

Abstract
Background/rationale
Amplification of the epidermal growth factor receptor (EGFR) gene represents one of the most frequent gene alterations in glioblastoma (GBM). In the current study, we evaluated gefitinib, a potent EGFR inhibitor, in the treatment of adults with newly diagnosed GBM.
Methods
98 patients (96 evaluable) were accrued from May 18, 2001 through August 2, 2002. All were newly diagnosed GBM patients who were clinically and radiographically stable/improved following radiation treatment (enrollment within 5 weeks of radiation completion). No prior chemotherapy was permitted. EGFR amplification/mutation, as assessed by FISH and immunohistochemistry, was not required for treatment with gefitinib but was studied when tissues were available. Gefitinib was administered at 500mg QD; for patients receiving dexamethasone and/or enzyme-inducing (CYP3A4) agents, dose was escalated to a maximum of 1000mg QD. Treatment cycles were repeated at 4-week intervals with brain MRI at 8-week intervals.
Results
Overall survival (OS; calculated from time of initial surgery) at 1 year (primary endpoint) with gefitinib was 54.2%, which was not statistically different compared to that of historical control population (48.9%, data from 3 previous phase III NCCTG studies of newly diagnosed GBM patients). Progression-free survival (PFS) at 1 year post-RT (16.7%) was also not significantly different to that of historical controls (30.3%). Clinical outcome was not affected by EGFR status (amplification and/or vIII mutation). Fatigue (41%), rash (62%), and loose stools (58%) constituted the most frequent adverse events, the majority of these being limited to Grade 1/2. Of note, the occurrence of drug-related adverse effects, such as loose stools was associated with improved OS.
Conclusions
In our evaluation of nearly one hundred patients with newly diagnosed GBM, treatment with adjuvant gefitinib post radiation was not associated with significant improvement in OS or PFS. However, patients who experienced gefitinib-associated adverse effects (rash/diarrhea) did demonstrate improved OS.
Key words for indexing: astrocytoma, glioblastoma, glioma, EGFR
Introduction
EGFR amplification represents the most common oncogene alteration in glioblastoma, and it is consistently reported in approximately 40-50% of these tumors (1-3). The amplification of the wild type EGFR gene appears to be a precursor event to subsequent mutations, most of which involve intragene deletions that may further augment the biologic activity of receptor signaling (4). The most thoroughly characterized EGFR mutation in human GBMs results in the synthesis of a protein lacking a portion of the extracellular domain (EGFRvIII or Δ6-273, indicative of the amino acids deleted), which results in constitutive, ligand independent receptor activity. Given the extensive literature supporting a role for EGFR in glioma biology, EGFR represents a logical target for therapeutics. In the current study, the EGFR inhibitor gefitinib was evaluated in newly diagnosed GBM patients to determine its potential effect on overall survival as well as to determine whether patient outcome was affected by EGFR status.
Gefitinib (ZD1839, Iressa; AstraZeneca, Wilmington, DE) is an oral low-molecular weight, adenosine triphosphate mimetic of the anilinoquinazoline family that reversibly inhibits the tyrosine kinase activity associated with EGFR (5;6). Gefitinib has demonstrated therapeutic activity in several systemic cancers (7;8), leading to its approval by the United States FDA for monotherapy in the treatment of patients with locally advanced or metastatic non-small-cell lung cancer after failure of both platinum-based and docetaxel chemotherapies.
Our current study was undertaken in the newly diagnosed GBM setting. As the study was initiated prior to the report demonstrating superiority of radiation in combination with temozolomide over radiation alone (9), patients in the current study were first treated with radiotherapy alone, following which they were started on gefitinib. The results of our study show that the benefit (improved overall survival) of gefitinib was restricted to those patients who experienced drug-related adverse effects, but that clinical benefit could not be correlated with EGFR status (amplification and/or vIII mutation).
Patients and Methods
Eligibility
Only newly diagnosed adult (≥18 years) GBM patients were eligible for enrollment. Following surgery or biopsy (histology centrally reviewed by CG or BWS), patients underwent what was then standard of care treatment (radiation therapy only, as this study started accrual prior to the report demonstrating superiority of radiation plus temozolomide over radiation alone). Patients were then deemed eligible if the post-radiation MRI/CT scan did not fulfill radiographic criteria for progression (>25% increase in the product of perpendicular diameters of the enhancing area and/or the appearance of new lesions). Patients were required to be ≥2 weeks but ≤5 weeks since the completion of radiation, and ≤15 weeks total since surgery/biopsy.
