3 RNAi plants. (XLSX 14 kb)
LC-MS metabolomic identification of lipids. The graphs depict the abundance of lipids (peak area/mg fresh weight) in the PttSCAMP3 RNAi lines compared to the wild type in the LC-MS metabolome analysis. Only metabolites having |p(CORR)| ≥ 0.6 were included. The full LC-MS metabolome dataset is listed in Additional file 5. DGDG, digalactosyldiacylglycerol; MGDG, monogalactosyldiacylglycerol; PA, phosphatidic acid. (PPTX 1263 kb)
Acknowledgements
We are grateful to Junko Takahashi Schmidt, Lorenz Gerber, and Veronica Bourquin for technical help, Stephanie Robert for discussions, and the plant cell wall and carbohydrate analytical facility at UPSC/SLU, supported by Bio4Energy and TC4F projects.
Funding
This work was supported by grants from the Swedish Research Council FORMAS (232–2009-1698), the Swedish Foundation for Strategic Research (RBP14–0011), the Kempe Foundation (SMK-1443), and the Swedish Governmental Agency for Innovation Systems Vinnova and the Swedish Research Council VR through the UPSC Berzelii Centre for Forest Biotechnology (2015–02290). The funding bodies did not influence in any way the design of the study, collection of the data, data analyses, interpretation of the data or writing of the manuscript.
Availability of data and materials
The metabolomic dataset supporting the conclusions of this article is available in the EMBL-EBI MetaboLights repository with the identifier MTBLS478.
The RNAseq dataset supporting the conclusions of this article is available in the European Nucleotide Archive with the wild-type samples under the accession PRJEB21452 and the transgenic samples under the accession PRJEB21466.
The mass spectrometry proteomics data supporting the conclusions of this article is available in ProteomeXchange via the PRIDE [57] partner repository with the dataset identifier PXD008266.
Abbreviations
- DGDG
Digalactosyldiacylglycerol
- GC-MS
Gas chromatography-mass spectrometry
- GPR
Glucose production rate
- HPAEC
High-performance anion-exchange chromatography
- LC-MS
Liquid chromatograph-mass spectrometry
- MGDG
Monogalactosyldiacylglycerol
- OnPLS
Orthogonal projections to latent structures
- PA
Phosphatidic acid
- PCA
Principal component analysis
- Py-GC/MS
Pyrolysis gas chromatography/mass spectrometry
- RNAi
RNA interference
- SCAMP
Secretory Carrier-Associated Membrane Proteins
- VST
Variance-stabilizing transformation
- WT
Wild type
Authors’ contributions
HT conceived the study, TM, TRH, GW, LJ and HT designed the experiments, TM, JT, TRH, GW, LJ and HT supervised the experiments, OO coordinated the various omics analyses and performed the metabolomic and proteomic analyses, NM performed the RNAseq analysis, TS and JT performed the OnPLS analysis, JB processed the proteomic data, INA performed the LC-MS metabolite identification, MLG performed the saccharification and monosaccharide assays, AP did the qPCR analysis, MA and TM processed the LC-MS data, and HT wrote the manuscript with contributions from all coauthors. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The transgenic hybrid aspen trees were produced and donated to us by the company SweTree Technologies. The Swedish legislation was followed for growing and storing genetically modified organisms. Umeå Plant Science Centre has a permit from the Swedish Board of Agriculture to grow genetically modified organisms under containment. Permits for growth and/or collection of plant material from the field are not applicable for this study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12864-017-4411-1) contains supplementary material, which is available to authorized users.
Contributor Information
Ogonna Obudulu, Email: ogondus@gmail.com.
Niklas Mähler, Email: niklas.mahler@umu.se.
Tomas Skotare, Email: tomas.skotare@umu.se.
Joakim Bygdell, Email: Joakim.Bygdell@umu.se.
Ilka N. Abreu, Email: ilka.abreu@slu.se
Maria Ahnlund, Email: Maria.Ahnlund@slu.se.
Madhavi Latha Gandla, Email: madhavi.latha.gandla@umu.se.
