

Abstract
Inflammation is a major risk factor for neonatal white matter injury (NWMI), which is associated with later development of cerebral palsy. Although recent studies have demonstrated maturation arrest of oligodendrocyte progenitor cells (OPCs) in NWMI, the identity of inflammatory mediators with direct effects on OPCs has been unclear. Here, we investigated downstream effects of pro-inflammatory IL-1β to induce cyclooxygenase-2 (COX2) and prostaglandin E2 (PGE2) production in white matter. First, we assessed COX2 expression in human fetal brain and term neonatal brain affected by hypoxic-ischemic encephalopathy. In the developing human brain, COX2 was expressed in radial glia, microglia, and endothelial cells. In human term neonatal hypoxic-ischemic encephalopathy cases with subcortical WMI, COX2 was strongly induced in reactive astrocytes with “A2” reactivity. Next, we show that OPCs express the EP1 receptor for PGE2, and PGE2 acts directly on OPCs to block maturation in vitro. Pharmacologic blockade with EP1-specific inhibitors (ONO-8711, SC-51089), or genetic deficiency of EP1 attenuated effects of PGE2. In an IL-1β-induced model of NWMI, astrocytes also exhibit “A2” reactivity and induce COX2. Furthermore, in vivo inhibition of COX2 with Nimesulide rescues hypomyelination and behavioral impairment. These findings suggest that neonatal white matter astrocytes can develop “A2” reactivity that contributes to OPC maturation arrest in NWMI through induction of COX2-PGE2 signaling, a pathway that can be targeted for neonatal neuroprotection.
Keywords: Cerebral palsy, neuroinflammation, astrocyte, white matter, oligodendrocyte, Cox2, prostaglandin
Graphical abstract
Introduction
Extremely low birth weight (ELBW) preterm infants show high rates of neurological impairment including cognitive, behavioral, neurosensory, and motor dysfunction as well as cerebral palsy (Moore et al., 2012; Serenius et al., 2013). Indeed, the prevalence of these conditions is increasing due to enhanced survival of ELBW preterm infants in the modern neonatal intensive care unit (Boyle et al., 2011; Guillen et al., 2015). Cerebral palsy in preterm infants is associated with neonatal white matter injury (NWMI), pathologic disturbances in myelination that can be focal or diffuse (Woodward et al., 2006; Northam et al., 2011; Fern et al., 2014) and often associated with gray matter abnormalities (Pierson et al., 2007). Magnetic resonance imaging (MRI) has aided detection of NWMI and is predictive of preterm infants at high risk of developing cerebral palsy during childhood (Woodward et al., 2006). Despite interventions that have dramatically improved ELBW infant survival, no neuroprotective therapy exists to prevent rising rates of cerebral palsy in developed countries.
The predominant form of NWMI is a diffuse injury to myelin tracts (Counsell et al., 2003) that involves inflammation and gliosis, a reactive response by microglia and astrocytes (Inder et al., 2005; Pekny and Nilsson, 2005; Riddle et al., 2011; Verney et al., 2012; Supramaniam et al., 2013) that can be triggered by systemic processes such as infection (Malaeb and Dammann, 2009; Deng, 2010; Deng et al., 2014; Hagberg et al., 2015). Increased markers of inflammation in the neonatal period are strongly associated with the development of cerebral palsy, NWMI and poor neurological outcomes (Dammann and Leviton, 1997; Leviton et al., 2016). While it had been thought that inflammation led to NWMI by depleting the oligodendrocyte progenitor cell (OPC) pool (Back, 2006), more recent histologic studies using markers of discrete stages of OPC development in NWMI reveal that OPCs are present but arrested in a pre-myelinating and immature state (Billiards et al., 2008; Buser et al., 2012; Verney et al., 2012).
Reactive astrogliosis is a hallmark of human NWMI (Khwaja and Volpe, 2007; Back and Miller, 2014; Back and Rosenberg, 2014) and can have either protective or deleterious effects (Williams et al., 2007; Sofroniew, 2015). While factors induced by reactive astrocytes such as hyaluronic acid (Back et al., 2005), BMP (Wang et al., 2011), endothelin-1(Hammond et al., 2014) can impair OPC maturation, STAT3-dependent astrocyte reactivity is also protective (Nobuta et al., 2012), suggesting functional hetereogeneity among reactive astrocytes. Reactive astrocytes have recently been subtyped as “A1” or “A2” based on distinct molecular markers (Liddelow et al., 2017). Reactive astrocytes expressing “A1” markers are found in multiple adult human neurodegenerative conditions and are thought to confer neurotoxic effects. In transcriptional assessments of reactive astrocyte subtypes in mouse models, induction of Cox2 was associated with the “A2” phenotype (Zamanian et al., 2012; Liddelow et al., 2017). However, the role of “A2” astrocytes in neuroinflammatory injury is unclear and human neuropathologic conditions associated with “A2” astrocytes have not been reported.
The pro-inflammatory cytokine IL-1β induces cyclooxygenase type 2 (COX2) and prostaglandin E2 (PGE2) production, and systemic IL-1β administration is sufficient to induce NWMI in a rodent model (Favrais et al., 2011). Prostaglandin E2 (PGE2) is a pro-inflammatory mediator that is derived from arachidonic acid through the rate-limiting cyclooxygenase (COX) enzymes and signals to the EP family of cell surface receptors (Legler et al., 2010). PGE2 can be released by activated microglia and reactive astrocytes in the immature brain (Molina-Holgado et al., 2000; Xu et al., 2003; Xia et al., 2015). PGE2 is elevated in the CSF of term and preterm neonates with culture-verified sepsis and meningitis (Siljehav et al., 2015), as well as neonates afflicted by perinatal asphyxia (Björk et al., 2013). Relevant to the observations of oligodendrocyte maturation arrest is that PGE2 can alter the fates of progenitor cell populations (Castellone et al., 2005; Goessling et al., 2009). In this study, we asked whether PGE2 could directly inhibit oligodendrocyte progenitor maturation and possibly be a therapeutic target to reduce inflammation-induced NWMI?