Patients were required to have a performance status of ECOG 2 or better, nonpregnant, and not breastfeeding. Normal laboratory measures of hepatic, renal, and bone marrow function were required as defined as an absolute neutrophil count ≥1,500/μL, a platelet count ≥100,000/μL, a creatinine level and total bilirubin less than 1.5× the upper limit of institutional normal, and plasma AST levels ≤3× the upper limit of institutional normal. Patients were deemed ineligible if they had received prior chemotherapy (including GliadelR), stereotactic radiosurgery, or brachytherapy. Other exclusion factors included uncontrolled infection, evidence of other active malignancy or other severe concurrent disease. 98 patients (96 evaluable) were accrued from May 18, 2001 through August 2, 2002.
Treatment Plan and Dose Modifications
Gefitinib was administered to all patients at an initial oral dose of 500 mg/d. Patients who received dexamethasone and/or enzyme inducing antiepileptic drugs (EIAEDs) and/or other CYP3A4-inducing agents without toxicities after 2 weeks of receiving gefitinib had the gefitinib dose escalated by 250 mg increments to a maximum daily dose of 1000 mg. If no adverse effects were noted after an additional 2 weeks, the dose was escalated to 1000 mg/d. Therapy was continued until disease progression, significant clinical decline, unacceptable toxicity, or patient decision. Toxicity was graded using the National Cancer Institute Common Toxicity Criteria, version 2.0
For toxicity of grade 2 or higher, treatment was held and re-evaluated at weekly intervals until improvement to ≤grade 1 and the gefitinib was re-instituted at the same dose. If toxicity recurred, then gefitinib was held till resolution and reinstated at daily dose that was 250 mg less that the dose that caused toxicity. Patients requiring a second dose reduction or those in whom toxicity did not resolve to ≤grade 1 were taken off the study.
Measurement of Effect
For those patients who had measurable disease on MRI/CT at time of enrollment, partial response (PR) was defined as ≥50% reduction in product of perpendicular diameters of contrast enhancement in the setting of stable or decreasing steroid dosing. Progression was defined as >25% increase in the product of perpendicular diameters of the enhancing area and/or the appearance of new lesions.
EGFR Analyses
Fluorescence in situ hybridization (FISH) as per previously published protocol was used to assess for EGFR amplification (4). Immunostaining for total EGFR was performed as previously described (10). Briefly, 5-μm-thick sections were mounted on positively charged slides, deparaffinized, and rehydrated in phosphate-buffered saline (PBS). Endogenous peroxidase activity was blocked with 3% hydrogen peroxide in PBS/0.05% Tween 20 for 20 min. Sections were then washed in PBS and blocked for 20 min in the appropriate serum from the same species as the secondary antibody diluted to 10% in PBS. For EGFR vIII staining, microwave antigen retrieval was performed by placing the slides in 50 mM citrate buffer (pH 6.0) and microwaving for 12 min at full power and 10 min at 20% power, followed by cooling for 15 min and two to three 5-min washes in PBS. For total EGFR staining, pretreatment consisted of 0.025% trypsin placed on the tissue followed by 30-min incubation at room temperature. Primary antibodies, diluted in PBS/10% serum, were applied to the sections in a humid chamber overnight at 4°C. After washing 2 to 3 times in PBS, secondary antibodies were applied using the Dako (Carpinteria, CA) Envision kit according to the manufacturer’s instructions. Detection of bound secondary antibody was performed with diaminobenzidine for 5 min. Sections were then counterstained with light hematoxylin and mounted. The total EGFR antibody used is mouse monoclonal clone 528 (Oncogene Science, Cambridge, MA; 1:50 dilution), which recognizes both wild-type and vIII EGFR. Anti vIII EGFR antibody is a rabbit polyclonal (1:1,200, Zymed, South San Francisco, CA).