Anna Petterle, Email: anna_petterle@hotmail.it.
Thomas Moritz, Email: Thomas.Moritz@slu.se.
Torgeir R. Hvidsten, Email: torgeir.r.hvidsten@nmbu.no
Leif J. Jönsson, Email: leif.jonsson@umu.se
Gunnar Wingsle, Email: Gunnar.Wingsle@slu.se.
Johan Trygg, Email: johan.trygg@umu.se.
Hannele Tuominen, Email: hannele.tuominen@umu.se.
References
- 1.Bruckner T, Bashmakov IA, Mulugetta Y, Chum H, de la Vega Navarro A, Edmonds J, et al. Energy Systems. In: Edenhofer O, Pichs-Madruga R, Sokona Y, Farahani E, Kadner S, Seyboth K, Adler A, Baum I, Brunner S, Eickemeier P, Kriemann B, Savolainen J, Schlömer S, von Stechow C, Zwickel T, Minx JC, editors. Climate change 2014: mitigation of climate change. Contribution of working group III to the fifth assessment report of the intergovernmental panel on climate change, vol. 2014. Cambridge: Cambridge University Press. p. 511–97.
- 2.W-J H, Harding SA, Lung J, Popko JL, Ralph J, Stokke DD, et al. Repression of lignin biosynthesis promotes cellulose accumulation and growth in transgenic trees. Nat Biotech. 1999;17:808–812. doi: 10.1038/11758. [DOI] [PubMed] [Google Scholar]
- 3.RL W, Remington DL, MacKay JJ, McKeand SE, O’Malley DM. Average effect of a mutation in lignin biosynthesis in loblolly pine. Theor Appl Genet. 1999;99:705–710. doi: 10.1007/s001220051287. [DOI] [PubMed] [Google Scholar]
- 4.Novaes E, Osorio L, Drost DR, Miles BL, Boaventura-Novaes CRD, Benedict C, et al. Quantitative genetic analysis of biomass and wood chemistry of Populus under different nitrogen levels. New Phytol. 2009;182:878–890. doi: 10.1111/j.1469-8137.2009.02785.x. [DOI] [PubMed] [Google Scholar]
- 5.Wagner A, Donaldson L, Kim H, Phillips L, Flint H, Steward D, et al. Suppression of 4-Coumarate-CoA Ligase in the coniferous gymnosperm Pinus radiata. Plant Physiol. 2009;149:370–383. doi: 10.1104/pp.108.125765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Voelker SL, Lachenbruch B, Meinzer FC, Kitin P, Strauss SH. Transgenic poplars with reduced lignin show impaired xylem conductivity, growth efficiency and survival. Plant Cell Environ. 2011;34:655–668. doi: 10.1111/j.1365-3040.2010.02270.x. [DOI] [PubMed] [Google Scholar]
- 7.Bonawitz ND, Chapple C. Can genetic engineering of lignin deposition be accomplished without an unacceptable yield penalty? Curr Opin Biotechnol. 2013;24:336–343. doi: 10.1016/j.copbio.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 8.Van Acker R, Leplé J-C, Aerts D, Storme V, Goeminne G, Ivens B, et al. Improved saccharification and ethanol yield from field-grown transgenic poplar deficient in cinnamoyl-CoA reductase. Proc Natl Acad Sci U S A. 2014;111:845–850. doi: 10.1073/pnas.1321673111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coleman HD, Yan J, Mansfield SD. Sucrose synthase affects carbon partitioning to increase cellulose production and altered cell wall ultrastructure. Proc Natl Acad Sci U S A. 2009;106:13118–13123. doi: 10.1073/pnas.0900188106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Park YW, Baba K, Furuta Y, Iida I, Sameshima K, Arai M, et al. Enhancement of growth and cellulose accumulation by overexpression of xyloglucanase in poplar. FEBS Lett. 2004;564:183–187. doi: 10.1016/S0014-5793(04)00346-1. [DOI] [PubMed] [Google Scholar]