Here, we show that astrogliosis in human neonatal white matter injury is associated with “A2” astrocytes that express COX2. In vivo systemic IL-1β treatment in a mouse model of neonatal hypomyelination also induces “A2” astrocyte reactivity. IL-1β upregulates COX2 and the production of PGE2, which directly inhibits OPC maturation in an EP1-receptor dependent manner. Moreover, systemic inhibition of COX2 in vivo reduced IL-1β–mediated effects on hypomyelination and OPC maturation arrest, suggesting a potential therapeutic approach.
Materials And Methods
Animals and treatments
Animal husbandry, protocols, and ethics were approved by the University of California, San Francisco and the Bichat and Robert Debre Hospital ethics committees; protocols were approved by and adhere to the European Union Guidelines for the Care and Use of Animals, and the Institutional Animal Care and Use Committee in the USA. EP1 (B6.129P2-Ptger1tm1Dgen/Mmnc) mice were obtained from the Mutant Mouse Resource and Research Centers at the University of North Carolina(MMRC/UNC); frozen sperm from a mixed strain background (129 and C57/Bl6) was re-derived onto the C57/BL6 background; all experiments involving EP1 mice utilized littermate controls. EP1 deficiency did not grossly affect brain morphology (data not shown). IL-1β (R&D Systems, Minneapolis, MN) injections at postnatal dates 1-5 (P1-P5) were conducted with male Swiss Webster mice as previously described (Favrais et al., 2011). Because the IL-1β-induced white matter model was conducted in male pups only, sex differences were not assessed. Briefly, on P1, litters were culled to approximately 10 pups, and all pups in a litter were allocated to a group (PBS or IL-1β). Mice received twice a day (morning and evening) from P1 to P4 and once on P5 (morning) a 5 μl intra-peritoneal injection of 10μg/kg/injection recombinant mouse IL-1β in phosphate buffered saline (PBS; R&D Systems) or PBS alone. Nimesulide (Sigma-Aldrich), a selective COX2 inhibitor, was intraperitoneally injected following the same schedule as IL-1β protocol. Nimesulide was diluted in a solution of DMSO (0.1%, Sigma) to achieve a dose of 1mg/kg/injection and injected at the same time with PBS or IL-1β, as previously described (Favrais et al., 2007). 0.1% DMSO alone had no effects (data not shown).
Oligodendrocyte progenitor cell and mixed glial cell cultures and treatments
Oligodendrocyte precursor cell cultures were obtained from mouse and rat pups through two separate methods. Mouse OPCs were immunopanned from P6-P8 mouse cortices as previously described (Fancy et al., 2011), plated on poly-D-lysine coverslips (Neuvitro; Vancouver, WA), and maintained in proliferation media containing the following growth factors: platelet-derived growth factor-AA (PDGF-AA), ciliary neurotrophic factor (CNTF), and neurotrophin-3(NT3) (Peprotech, Rocky Hill, NJ) at 10% CO2 and 37°C. Purified cell preparations were >95% Olig2+, <1% Iba1+ and <4% GFAP+ as assessed by IHC (data not shown). After 1-2 days in proliferation media, differentiation was induced by changing media to contain CNTF and triiodothyronine (T3; Sigma, St. Louis, MO). PGE2 (Sigma), Wnt3a (Peprotech), IL-1β (R&D systems), ONO-8711 (Cayman Chemicals, Ann Arbor, MI), and DMOG (Sigma) were added with differentiation media.
Rat OPC cultures were obtained from the McCarthy and DeVellis' modified protocol (McCarthy and de Vellis, 1980). Briefly, cortices from P0-P2 Sprague-Dawley rat pups were used to obtain mixed glial cultures for 10 days in MEM medium (Sigma) with 20% Fetal Bovine Serum (FBS). At day 11, a 2-step-shaking (260 RPM, 37°C, ambient air) was performed with a first short shaking for 1.5 hours to remove microglial cells and a second one for 18 hours to harvest oligodendrocytes. Then, OPC proliferation was induced by a medium enriched in PDGF-AA (10 ng/ml; Peprotech) and basic Fibroblastic growth factor (bFGF, 10 ng/ml; Sigma) for 5 days. OPC purity had been assessed > 90% at day 4 (data not shown). At day 4 of proliferation phase, PGE2 (Sigma) was added to the medium diluted in 0.1% DMSO (Sigma) from 1nM to 1mM for 24 hours. SC-51089 (10 μM, Tocris Biosciences), a selective EP1 receptor antagonist, was applied to rat OPC cultures with or without 10 μM PGE2. At day 5, PDGF-AA, b-FGF, PGE2 and SC-51089 were removed of the medium to initiate OPC differentiation. Myelin basic protein (MBP) immunostaining was performed at day 3 of maturation phase. Counting of MBP+ cells was based on counting in 5 random fields in duplicate and from at least 3 independent experiments. Mixed glial cultures were prepared as previously described (Schildge et al., 2013) and plated on poly-D-lysine (EMD Millipore, Darmstadt, Germany) coated plates. Cells were stimulated with IL-1β and Nimesulide for assays 7-10 days after plating. Cells were collected for western blot analysis or medium was collected for measurement of PGE2 concentration.
Antibody-coupled magnetic cell isolation of glia
Cells positive for CD11b (microglia and macrophages), O4 (pan-oligodendrocytes) or GLAST (astrocytes), were extracted using the antibody-coupled magnetic bead system (MACS) following the manufacturer's recommendations (Miltenyi Biotec, Bergisch Gladbach, Germany) and as previously reported (Schang et al., 2014). Cells were from cortices isolated at P5, 4 hours after the final injection of PBS or IL-1β. The purity of fractions was verified using qRT-PCR for glial fibrillary acid protein (Gfap), neuronal nuclear antigen (Rbfox3, NeuN), ionizing calcium binding adapter protein (Aif1, Iba1), and oligodendrocyte differentiation factor 2 (Olig2).