Statistical considerations
This was a phase-II trial designed to assess (a) the ability of gefitinib to extend survival in GBMs, as measured by 52-week survival after biopsy or definitive surgery. The single-stage 0.10-level procedure tested the null hypothesis, in that the true ‘success’ probability was ≤50% (i.e., that the median ‘post-surgery survival’ is ≤52 weeks) versus the alternative hypothesis that it was >50%). With a sample size of at least 84 patients, this procedure had 90% power to detect a true ‘success’ probability of 65%, which corresponds to a median ‘post-surgery survival’ of 83.67 weeks, assuming an exponential survival distribution. Two interim analyses were performed. The first was when 35 patients had been followed for at least 53 weeks post surgery/biopsy, and the second was when 60 patients had been followed for 52 weeks post surgery/biopsy. Neither of the interim analyses demonstrated evidence to terminate the study.
Results
Patients and Eligibility
Over 20 member institutions of the North Central Cancer Treatment Group (NCCTG) contributed patients to this study between May 18, 2001 and August 2, 2002. Of the ninety-eight patients enrolled, 1 patient cancelled participation in the study and 1 patient was deemed ineligible upon further screening. Table 1 reflects the 96 eligible patients. Performance status was ECOG 1 or better in 86% of patients, and ECOG 2 in the remaining 14% of patients. The majority of patients (92%) underwent either subtotal (45%) or gross total (47%) resection.
Table 1.
Baseline Characteristics
| Variable | Summary |
|---|---|
| Age (years) | |
| mean ± SD | 57 ± 11 |
| median (min, max) | 57 (19, 79) |
| Gender, n(%) | |
| female | 32 (33%) |
| male | 64 (67%) |
| ECOG performance score | |
| 0 | 32 (33%) |
| 1 | 51 (53%) |
| 2 | 13 (14%) |
| Extent of resection | |
| biopsy | 8 (8%) |
| subtotal | 43 (45%) |
| gross total | 45 (47%) |
| EIAC | |
| yes | 65 (68%) |
| no | 31 (32%) |
Gefitinib treatment was continued until disease progression. In 15 patients in whom the follow-up MRI remained progression-free at 1 year, the decision to continue gefitinib was made by the treating physician. 68% of patients were treated with enzyme-inducing anticonvulsant medication, while 32% either received non-enzyme inducing medication or no anticonvulsant medication.
Toxicity
The most frequently observed adverse effects (AE) were rash and loose stool (Tables 2 and 3). Of the patients who developed rash, most were grade 1 to 2 (81%). The rash generally occurred early; 55 patients developed the rash during the first cycle, 4 during the second cycle, and 4 thereafter. Doses at which rash started were 500 mg for 57 patients, 750 mg in 2 patients, and 1,000 mg in 4 patients.
Table 2.
Most Frequent Toxicities Regardless of Attribution
| Grade 1 | Grade 2 | Grade 3 | Grade 4 | Total | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Toxicity | No. of Patients | % | No. of Patients | % | No. of Patients | % | No. of Patients | % | No. of Patients | % |
| Rash | 28 | 29.2 | 23 | 24.0 | 11 | 11.5 | 62 | 64.6 | ||
| Diarrhea | 42 | 43.8 | 13 | 13.5 | 9 | 9.4 | 64 | 66.7 | ||
| Fatigue | 17 | 17.7 | 17 | 17.7 | 7 | 7.3 | 41 | 42.7 | ||
| Dry Skin | 28 | 29.2 | 8 | 8.3 | 36 | 37.5 | ||||
| Nausea | 19 | 19.8 | 10 | 10.4 | 29 | 30.2 | ||||
| Pruritis | 15 | 15.6 | 13 | 13.5 | 28 | 29.2 | ||||
| Seizure | 11 | 11.5 | 9 | 9.4 | 3 | 3.1 | 23 | 24.0 | ||
| Neuro-Motor | 3 | 3.1 | 7 | 7.3 | 11 | 11.5 | 1 | 1.0 | 22 | 22.9 |
| SGOT (AST) | 13 | 13.5 | 4 | 4.2 | 4 | 4.2 | 1 | 1.0 | 22 | 22.9 |
| Headache | 12 | 12.5 | 7 | 7.3 | 2 | 2.1 | 21 | 21.9 | ||
Table 3.