- 11.Taniguchi T, Konagaya K, Kurita M, Takata N, Ishii K, Kondo T, et al. Growth and root sucker ability of field-grown transgenic poplars overexpressing xyloglucanase. J Wood Sci. 2012;58:550–556. doi: 10.1007/s10086-012-1281-7. [DOI] [Google Scholar]
- 12.Colmsee C, Mascher M, Czauderna T, Hartmann A, Schlüter U, Zellerhoff N, et al. OPTIMAS-DW: a comprehensive transcriptomics, metabolomics, ionomics, proteomics and phenomics data resource for maize. BMC Plant Biol. 2012;12:245. doi: 10.1186/1471-2229-12-245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thao NP. Tran VL-SP. enhancement of plant productivity in the post-genomics era. Curr Genomics. 2016;17:295–296. doi: 10.2174/138920291704160607182507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Srivastava V, Obudulu O, Bygdell J, Löfstedt T, Rydén P, Nilsson R, et al. OnPLS integration of transcriptomic, proteomic and metabolomic data shows multi-level oxidative stress responses in the cambium of transgenic hipI- superoxide dismutase Populus plants. BMC Genomics. 2013;14:893. doi: 10.1186/1471-2164-14-893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Decourcelle M, Perez-Fons L, Baulande S, Steiger S, Couvelard L, Hem S, et al. Combined transcript, proteome, and metabolite analysis of transgenic maize seeds engineered for enhanced carotenoid synthesis reveals pleotropic effects in core metabolism. J Exp Bot. 2015;66:3141–3150. doi: 10.1093/jxb/erv120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ali MB, Howard S, Chen S, Wang Y, Yu O, Kovacs LG, et al. Berry skin development in Norton grape: distinct patterns of transcriptional regulation and flavonoid biosynthesis. BMC Plant Biol. 2011;11:7. doi: 10.1186/1471-2229-11-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Löfstedt T, Trygg J. OnPLS-a novel multiblock method for the modelling of predictive and orthogonal variation. J Chemom. 2011;25:441–455. [Google Scholar]
- 18.Löfstedt T, Hanafi M, Mazerolles G, Trygg J. OnPLS path modelling. Chemom Intell Lab Syst. 2012;118:139–149. doi: 10.1016/j.chemolab.2012.08.009. [DOI] [Google Scholar]
- 19.Law AHY, Chow C-M, Jiang L. Secretory carrier membrane proteins. Protoplasma. 2012;249:269–283. doi: 10.1007/s00709-011-0295-0. [DOI] [PubMed] [Google Scholar]
- 20.Wang H, Tse YC, Law AHY, Sun SSM, Sun Y, Bin XZF, et al. Vacuolar sorting receptors (VSRs) and secretory carrier membrane proteins (SCAMPs) are essential for pollen tube growth. Plant J. 2010;61:826–838. doi: 10.1111/j.1365-313X.2009.04111.x. [DOI] [PubMed] [Google Scholar]
- 21.Nilsson O, Aldén T, Sitbon F, Little CHA, Chalupa V, Sandberg G, et al. Spatial pattern of cauliflower mosaic virus 35S promoter-luciferase expression in transgenic hybrid aspen trees monitored by enzymatic assay and non-destructive imaging. Transgenic Res. 1992;1:209–220. doi: 10.1007/BF02524751. [DOI] [Google Scholar]
- 22.Celedon PAF, de Andrade A, Meireles KGX, Cruz Gallo d, de Carvalho MC, Caldas DGG, Moon DH, et al. Proteomic analysis of the cambial region in juvenile Eucalyptus grandis at three ages. Proteomics. 2007;7:2258–2274. doi: 10.1002/pmic.200600989. [DOI] [PubMed] [Google Scholar]
- 23.Delhomme N, Mähler N, Schiffthaler B, Sundell D. Guidelines for RNA-Seq data analysis. Epigenesys Protoc. 2014:1–24.