RNA isolation and quantitative real-time PCR
RNA was extracted from samples in Trizol (Life Technologies, Carlsbad, CA) with phenol-chloroform followed by RNeasy Mini Kit (Qiagen, Hilden, Germany), and cDNA generated by High-Capacity RT-PCR kit (Applied Biosystems, Foster City, CA) or iScript cDNA synthesis kit (Bio-Rad, Hercules, CA). qPCR using Sybr Green (Roche, Basel, Switzerland; or Biorad) was conducted on a LightCycler480 (Roche) or a CFX384 (Biorad). Primers for qPCR include: Hprt (forward – TGGTGAAAAGGACCTCTCGAA, reverse – TCAAGGGCATATCCAACAACA), EP1/Ptger1 (forward – GGGCTTAACCTGAGCCTAGC, reverse – GTGATGTGCCATTATCGCCTG), EP2/Ptger2 (forward - GGAGGACTGCAAGAGTCGTC, reverse – GCGATGAGATTCCCCAGAACC), EP3/Ptger3 (forward – CCGGAGCACTCTGCTGAAG, reverse – CCCCACTAAGTCGGTGAGC), and EP4/Ptger4 (forward – ACCATTCCTAGATCGAACCGT, reverse – CACCACCCCGAAGATGAACAT), Rpl13 (forward - ACA GCC ACT CTG GAG GAG AA, reverse - GAG TCC GTT GGT CTT GAG GA), Ptgs2 (forward – TCATTCACCAGACAGATTGCT, reverse – AAGCGTTTGCGGTACTCATT), Cd109 (forward – TCCCACTGTGAGAGACTACAAA, reverse - ACCTGGGTGTTGTAGCTTCG), S100a10 (forward – GTTTGCAGGCGACAAAGACC, reverse - ATTTTGTCCACAGCCAGAGG), Emp1 (forward – CTCCCTGTCCTACGGCAATG, reverse - GAGCTGGAACACGAAGACCA), Fbln5 (forward – AGCAACAACCCGATACCCTG, reverse - GGCACTGATAGGCCCTGTTT), Amigo2 (forward – CCGATAACAGGCTGCTGGAG, reverse - AGAATATACCCCGGCGTCCT), Serping1 (forward – GCCTCGTCCTTCTCAATGCT, reverse - CGCTACTCATCATGGGCACT), Cxcl10 (forward – GCTGCAACTGCATCCATATC, reverse - GGATTCAGACATCTCTGCTCAT), Sphk1 (forward – TCCAGAAACCCCTGTGTAGC, reverse - CAGCAGTGTGCAGTTGATGA), and Gfap (forward – AAGCCAAGCACGAAGCTAAC, reverse - CTCCTGGTAACTGGCCGACT).
Rodent immunohistochemistry and immunofluorescence
Coverslips were fixed in 4% PFA and immunostained with rabbit anti-Olig2 (EMD Millipore, Billerica, MA), rat anti-MBP (Biorad), mouse anti-phospho-histone 3 (Cell Signaling, Danvers, MA), or mouse anti-Nk×2.2 (Developmental Hybridoma Bank, University of Iowa). Secondary fluochrome-tagged antibodies were obtained from (Invitrogen/Thermo Fisher, Waltham, MA). Images were obtained on an Axioimager Z1 microscope (Zeiss, Oberkochen, Germany). Concerning ex-vivo experiments, P5 and P30 mouse brains were collected in the 4 experimental groups designed (PBS, Nimesulide, IL-1β, IL-1β+Nimesulide) and fixed to obtain 10μm thick coronal sections. Immunostainings with rabbit anti-NG2 (Millipore) on P5 brains to quantify OPCs and mouse anti-MBP (Millipore) antibodies on P30 brains for myelinated axons were performed as previously described (Favrais et al., 2011). NG2+ cells were counted within the white matter tracts of the external capsule using ImageJ software (NIH, Bethesda, MD). MBP immunostaining intensity was assessed by ImageJ densitometry analysis at the level of the sensory-motor cortex.
Human tissue and immunofluorescence
All human post-mortem tissue was acquired with prior ethical approval from The French Agency of Biomedicine (Agence de Biomédicine; approval PFS12-0011) or in accordance with guidelines established by the University of California, San Francisco Committee on Human Research (H11170-19113-07). All tissues were collected following the provision of informed consent.
Post-mortem fetal human brain sections were obtained from three cases of 27-, 30- and 31-weeks gestational age that did not have overt brain damage (Supplementary Table 1). Tissue was fixed with 4% paraformaldehyde, frozen and sections cut at 12 μm. Staining was performed for goat anti-Iba1 (Abcam, Cambridge, UK), rabbit anti-Nestin (EMD Millipore), mouse anti-CD34 (Biorad) and rabbit anti-COX2 (Abcam). Sections mounted on glass slides were rehydrated in PBS and pre-incubated in PBS with 0.2% gelatin and 0.25% Triton X-100 (PBS-T-gelatin) for 15 minutes followed by overnight incubation with primary antibodies diluted in PBS-T-gelatin. The sections were rinsed with PBS-T-gelatin and incubated with secondary antibodies diluted in PBS-T-gelatin for 1.5 hours. In order to perform COX2/Nestin double labeling, we employed the Tyramide Signal Amplification (TSA) Systems (PerkinElmer). Briefly, Nestin labeling was revealed with TSA-Cy3 as described by manufacturer's instructions. Then, sections were treated at 94 °C in buffer citrate (1.8 mM acid citric, 8.2 mM sodium citrate, pH6) for 15 minutes. After three washes in PBS sections were incubated overnight with anti-COX2 antibody and revealed as described above. Sections were then rinsed with PBS and incubated with DAPI for 5 minutes for nuclear counterstaining. All incubations were performed at room temperature, protected from light in a humidified chamber. Finally, the sections were rinsed with PBS, coverslipped with Fluoromount (Southern Biotech) and stored at 4°C for subsequent confocal microscopic analysis.