Most Frequent Treatment Related Toxicities
| Grade 1 | Grade 2 | Grade 3 | Grade 4 | Total | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Toxicity | No. of Patients | % | No. of Patients | % | No. of Patients | % | No. of Patients | % | No. of Patients | % |
| Rash | 26 | 27.1 | 23 | 24.0 | 11 | 11.5 | 60 | 62.5 | ||
| Diarrhea | 31 | 32.3 | 12 | 12.5 | 9 | 9.4 | 52 | 54.2 | ||
| Dry Skin | 25 | 26.0 | 8 | 8.3 | 33 | 34.4 | ||||
| Fatigue | 14 | 14.6 | 13 | 13.5 | 5 | 5.2 | 32 | 33.3 | ||
| Pruritis | 13 | 13.5 | 13 | 13.5 | 26 | 27.1 | ||||
| Nausea | 19 | 19.8 | 5 | 5.2 | 24 | 25.0 | ||||
| SGOT (AST) | 10 | 10.4 | 2 | 2.1 | 4 | 4.2 | 1 | 1.0 | 17 | 17.7 |
| Anorexia | 8 | 8.3 | 6 | 6.3 | 14 | 14.6 | ||||
| SGPT (ALT) | 3 | 3.1 | 3 | 3.1 | 2 | 2.1 | 2 | 2.1 | 10 | 10.4 |
| Vomiting | 6 | 6.3 | 3 | 3.1 | 9 | 9.4 | ||||
Of the patients with diarrhea, most were grade 1 to 2 (84%). Of the patients with diarrhea, the majority (86%) developed diarrhea during cycle 1, 3 (5%) during cycle 2, 3 during cycle 3, and 3 thereafter. The diarrhea occurred at a dose of 250 mg in 2 patients (those 2 patients experienced diarrhea at cycle 2, while they experienced rash in cycle 1 thus their dose level in cycle 2 was reduced to 250mg), 500 mg in 56 patients, 750 mg in 4 patients, and 1,000 mg in 2 patients. The dose level when rash occurred is not a function of EIAEDs, but the dose level when diarrhea occurred is related to patients EIAED status (mean: 476 mg for non-EIAED and 547 mg for EIAED, p-value = 0.01) Rash improved in most patients with either dose modifications of gefitinib and/or the use of topical medications. Loose stool was treated effectively with lomotil; for those patients in whom lomotil was insufficient, gefitinib dose was reduced. Other toxicities that were encountered that were felt to be possibly related to gefitinib use included conjunctivitis, anorexia, weight loss, and AST and ALT elevation. No Grade 5 toxicity was observed.
Several patients experienced adverse events that were felt to be secondary to their cancer rather than gefitinib treatment, including seizure, cerebral edema, CNS hemorrhage, lower extremity deep vein thrombosis, confusion, muscle weakness, and incontinence. Most of the neurologic events occurred within the context of a progressive tumor. No patient experienced pulmonary toxicity, cellulitis, nausea, vomiting, electrolyte changes, or renal dysfunction.
Measurement of Therapeutic Effect, Time to Progression, and Survival
For the primary outcome of overall survival as compared to a historical control population derived from prior NCCTG studies of newly diagnosed GBM patients, no difference was observed, with a median survival of approximately 12 months (Fig. 1). Similarly, there was no difference in regards to progression-free survival (Fig. 2).
Figure 1. Overall survival: Gefitinib vs. Historical Data.

Compared to historical control population of newly diagnosed glioblastoma patients (previous NCCTG clinical trials), treatment with gefitinib post-radiation was not associated with any appreciable improvement in overall survival.
Figure 2. Progression Free Survival: Gefitinib vs. Historical Data.

Compared to historical control population of newly diagnosed glioblastoma patients (previous NCCTG clinical trials), treatment with gefitinib post-radiation was not associated with a statistically significant change in progression-free survival.
55 tumor samples were evaluable for EGFR status – for all samples, both FISH and IHC techniques were utilized and a high concordance between the two methods was observed. In comparing results from FISH and IHC methods, EGFR gene amplification (as assessed by FISH) was never observed in tumors that showed no EGFR protein expression (as assessed by IHC), which is similar to a previous report indicating that FISH may not be necessary when IHC shows no EGFR protein expression (11). Hence, for the survival analysis as a function of EGFR status, IHC results were used. Of note, when the FISH results were used for the survival analysis, the survival results were no different from those obtained from IHC data. We then compared outcome of patients with tumors demonstrating EGFR overexpression (20 of 55 evaluable specimens) on IHC versus those whose tumors were negative for EGFR immunoreactivity. As shown in Fig. 3, there was no significant difference in overall survival between patients whose tumors demonstrated EGFR immunoreactivity (e.g., EGFR overexpression/amplification) versus those whose tumors did not show EGFR immunoreactivity. Of note, when the FISH results were used for the survival analysis, the survival results were no different from those obtained from IHC data. Of 20 tumor samples found to harbor EGFR amplification, 12 demonstrated EGFRvIII mutation. When comparing patients with EGFRvIII mutation versus amplification of wild-type EGFR, no difference was observed for overall – or progression free survival (data not shown). In summary, EGFR status did not correlate with clinical outcome in our study.