- 24.Tuskan GA, DiFazio S, Jansson S, Bohlmann J, Grigoriev I, Hellsten U, et al. The genome of black cottonwood, Populus trichocarpa (Torr. & gray) Science. 2006;313:1596–1604. doi: 10.1126/science.1128691. [DOI] [PubMed] [Google Scholar]
- 25.Goodstein DM, Shu S, Howson R, Neupane R, Hayes RD, Fazo J, et al. Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 2012;40:D1178–D1186. doi: 10.1093/nar/gkr944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nordberg H, Cantor M, Dusheyko S, Hua S, Poliakov A, Shabalov I, et al. The genome portal of the Department of Energy Joint Genome Institute: 2014 updates. Nucleic Acids Res. 2014;42:D26–D31. doi: 10.1093/nar/gkt1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anders S, Pyl PT, Huber W. HTSeq—a python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brunner AM, Yakovlev IA, Strauss SH. Validating internal controls for quantitative plant gene expression studies. BMC Plant Biol. 2004;4:14. doi: 10.1186/1471-2229-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3:R34. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Masuda T, Tomita M, Ishihama Y. Phase transfer surfactant-aided Trypsin digestion for membrane proteome analysis. J Proteome Res. 2008;7:731–740. doi: 10.1021/pr700658q. [DOI] [PubMed] [Google Scholar]
- 33.Obudulu O, Bygdell J, Sundberg B, Moritz T, Hvidsten TR, Trygg J, et al. Quantitative proteomics reveals protein profiles underlying major transitions in aspen wood development. BMC Genomics. 2016;17:119. doi: 10.1186/s12864-016-2458-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jonsson P, Johansson AI, Gullberg J, Trygg J, A J, Grung B, et al. High-throughput data analysis for detecting and identifying differences between samples in GC/MS-based Metabolomic analyses. Anal Chem 2005;77:5635–5642. [DOI] [PubMed]
- 35.Dobrowolska I, Businge E, Abreu IN, Moritz T, Egertsdotter U. Metabolome and transcriptome profiling reveal new insights into somatic embryo germination in Norway spruce (Picea abies) Tree Physiol. 2017;28:1–15. doi: 10.1093/treephys/tpx078. [DOI] [PubMed] [Google Scholar]
- 36.Nording ML, Yang J, Georgi K, Hegedus Karbowski C, German JB, Weiss RH, et al. Individual variation in Lipidomic profiles of healthy subjects in response to Omega-3 fatty acids. PLoS One. 2013;8:e76575. doi: 10.1371/journal.pone.0076575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Latha Gandla M, Derba-Maceluch M, Liu X, Gerber L, Master ER, Mellerowicz EJ, et al. Expression of a fungal glucuronoyl esterase in Populus: effects on wood properties and saccharification efficiency. Phytochemistry. 2015;112:210–220. doi: 10.1016/j.phytochem.2014.06.002. [DOI] [PubMed] [Google Scholar]
- 38.Gerber L, Eliasson M, Trygg J, Moritz T, Sundberg B. Multivariate curve resolution provides a high-throughput data processing pipeline for pyrolysis-gas chromatography/mass spectrometry. J Anal Appl Pyrolysis. 2012;95:95–100. doi: 10.1016/j.jaap.2012.01.011. [DOI] [Google Scholar]
- 39.Bylesjö M, Eriksson D, Kusano M, Moritz T, Trygg J. Data integration in plant biology: the O2PLS method for combined modeling of transcript and metabolite data. Plant J. 2007;52:1181–1191. doi: 10.1111/j.1365-313X.2007.03293.x. [DOI] [PubMed] [Google Scholar]
- 40.Wold S. Cross-Validatory estimation of the number of components in factor and principal components models. Technometrics. 1978;20:397–405. doi: 10.1080/00401706.1978.10489693. [DOI] [Google Scholar]
- 41.Wiklund S, Johansson E, Sjöström L, Mellerowicz EJ, Edlund U, Shockcor JP, et al. Visualization of GC/TOF-MS-based Metabolomics data for identification of biochemically interesting compounds using OPLS class models. Anal Chem. 2008;80:115–122. doi: 10.1021/ac0713510. [DOI] [PubMed] [Google Scholar]