Tissue from term hypoxic-ischemic encephalopathy and control cases (Table 1) were immersed in PBS with 4% paraformaldehyde for 3 days. On day 3, the brain was cut in the coronal plane at the level of the mammillary body and immersed in fresh 4% paraformaldehyde/PBS for an additional 3 days. After fixation, all tissue samples were equilibrated in PBS with 30% sucrose for at least 2 days. Following sucrose equilibration, tissue was placed into molds and embedded with OCT for 30 – 60 minutes at room temperature or 4°C followed by freezing in dry ice-chilled ethanol or methyl butane. The diagnosis of hypoxic ischemic encephalopathy (HIE) requires clinical and pathological correlations. With respect to the pathological features, all HIE cases in this study showed consistent evidence of diffuse white matter injury, including astrogliosis and macrophage infiltration using GFAP and CD68 staining. All brain samples were examined and classified by an experienced neuropathologist. While some control samples included infants with congenital diaphragmatic hernia, which may result in hypoxemia, all brain samples were examined and classified by an experienced neuropathologist and control samples did not exhibit evidence of astrogliosis or macrophage infiltration. Slides were blocked with avidin and biotin (Vector Labs Burlingame, CA), and 10% goat serum, then permeabilized with TritonX-100 0.05%, and incubated overnight with primary antibodies at room temperature: mouse anti-S100A10 (Invitrogen; MA5-15326), rat anti-GFAP (Invitrogen; MA5-12023), or rabbit anti-COX2 (Abcam). COX2 was signal amplified with biotinylated goat anti-rabbit secondary followed by avidin-peroxidase complex (Vectastain ASBC, Vector). Fluorescence staining was performed with fluorochromes tagged to streptavidin or goat secondary antibodies (Invitrogen).
Table 1. Hypoxic-ischemic encephalopathy cases and controls.
| Case Number | Postnatal Age | Sex | Diagnosis | Postmortem interval (hours) |
|---|---|---|---|---|
| UCSF 2010-005 | 0 day | Male | Control (Congenital Diaphragmatic Hernia) | 36 |
| UCSF 2010-008 | 0 day | Female | Control (Congenital Diaphragmatic Hernia) | 18 |
| UCSF 2010-013 | 2 day | Female | Control (Pneumothorax) | 21 |
| UCSF 2011-005 | 3 day | Male | HIE (Placental abruption) | 47 |
| UCSF 2012-001 | 3 day | Male | HIE (Perinatal asphyxia) | 9 |
| UCSF 2012-024 | 1 day | Female | Control (Anhydramnios) | 25 |
| UCSF 2013-008 | 1 day | Male | HIE (Fetal bradycardia) | 98 |
BrdU, LDH and PGE2 measurements
Oligodendrocyte proliferation or death were observed just after the PGE2 or vehicle removal by BrdU (Cell Signaling) or Lactate Dehydrogenase (LDH) (Sigma) colorimetric assays (absorbance 450 nm), respectively. Proliferation immunoassay was performed on cells previously coated in 96 well-plate, whereas cell death was assessed through the measurement of LDH release in the medium following the manufacturer's instructions. Measurements were been performed in duplicate and counts collected from at least from 2 independent experiments. PGE2 levels in mixed glial culture media were measured by ELISA (Abcam).
Signaling pathway ELISAs
To explore PGE2 signaling pathway, cellular inflammation proteins were measured using a multi-target sandwich ELISA focusing on phospho-p38MAPK, phospho-p65NFκB, phospho-SAPK/JNK, phospho-IκBα and phospho-STAT3 (Cell Signaling, PathScan inflammation). Total cell proteins were extracted at the end of PGE2 24h-exposure. Lysis buffer contained 4-hydroxybutyl-acrylate with 1% Triton-X (Sigma), 1% protein inhibitor cocktail (Sigma) and 5 nm sodium fluoride (Sigma). OPCs were lysed on ice and froze at -20°C until use. After defrosting on ice, 10 second-sonication was performed followed by a centrifugation (14000 rpm) for 15 minutes at 4°C. Then, the supernatant was collected, and protein concentration was measured based on Bradford method using Bovine Serum Albumin (BSA) standard curve and colorimetric assay (Biorad, Bradford protein assay). Then, ELISA assay was performed on a 96 pre-coated well-plate with 4 samples per experimental groups (DMSO 0.1% for 24 hours versus PGE2 10 μM for 24 hours) in duplicate following the manufacturer's instructions.
Western blot
Cells were lysed with RIPA buffer directed on tissue culture plates, scraped, vortex and centrifuged to clarify lysates. Lysate protein concentrations were measured by BCA (Biorad). Lysates were resolved on Bolt gels (Invitrogen) using MOPS buffer, transferred to PVDF-F (EMD Millipore) and imaged with Odyssey luminescence (LI-COR Biosciences, Lincoln, NE). Primary antibodies: rabbit COX2 (Abcam), rabbit HIF1α (Cayman Chemicals), mouse active β-catenin (EMD Millipore), phospho-Akt (Cell Signaling), pan-Akt (Cell Signaling) and GAPDH (Sigma) and rabbit total β-catenin (Cell Signaling). IRDye-conjugated secondary antibodies were from Licor. Fluorimetric analysis and imaging were performed with Odyssey luminescence (LI-COR Biosciences, Lincoln, NE).
Behavioral Assessment
Temporal and spatial memory functions were assessed at P29 and P30 through the novel object recognition (NOR) and the object location memory (OLM) tests, respectively. For these tests, the exploration time of two objects placed in 36 × 36 × 10 cm box arena was measured twice for 4 minutes and 3 minutes apart. First, two identical objects were placed in two distinct corners of the box. Second, one of the two objects were either displaced or replaced by a new one for OLM or NOR assessments, respectively. Exploration time was defined as the duration an animal spend either pointing its nose towards the object at a distance of <1 cm and/or touching it with the nose; turning around, climbing, and sitting on the object were not considered as exploration. Recognition of the familiar object was scored by preferential exploration of the novel object using a discrimination index (novel object interaction/total interaction with both objects, range from 0 to 100%; 50% = no preference).
Statistics
Data are presented as means +/- SEM. Unpaired two-tailed t-tests or Mann Whitney U tests were performed for two group analyses based on the outcome of normality testing, or a one-way Anova for 3 or more group analyses, as indicated in the text and figure legends. Analyses were performed using Graphpad Prism (Graphpad Software, San Diego, CA) and Excel (Microsoft, Redmond, WA)
Results
COX2 protein is expressed in glial cells of the 3rd trimester human fetal brain
To determine whether COX2 was normally expressed in the developing human brain, we undertook immunohistochemical (IHC) analysis using a collection of human fetal brain samples (27, 30, and 31 gestational week cases; Supplementary Table 1). In the three cases, IHC staining revealed COX2 expression in Iba1-positive microglia (Fig. 1 A), Nestin-positive putative radial glia (including a subset of immature astrocytes) (Fig. 1 B), and CD34-positive endothelial cells (Fig. 1 C) within the sub-ventricular zone.