Figure 3. Overall Survival: EGFR amplified vs. not amplified.

EGFR status was available in 55 patient samples (27 EGFR amplification positive, 28 EGFR amplification negative). Between the two groups, there was no difference in overall survival.
Drug-attributable adverse effects correlate with improved overall survival
While patient outcome was no different between gefitinib treated patients and historical controls (Figs. 1 and 2), we did observe an increase in overall survival in patients who experienced loose/watery stool (gefitinib-associated adverse effect), as compared with those study patients who did not experience such AEs (Fig. 4). PFS was also superior in patients who experienced diarrhea (median PFS of 7.1 months versus 5.5 months, p=0.047; Fig. 5). The improved outcome associated with drug-attributable systemic AEs is similar to that reported by Rich et al. in their evaluation of gefitinib in the setting of recurrent high grade gliomas (12). Among the patients in our study who experienced diarrhea, the severity of diarrhea did not correlate with patient outcome and there was no difference in outcome as a function of EGFR status. Furthermore, the occurrence of diarrhea was not associated with other prognostic variables such as age, performance status, tumor location, or extent of surgical resection.
Figure 4. Overall Survival: Patients experienced diarrhea versus not.

Patients who experienced gefitinib-associated adverse effects had an overall survival that was superior to those patients who did not experience such adverse effects. The two-year survival for the patients who experienced loose/watery stool was approximately 27%, compared to the 2 year survival of 10% in the historical control group.
Figure 5. Progression-free survival (PFS) for patients experienced diarrhea versus not.
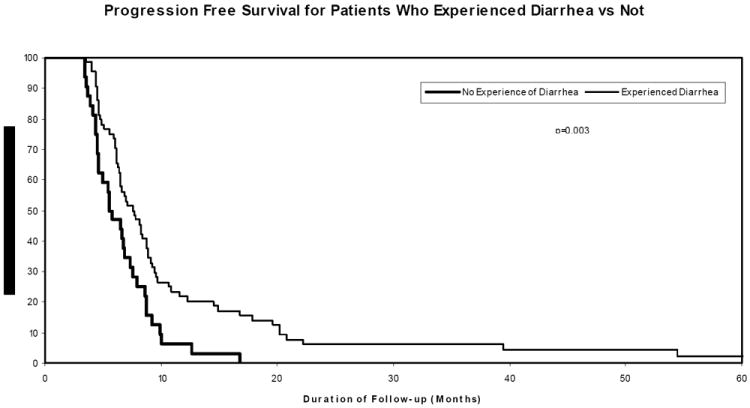
The PFS of patients who experienced loose/watery stool, an adverse effect attributable to gefitinib, was superior to that of patients who did not experience such adverse effects.
Of the 63 patients who experienced diarrhea, the great majority (54 patients, 86%) had this AE occur during the first cycle of gefitinib (Table 2) when they were on the starting dose (500 mg). Diarrhea occurred at a dose of 750 mg in 4 patients and 1,000 mg in 2 patients.
Discussion
Based upon extensive data that implicates EGFR and its downstream pathways in the biology of malignant glioma, encompassing proliferation, invasion, and angiogenesis, EGFR represents a logical target for therapeutic trials in this tumor for which treatment options are severely limited. Our current study was undertaken in the newly diagnosed GBM setting. As the study was initiated prior to the report demonstrating superiority of radiation in combination with temozolomide over radiation alone (9), patients in the current study were first treated with radiotherapy alone, following which they were started on gefitinib.