- 42.Tulipani S, Llorach R, Jáuregui O, López-Uriarte P, Garcia-Aloy M, Bullo M, et al. Metabolomics unveils urinary changes in subjects with metabolic syndrome following 12-week nut consumption. J Proteome Res. 2011;10:5047–5058. doi: 10.1021/pr200514h. [DOI] [PubMed] [Google Scholar]
- 43.Llorach R, Garrido I, Monagas M, Urpi-Sarda M, Tulipani S, Bartolome B, et al. Metabolomics study of human urinary Metabolome modifications after intake of almond (Prunus Dulcis (mill.) D.A. Webb) skin Polyphenols. J Proteome Res. 2010;9:5859–5867. doi: 10.1021/pr100639v. [DOI] [PubMed] [Google Scholar]
- 44.Llorach R, Urpi-Sarda M, Jauregui O, Monagas M, Andres-Lacueva C. An LC-MS-based Metabolomics approach for exploring urinary Metabolome modifications after cocoa consumption. J Proteome Res. 2009;8:5060–5068. doi: 10.1021/pr900470a. [DOI] [PubMed] [Google Scholar]
- 45.Llorach R, Medina S, García-Viguera C, Zafrilla P, Abellán J, Jauregui O, et al. Discovery of human urinary biomarkers of aronia-citrus juice intake by HPLC-q-TOF-based metabolomic approach. Electrophoresis. 2014;35:1599–1606. doi: 10.1002/elps.201300565. [DOI] [PubMed] [Google Scholar]
- 46.Toyooka K, Goto Y, Asatsuma S, Koizumi M, Mitsui T, Matsuoka KA. Mobile Secretory vesicle cluster involved in mass transport from the Golgi to the plant cell exterior. Plant Cell. 2009;21:1212–1229. doi: 10.1105/tpc.108.058933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sundell D, Street NR, Kumar M, Mellerowicz EJ, Kucukoglu M, Johnsson C, et al. AspWood: high-spatial-resolution transcriptome profiles reveal uncharacterized modularity of wood formation in Populus tremula. Plant Cell. 2017;29:1585–1604. doi: 10.1105/tpc.17.00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morreel K, Ralph J, Kim H, Lu F, Goeminne G, Ralph S, et al. Profiling of Oligolignols reveals Monolignol coupling conditions in lignifying poplar xylem. Plant Physiol. 2004;136:3537–3549. doi: 10.1104/pp.104.049304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lin C-Y, Wang JP, Li Q, Chen H-C, Liu J, Loziuk P, et al. 4-Coumaroyl and Caffeoyl Shikimic acids inhibit 4-Coumaric acid:coenzyme a Ligases and modulate metabolic flux for 3-hydroxylation in Monolignol biosynthesis of Populus trichocarpa. Mol Plant. 2015;8:176–187. doi: 10.1016/j.molp.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 50.Trygg J, Wold S. O2-PLS, a two-block (X-Y) latent variable regression (LVR) method with an integral OSC filter. J Chemom. 2003;17:53–64. doi: 10.1002/cem.775. [DOI] [Google Scholar]
- 51.Trygg J. O2-PLS for qualitative and quantitative analysis in multivariate calibration. J Chemom. 2002;16:283–293. doi: 10.1002/cem.724. [DOI] [Google Scholar]
- 52.Lam SK, Siu CL, Hillmer S, Jang S, An G, Robinson DG, et al. Rice SCAMP1 defines Clathrin-coated, trans-Golgi–located tubular-vesicular structures as an early Endosome in tobacco BY-2 cells. Plant Cell. 2007;19:296–319. doi: 10.1105/tpc.106.045708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Samuels L, Kunst L, Jetter R. Sealing plant surfaces: Cuticular wax formation by epidermal cells. Annu Rev Plant Biol. 2008;59:683–707. doi: 10.1146/annurev.arplant.59.103006.093219. [DOI] [PubMed] [Google Scholar]
- 54.Miao Y-C, Liu C-J. ATP-binding cassette-like transporters are involved in the transport of lignin precursors across plasma and vacuolar membranes. Proc Natl Acad Sci U S A. 2010;107:22728–22733. doi: 10.1073/pnas.1007747108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Alejandro S, Lee Y, Tohge T, Sudre D, Osorio S, Park J, et al. AtABCG29 is a Monolignol transporter involved in lignin biosynthesis. Curr Biol. 2012;22:1207–1212. doi: 10.1016/j.cub.2012.04.064. [DOI] [PubMed] [Google Scholar]