Figure 1. COX2 immunohistochemistry in the human third trimester brain.
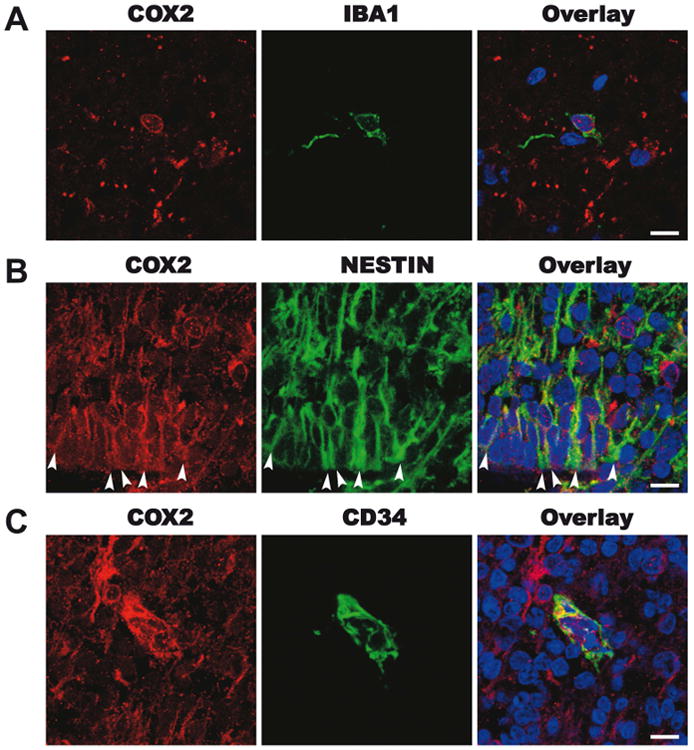
Representative images from the dorsal cortex of a 30-week human fetal brain. A. In the subplate, red COX2, green IBA1+ microglia and an overlay panel including DAPI positive nuclear staining. B. in the subventricular zone, red COX2, green nestin+ putative radial glia and astrocytes and an overlay panel including DAPI+ nuclear staining. C. In the subventricular zone, red COX2, green CD34+ endothelia cell and an overlay panel including DAPI positive nuclear staining. Scale bar = 10μm.
COX2 protein is induced by human reactive astrocytes in neonatal white matter
To further investigate the expression of COX2 in neonatal white matter pathology, we performed immunohistochemistry on subcortical white matter samples of the cingulate cortex (Fig. 2 A) from post-mortem samples in a collection of term infant cases that suffered from hypoxic-ischemic encephalopathy (HIE) and matched controls (Table 1). We found that COX2 expression was substantially increased in reactive GFAP+ white matter astrocytes (Fig. 2 B and C). When we enumerated the number of GFAP+ astrocytes and CD45+ immune cells expressing COX2 in control and HIE cases, we found that GFAP+ astrocytes exhibited a significant increase in total numbers and COX2 expression. In comparison, CD45+ cells were unchanged in total numbers or COX2 expression (Fig. 2 D). While the total number of COX2+ cells also includes endothelial cells (see above and Fig. 1 C) and immune cells (including microglia and peripherally-derived myeloid cells), the increase within GFAP+ cells accounts for the overall rise in COX2+ cells in HIE cases.
Figure 2. COX2 immunohistochemistry of subcortical white matter from human hypoxic ischemic encephalopathy (HIE) cases.
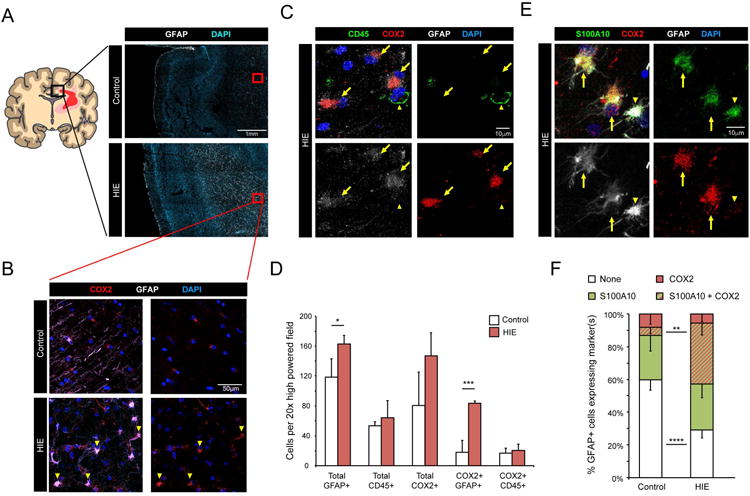
A Cartoon illustrating affected white matter areas in human term HIE. Black box represents cingulate region used for analysis. Red boxes are examples of subcortical white matter regions used for analysis. HIE cases exhibit increased GFAP (white) immunoreactivity. B. Representative images from term infants with or without HIE, stained for COX2 (red) and GFAP (white). Arrowheads mark COX2+ GFAP+ astrocytes. C. Representative images of white matter expression of COX2 in GFAP+ astrocytes. Arrows mark COX2+ GFAP+ astrocytes. Arrowhead marks a COX2- CD45+ microglia/myeloid cell. D. Quantification of indicated cell types in control and HIE white matter. E & F. Representative images (E) and quantification (F) of S100A10 co-expression with COX2 in white matter GFAP+ astrocytes. Arrows mark GFAP+ astrocytes co-expressing S100A10 and COX2. Arrowhead marks a GFAP+ astrocytes expressing only S100A10. Data from n=4 control and n=3 HIE cases. p-values calculated from two-tailed unpaired t-tested. * p <0.05, ** p<0.01, *** p<0.005, **** p<0.001
Reactive astrocytes have recently been delineated as “A1” or “A2” subtypes based on distinct expression patterns of molecular markers (Liddelow et al., 2017). In transcriptional assessments of these reactive astrocyte subtypes, COX2 (Ptgs2) upregulation was reported to be associated with the “A2” phenotype (Zamanian et al., 2012; Liddelow et al., 2017). Therefore, we also looked for expression of the “A2” associated marker, S100A10, and found strong co-expression within GFAP+ white matter astrocytes in HIE cases (Fig. 2 E and F). These findings show that COX2 is strongly induced in human HIE white matter within reactive astrocytes of an “A2”-associated phenotype, and suggests that “A2” reactive astrocytes may be an important source of PGE2 in human NWMI.