Despite the scientific logic of targeting EGFR, our study did not show a benefit of gefitinib compared to historical control group comprised of prior studies in newly diagnosed GBM patients (Fig. 1). Moreover, as in other cancer types, EGFR status (amplification and/or EGFRvIII expression) was not associated with an increased sensitivity to gefitinib nor did the absence of EGFR expression preclude tumor control in this study (Fig. 3) (12). In regards to recent reports that demonstrate a correlation between PTEN/AKT status and response to EGFR inhibitor therapy (13;14), we did not evaluate PTEN/AKT status on our tumor samples. In those two retrospective studies, EGFR status (EGFR amplification, EGFRvIII expression) and increased AKT pathway activity (by way of either diminished PTEN or increased AKT phosphorylation) were associated with a higher likelihood of radiographic response to EGFR inhibitor therapy. However, such a correlation between outcome and EGFR/AKT status could not be confirmed in a subsequent prospective randomized phase II study of erlotinib and standard alkylators (15). Similarly, additional reports have also shown that EGFR amplification and/or the presence of EGFRvIII was not a prognostic or predictive factor in GBM patients (16;17). Hence, while the laboratory data pertaining to EGFR would indicate that EGFR status should impact upon patient outcome, the influence of EGFR status as a single molecular variable on overall survival remains unresolved. Rather, the emerging data would indicate that perhaps EGFR status in combination with other molecular variables, such as YKL-40 (18;19), may correlate with patient outcome.
While patient outcome was no different between gefitinib-treated patients of this study compared to our historical control previous GBM clinical studies, we did observe a significant increase in overall survival in those patients who experienced loose/watery stool, an AE attributable to EGFR inhibitors such as gefitinib (Fig. 4). In this regard, the two-year survival for the patients who experienced loose/watery stool was approximately 27%, compared to the 2 year survival of 10% in the historical control group. This 28% 2 year survival in patients who experienced loose/watery stool in our study is similar to the 2 year survival observed with the combination of temozolomide concurrent with radiation followed by adjuvant temozolomide (9) as reported by Stupp et al.
Despite the association between diarrhea and improved outcome in our study, the mechanistic relationship between this AE and outcome remains unclear. In the setting of recurrent glioma patients treated with gefitinib, the occurrence of diarrhea correlated with improved PFS and OS (12). In the context of newly diagnosed GBM, the UCSF group reported an overall survival benefit for patients treated with another EGFR inhibitor (erlotinib) given with temozolomide during and after radiation compared to historical controls who received radiation/temozolomide (20). However, in our own experience with study of erlotinib (NCCTG study N0177), the addition of erlotinib to radiation/temozolomide was not associated with improved survival (21); furthermore, in that study, the occurrence of diarrhea did not correlate with improved outcome. The UCSF and NCCTG evaluations of erlotinib were very similar studies, except that the NCCTG study had a somewhat higher number of patients (98) compared to the 65 patients in the UCSF study, which may in part account for differences in outcome results between the two studies of erlotinib given with radiation/temozolomide.
While it is possible that diarrhea may be a systemic surrogate index or pharmacodynamic marker of treatment efficacy, this premise remains speculative. In the current study, pharmacokinetic data were not collected, but there was no correlation between survival and dose of gefitinib. Furthermore, it appears as though it is not a case that individuals were afflicted with diarrhea simply as the result of a cumulative dose (if that were the case, it would explain why patients with diarrhea lived longer – that is, they lived long enough to get multiple doses and multiple doses caused diarrhea), as the great majority (86%) of patients who experienced diarrhea had this AE occur during the first cycle of gefitinib treatment. As to whether diarrhea may be associated with other outcome variables to account for the observed survival difference, we did not observe any association between this AE and prognostic variables such as age, performance status, tumor location, or extent of resection, or EGFR status.
In summary, while gefitinib treatment post radiation did not improve outcome compared to historical control group of GBM patients, there was an improved overall survival in patients who experienced loose/watery stools, indicating that gefitinib may have some activity in a subset of GBM patients. Future studies may draw upon emerging laboratory data to guide appropriate use of EGFR inhibitors in combination with agents that target downstream signaling components, such as AKT-dependent and AKT-independent intermediaries (22) in an effort to augment the benefit of EGFR-targeted therapeutics.
Acknowledgments
We acknowledge the support of AstraZeneca for this study.
Abbreviations
- EGFR
epidermal growth factor receptor
- GBM
glioblastoma multiforme
- FISH
fluorescence in situ hybridization
Footnotes
This study was conducted as a collaborative trial of the North Central Cancer Treatment Group and Mayo Clinic and was supported in part by Public Health Service grants: CA-25224, CA-37404, CA-35195, CA-35101, CA-37417, CA-63849, CA-35113, CA-35267, CA-35269, CA-35103, CA-63848, CA-35415, CA-35431, CA-52352, and CA-35448 from the National Cancer Institute, Department of Health and Human Services. The content is solely the responsibility of the authors and does not necessarily represent the views of the National Cancer Institute or the National Institute of Health.