- 56.DeBolt S, Scheible W-R, Schrick K, Auer M, Beisson F, Bischoff V, et al. Mutations in UDP-glucose:sterol Glucosyltransferase in Arabidopsis cause transparent Testa phenotype and Suberization defect in seeds. Plant Physiol. 2009;151:78–87. doi: 10.1104/pp.109.140582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vizcaíno JA, Csordas A, Del-Toro N, Dianes JA, Griss J, Lavidas I, et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016;44:11033. doi: 10.1093/nar/gkw880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Whelan S, Goldman NA. General empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol Biol Evol. 2001;18:691–699. doi: 10.1093/oxfordjournals.molbev.a003851. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Coexpression analysis of PttSCAMP3 in AspWood. Closest neighbours of PttSCAMP3 were identified in AspWood database (http://aspwood.popgenie.org/aspwood-v3.0/) using the default settings. (XLSX 21 kb)
The multivariate modelling of the data. A. PCA scores overview of the OnPLS model for the wild type (WT) and transgenic PttSCAMP3 RNAi lines. PCA is based on all variation (global, local and unique variation) in the OnPLS model. B. Contribution of the PCA loadings for the different datasets to the overall variation between the transgenic lines and the wild type in the OnPLS model. The datasets are the following: LCMS, LC-MS metabolomic dataset; Trans; transcriptomic dataset; Prot, proteomic dataset; GC, GC-MS metabolomic dataset; MSPy, Pyrolysis-gas chromatography/mass spectrometry dataset. The numbers 1–7 refer to the number of PCA component for each particular dataset. (PPTX 126 kb)
RNAseq analysis of wild type and PttSCAMP3 RNAi plants. (XLSX 1841 kb)
Proteomic analysis of wild type and PttSCAMP3 RNAi plants. 4.1. List of all peptides identified in the proteomic study. Statistical (OnPLS) analysis was done at the peptide level to compare the data from the wild type and the combined data from the three transgenic lines. The statistical analysis on the protein level is based on the p(CORR) values of the cognate peptides. 4.2. List of all proteins identified in the proteomic study on the basis of the cognate unique peptides. The p(CORR) value is a median for the p(CORR) values of the unique peptides corresponding to each of the proteins, and depict statistical significance between the wild type and the transgenic lines. (XLSX 698 kb)
LC-MS metabolomic analysis of wild type and PttSCAMP3 RNAi plants. (XLSX 64 kb)
GC-MS metabolomic analysis of wild type and PttSCAMP3 RNAi plants. (XLSX 19 kb)
Py-GC/MS analysis of wild type and PttSCAMP3 RNAi plants. (XLSX 14 kb)
LC-MS metabolomic identification of lipids. The graphs depict the abundance of lipids (peak area/mg fresh weight) in the PttSCAMP3 RNAi lines compared to the wild type in the LC-MS metabolome analysis. Only metabolites having |p(CORR)| ≥ 0.6 were included. The full LC-MS metabolome dataset is listed in Additional file 5. DGDG, digalactosyldiacylglycerol; MGDG, monogalactosyldiacylglycerol; PA, phosphatidic acid. (PPTX 1263 kb)
Data Availability Statement
The metabolomic dataset supporting the conclusions of this article is available in the EMBL-EBI MetaboLights repository with the identifier MTBLS478.
The RNAseq dataset supporting the conclusions of this article is available in the European Nucleotide Archive with the wild-type samples under the accession PRJEB21452 and the transgenic samples under the accession PRJEB21466.
The mass spectrometry proteomics data supporting the conclusions of this article is available in ProteomeXchange via the PRIDE [57] partner repository with the dataset identifier PXD008266.