Astrocytes exhibit “A2” reactivity with systemic IL-1β treatment
We have previously reported that P1-P5 systemic administration of IL-1β impairs OPC maturation and results in myelination defects that mimic human preterm deficits (Favrais et al., 2011). To further investigate the ability of microglia and/or astrocytes to generate prostaglandin in vivo, we isolated these cells from mouse pups treated with systemic IL-1β. As shown (Fig. 3 A), both CD11b+ microglia and GLAST+ astrocytes isolated from IL-1β treated animals expressed elevated levels of COX2 transcript compared to controls.
Figure 3. GLAST+ astrocytes isolated from IL-1β treated mice induce Cox2 (Ptgs2) and express markers of “A2” reactivity.
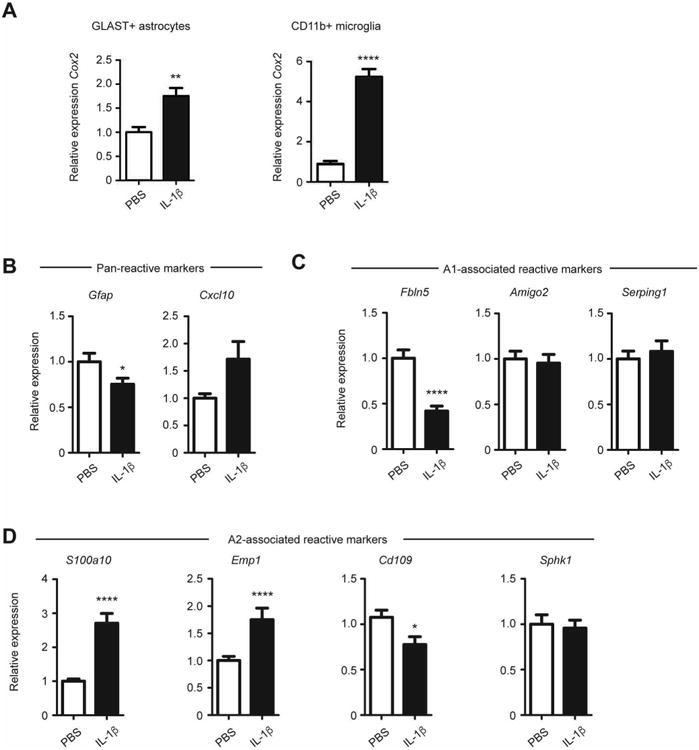
Transcriptional analysis of cells isolated by magnetic bead purification from P5 mice treated with PBS or IL-1β A. Transcriptional induction of Cox2 in GLAST+ astrocytes and CD11b+ microglia isolated. B. Expression of pan-reactive markers (Gfap, Cxcl10) in GLAST+ astrocytes. C. Expression of A1-associated markers (Fbln5, Amigo2, Serping1) in GLAST+ astrocytes. D. Expression of A2- associated markers (Ptgs2, S100a10, Emp1, Cd109, Sphk1) in GLAST+ astrocytes. Data representative of n=13 per group. * p<0.05, ** p<0.01, **** p<0.001; analysis by Mann-Whitney test.
Our findings in human NMWI indicate that robust COX2 induction occurs in reactive astrocytes with an “A2” phenotype. Therefore, we asked whether reactive astrocytes following systemic IL-1β exposure also exhibit an “A2” transcriptional profile of reactivity. GLAST+ cells were isolated at P5 following P1-P5 systemic IL-1β treatment and assessed for markers of pan reactivity (Fig. 3 B), A1-associated reactivity (Fig. 3 C), and A2-associated reactivity (Fig. 3 D). Together, the expression pattern shows a differential increase the A2-associated markers S100a10 and Emp1 but lack of induction for A1-associated markers (Fbln5, Amigo2, Serping1). These findings indicate that astrocytes in the IL-1β model of NWMI develop an A2-associated reactivity, reflecting the white matter astrocyte phenotype seen in neonatal human pathology.
IL-1β induces COX2-dependent production of Prostaglandin E2
We next confirmed that IL-1β induction of COX2 results in PGE2 production. Mixed glial cultures containing microglia and astrocytes were stimulated with IL-1β. As shown (Supplementary Figure. 1 A), IL-1β stimulation of mixed glial cultures resulted in elevated COX2 protein consistent with previously published work that COX2 could be induced by astrocytes or microglia (Katsuura et al., 1989; Molina-Holgado et al., 2000). IL-1β-stimulated mixed glial cultures also produced PGE2 and this was inhibited by Nimesulide, which specifically targets COX2 (Supplementary Figure. 1 B). In contrast, we found that direct IL-1β treatment of purified OPCs did not induce COX2 or lead to OPC maturation arrest (Supplementary Figure. 1 A and C), consistent with a previous study (Vela, 2002). Taken together, these findings indicate that IL-1β activates astrocytes and microglia, but not OPCs, to produce PGE2 in a COX2-dependent manner.
Prostaglandin E2 arrests OPC maturation
To test whether PGE2 had a direct effect on OPCs, cells were isolated from neonatal mouse cortices using anti-PDGFRα immunopanning (Emery and Dugas, 2013). Upon T3 hormone maturation treatment, OPCs differentiate and express MBP while expression of the immature OPC marker Nk×2.2+ decreases (Qi et al., 2001) (Fig. 4 A). PGE2 treatment resulted in a robust and dose-dependent suppression of this T3 induced MBP expression (Fig. 4 B and C). We confirmed that PGE2 blocked OPC maturation by monitoring persistent expression of immature OPC marker Nk×2.2 (Fig. 4 D). PGE2 had no effect on overall Olig2+ cell numbers, consistent with an alteration of OPC differentiation as compared to proliferation or OPC death (Fig. 4 E). In parallel, purified rat OPCs were also treated with PGE2 and found to have a dose dependent blockade in MBP expression at maturation day 3 (Fig. 4 F and G). An assessment of BrdU incorporation (Fig. 4 H) and histone-3 phosphorylation (Fig. 4 I) showed no difference between PGE2 and control treated cells. Furthermore, a cytotoxicity assay also showed no difference in LDH release (Fig. 4 J). Thus, PGE2 is a potent inhibitor of mouse and rat OPC maturation in vitro, but does not affect OPC proliferation or survival.