Conflict of Interest Statement: None of the authors have any conflicts of interests to disclose.
Additional participating institutions include: Missouri Valley Cancer Consortium, Omaha, NE 68106 (Gamini S. Soori, M.D.); Mayo Clinic Arizona, Scottsdale, AZ 85259-5404 (Tom R. Fitch, M.D.); CentraCare Clinic, Duluth CCOP, Duluth, MN 55805 (Daniel A. Nikcevich, M.D.); Siouxland Hematology-Oncology Associates, Sioux City, IA 51105 (Donald B. Wender, M.D.); Michigan Cancer Research Consortium, Ann Arbor, MI 48106 (Philip J. Stella, M.D.); Carle Cancer Center CCOP, Urbana, IL 61801 (Kendrith M. Rowland, Jr, M.D.); Cedar Rapids Oncology Project CCOP, Cedar Rapids, IA 52403 (Martin Wiesenfeld, M.D.); Meritcare Hospital CCOP, Fargo, ND 58122 (Preston D. Steen, M.D.); Medcenter One Health Systems, Bismarck, ND 58506 (Mid Dakota Clinic, Bismarck, ND 58501 (Edward J. Wos, M.D.); Geisinger Clinic & Medical Center CCOP, Danville, PA 17822 (Albert M. Bernath, Jr., M.D.); Altru Health Systems, Grand Forks, ND 58201 (Todor Dentchev, M.D.); Illinois Oncology Research Assn, CCOP, Peoria, IL 61615-7828 (John W. Kugler, M.D.); Rapid City Regional Oncology Group, Rapid City, SD 57709 (Richard C. Tenglin, M.D.); Saskatchewan Cancer Centre, Saskatoon, Saskatchewan, CANADA S7N 4H4 (Muhammad Salim, M.D.); Sioux Community Cancer Consortium, Sioux Falls, SD 57105 (Loren K. Tschetter, M.D.).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Libermann TA, Nusbaum HR, Razon N, et al. Amplification, enhanced expression and possible rearrangement of EGF receptor gene in primary human brain tumours of glial origin. Nature. 1985 Jan 10;313(5998):144–7. doi: 10.1038/313144a0. [DOI] [PubMed] [Google Scholar]
- 2.Wong AJ, Bigner SH, Bigner DD, et al. Increased expression of the epidermal growth factor receptor gene in malignant gliomas is invariably associated with gene amplification. Proc Natl Acad Sci USA. 1987 Oct;84(19):6899–903. doi: 10.1073/pnas.84.19.6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ekstrand AJ, James CD, Cavenee WK, et al. Genes for epidermal growth factor receptor, transforming growth factor alpha, and epidermal growth factor and their expression in human gliomas in vivo. Cancer Res. 1991 Apr 15;51(8):2164–72. [PubMed] [Google Scholar]
- 4.Frederick L, Wang XY, Eley G, James CD. Diversity and frequency of Epidermal Growth Factor Receptor mutations in human glioblastomas. Cancer Res. 2000 Mar 1;60(5):1383–7. [PubMed] [Google Scholar]
- 5.Culy CR, Faulds D. Gefitinib. Drugs. 2002;62(15):2237–48. doi: 10.2165/00003495-200262150-00008. [DOI] [PubMed] [Google Scholar]
- 6.Ciardiello F, Tortora G. A novel approach in the treatment of cancer: targeting the epidermal growth factor receptor. Clin Cancer Res. 2001 Oct;7(10):2958–70. [PubMed] [Google Scholar]
- 7.Cohen EE, Rosen F, Stadler WM, et al. Phase II trial of ZD1839 in recurrent or metastatic squamous cell carcinoma of the head and neck. J Clin Oncol. 2003 May 15;21(10):1980–7. doi: 10.1200/JCO.2003.10.051. [DOI] [PubMed] [Google Scholar]
- 8.Fukuoka M, Yano S, Giaccone G, et al. Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer (The IDEAL 1 Trial) [corrected] J Clin Oncol. 2003 Jun 15;21(12):2237–46. doi: 10.1200/JCO.2003.10.038. [DOI] [PubMed] [Google Scholar]