Figure 4. Prostaglandin E2 inhibits oligodendrocyte progenitor cell maturation.
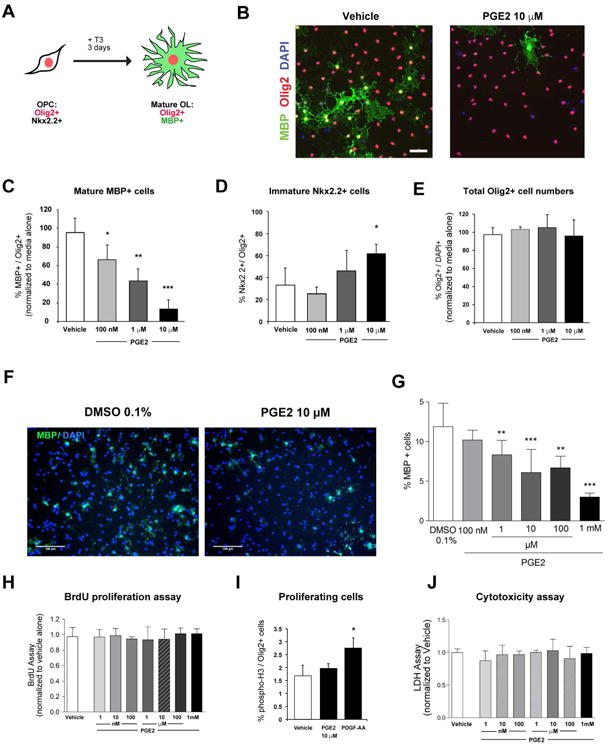
A Schematic of oligodendrocyte maturation assay. Nk×2.2 marks immature progenitors and MBP marks maturing oligodendrocytes. B. Representative images of cells, stained for Olig2 and MBP, after 3 days of differentiation with or without PGE2 (scale bar, 25 μm). C & D. Quantification of MBP+ (C) and Nk×2.2+ cells (D) exposed to indicated doses of PGE2. E. Total Olig2+ cell numbers following exposure to indicated doses of PGE2. F & G. Representative images (scale bar, 100μm) and quantification of MBP staining following treatment of rat OPC cells with or without PGE2 from 100nM to 1mM (n=6 per group). H. BrdU incorporation in OPCs exposed to PGE2 from 1nM to 1mM for 24 hours (n=4 per group). I. Phospho-histone 3 expression in OPCs exposed to PGE2. J. LDH release from OPCs exposed to PGE2 from 1nM to 1mM for 24 hours (n=3 per group). * p-value <0.05, ** p-value <0.01, *** p-value <0.005 Data shown compiled from at least 3 independent experiments.
PGE2 inhibits oligodendrocyte progenitor cell maturation through the prostaglandin E receptor 1 (EP1 receptor)
Prostaglandin E2 signals through four G-protein coupled receptors: EP1-EP4. RNA transcriptome profiling of cellular subsets in culture (Sharma et al., 2015) and from the postnatal mouse cortex (Zhang et al., 2014) indicated that EP1 is the predominant receptor in the oligodendrocyte lineage. We confirmed by qPCR that EP1 is expressed on immunopanned mouse OPCs (Fig. 5 A). We also performed transcriptional analysis of O4+ oligodendrocyte lineage cells isolated from of P5 and P10 mouse cortices and found that EP1 was the predominantly expressed receptor at these two separate time points (Fig. 5 B).
Figure 5. PGE2 maturation arrest of oligodendrocyte progenitor cells through EP1 receptor.
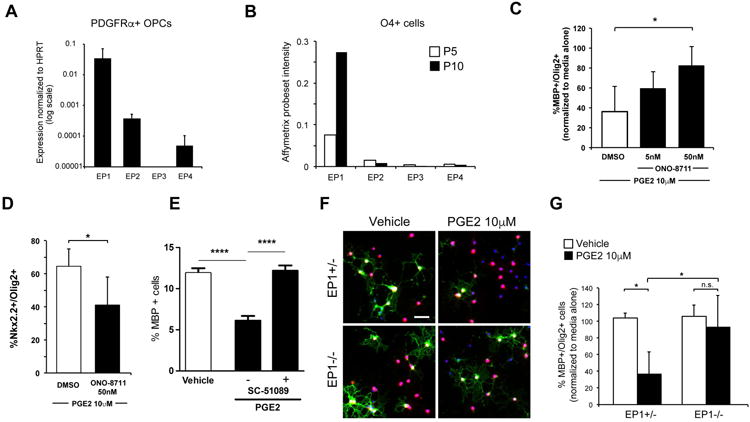
A Quantitative PCR expression of PGE2 EP1-EP4 receptors in immunopurified mouse OPCs. B. Microarray transcript levels of EP1-EP4 in O4+ isolated cells from P5 and P10 mouse cortices. C & D. Quantification of MBP+ (C) and Nkx2.2+ cells (D) exposed to PGE2 and EP1-specific inhibitor ONO-8711. E. Quantification of MBP+ cells after exposure to vehicle (0.1% DMSO), PGE2 10μM, or PGE2 10μM and EP1 inhibitor (SC-51089 10μM) in rat oligodendrocyte culture (n=10 per group). F & G. Representative images (F) and quantification of (G) OPC isolated from EP1-/- or control pups treated with PGE2 (scale bar, 20 μm). * indicates p-value <0.05 and **** p values <0.001. Data shown compiled from at least 3 independent experiments.