- 9.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005 Mar 10;352(10):987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 10.Aldape KD, Ballman K, Furth A, et al. Immunohistochemical detection of EGFRvIII in high malignancy grade astrocytomas and evaluation of prognostic significance. J Neuropathol Exp Neurol. 2004 Jul;63(7):700–7. doi: 10.1093/jnen/63.7.700. [DOI] [PubMed] [Google Scholar]
- 11.Guillaudeau A, Durand K, Pommepuy I, et al. Determination of EGFR status in gliomas: usefulness of immunohistochemistry and fluorescent in situ hybridization. Appl Immunohistochem Mol Morphol. 2009 May;17(3):220–6. doi: 10.1097/pai.0b013e31818db320. [DOI] [PubMed] [Google Scholar]
- 12.Rich JN, Reardon DA, Peery T, et al. Phase II trial of gefitinib in recurrent glioblastoma. J Clin Oncol. 2004 Jan 1;22(1):133–42. doi: 10.1200/JCO.2004.08.110. [DOI] [PubMed] [Google Scholar]
- 13.Haas-Kogan DA, Prados MD, Tihan T, et al. Epidermal growth factor receptor, protein kinase B/Akt, and glioma response to erlotinib. J Natl Cancer Inst. 2005 Jun 15;97(12):880–7. doi: 10.1093/jnci/dji161. [DOI] [PubMed] [Google Scholar]
- 14.Mellinghoff IK, Wang MY, Vivanco I, et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med. 2005 Dec 10;353(19):2012–24. doi: 10.1056/NEJMoa051918. [DOI] [PubMed] [Google Scholar]
- 15.van den Bent MJ, Brandes AA, Rampling R, Kouwenhoven MC, Kros JM, Carpentier AF, et al. Randomized phase II trial of erlotinib versus temozolomide or carmustine in recurrent glioblastoma: EORTC brain tumor group study 26034. J Clin Oncol. 2009 Mar 10;27(8):1268–74. doi: 10.1200/JCO.2008.17.5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu L, Backlund LM, Nilsson BR, Grander D, et al. Clinical significance of EGFR amplification and the aberrant EGFRvIII transcript in conventionally treated astrocytic gliomas. J Mol Med. 2005 Nov;83(11):917–26. doi: 10.1007/s00109-005-0700-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heimberger AB, Hlatky R, Suki D, et al. Prognostic effect of epidermal growth factor receptor and EGFRvIII in glioblastoma multiforme patients. Clin Cancer Res. 2005 Feb 15;11(4):1462–6. doi: 10.1158/1078-0432.CCR-04-1737. [DOI] [PubMed] [Google Scholar]
- 18.Pelloski CE, Mahajan A, Maor M, et al. YKL-40 expression is associated with poorer response to radiation and shorter overall survival in glioblastoma. Clin Cancer Res. 2005 May 1;11(9):3326–34. doi: 10.1158/1078-0432.CCR-04-1765. [DOI] [PubMed] [Google Scholar]
- 19.Pelloski CE, Ballman KV, Furth AF, et al. Epidermal growth factor receptor variant III status defines clinically distinct subtypes of glioblastoma. J Clin Oncol. 2007 Jun 1;25(16):2288–94. doi: 10.1200/JCO.2006.08.0705. [DOI] [PubMed] [Google Scholar]
- 20.Prados MD, Chang SM, Butowski N, et al. Phase II Study of Erlotinib Plus Temozolomide During and After Radiation Therapy in Patients With Newly Diagnosed Glioblastoma Multiforme or Gliosarcoma. J Clin Oncol. 2009 Feb 1;27(4):579–84. doi: 10.1200/JCO.2008.18.9639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown PD, Krishnan S, Sarkaria JN, et al. Phase I/II trial of erlotinib and temozolomide with radiation therapy in the treatment of newly diagnosed glioblastoma multiforme: North Central Cancer Treatment Group Study N0177. J Clin Oncol. 2008 Dec 1;26(34):5603–9. doi: 10.1200/JCO.2008.18.0612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fan Q-W, Cheng C, Knight ZA, et al. EGFR signals to mTOR through PKC and independently of Akt in glioma. Science Signaling. 2009;2(55):ra4. doi: 10.1126/scisignal.2000014. [DOI] [PMC free article] [PubMed] [Google Scholar]