To determine whether PGE2 acts through EP1 to interfere with OPC maturation, we employed both pharmacologic and genetic approaches. ONO-8711 is an EP1-specific inhibitor (Watanabe et al., 1999) and co-treatment of ONO-8711 reversed effects of PGE2 on MBP expression and maintained Nk×2.2 (Fig. 5 C and D). In parallel, similar result was observed with rat OPC cultures in presence of SC-51089 (Hallinan et al., 1993), another specific EP1 inhibitor (Fig. 5 E). Secondly, we compared the effects of PGE2 on OPCs purified from EP1-/- or littermate EP1+/- control pups. In contrast to control cells, EP1-/- OPCs were resistant to the effects of PGE2 (Fig. 5 F and G).
While PGE2 effects have been associated with interactions with Wnt or HIF1α signaling (Goessling et al., 2009; Ji et al., 2010), we found no evidence of β-catenin activation or HIF1α stabilization in OPCs following PGE2 exposure (Supplementary Figure. 2 A and B). In addition, we found no evidence for activation of p38MAPK, which has been reported to modulate OPC maturation (Chew et al., 2010). We also found no differences in inflammatory pathway effectors JNK, p65NFκB, IκBα, or STAT3 (Supplementary Figure. 2 C). We also assessed Akt, which regulates oligodendrocyte maturation (Luo et al., 2014) and brain inflammation with reports PGE2 interactions, albeit through the EP4/PI3K pathway (Shi et al., 2010). Akt exhibited no change in protein expression between 6 hours and 4 days following 24 hours of PGE2 exposure in rat culture (Supplementary Figure. 2 D). These results demonstrate that PGE2 directly inhibits OPC maturation in vitro through EP1 receptor engagement.
Inhibition of COX2 attenuates systemic IL-1β induced hypomyelination
To investigate whether COX2 inhibition could prevent the effects of neonatal exposure to IL-1β, we co-treated mice with IL-1β and Nimesulide between P1 and P5 (Fig. 6 A). Notably, we observed a significant increase of Ep1 transcript at P5 in cerebral tissue of mice following systemic administration of IL-1β (Fig. 6 B). Nimesulide prevented the IL-1β-induced increase of NG2 + cells at P5 and the decrease in MBP staining density within the sensory-motor cortex at P30 (Fig. 6 D-F). In addition, we performed testing of treated mice to determine whether COX2 inhibition could reverse behavioral deficits we had previously observed in mice exposed to neonatal IL-1β (Favrais et al., 2011). In novel object recognition and object location memory tests performed at P29 and P30, animals co-treated with IL-1β and Nimesulide performed as controls while animals treated with IL-1β alone showed memory deficits (Fig. 6 G). These findings suggest that inhibition of COX2 is protective against IL-1β mediated effects on neonatal brain.
Figure 6. Cyclooxygenase-2 inhibition prevents hypomyelination and memory deficits.
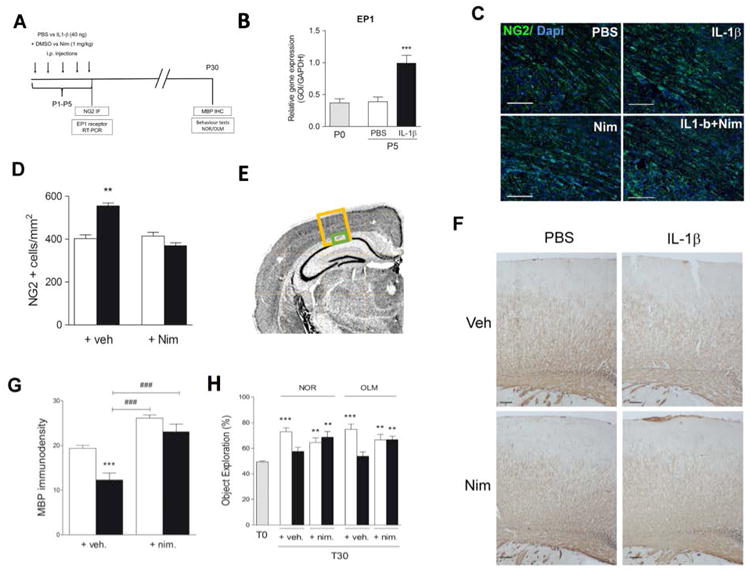
A Timeline of postnatal intraperitoneal treatment by PBS (PBS + Veh.) or IL-1β (IL-1β + Veh.) or PBS with nimesulide (PBS + nim.) or IL-1β with nimesulide (IL-1β + nim.) from P1 to P5 and assessments performed. White bars correspond to PBS treatment, black bars to IL-1β treatment and grey bars to postnatal day 0 previous to i.p. injections B. Ep1 expression measured by RT-PCR at P5 (n=5 per group). C & D. Representative images and graph of NG2 staining within external capsule at P5. (scale bar, 25μm; n=5 per group). E. Image of anatomical areas where NG2 (green box) and MBP (yellow box) were quantified. F. Representative images of MBP immunostaining within the sensory-motor cortex of P30 aged mice (scale bar, 100μm). G. Optical densities of MBP staining within the sensory-motor cortex of P30 mice (n=6 per group). H. Mice were subjected to NOR and OLM tests at P30 (n=10-18 per group). First round = T0 (gray bar), second round = T30. Results are expressed in means +/- SEM. Asterisks indicate statistically significant differences from white bar, ** p<0.01, **** p< 0.001 in Mann-Whitney or One-Way ANOVA tests and ### p< 0.001 in comparison with IL-1β group.
Discussion
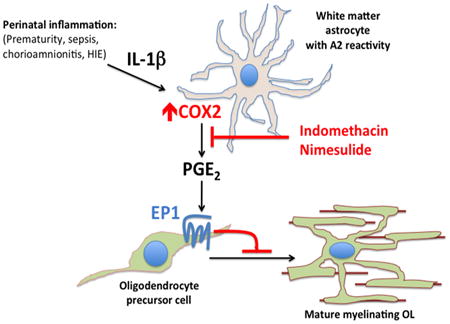
Despite interventions that have dramatically improved ELBW infant survival, no neuroprotective therapy exists for preterm infants in the neonatal intensive care unit to prevent rising rates of cerebral palsy. In this study, we find that PGE2 can act directly on OPCs to inhibit their maturation and, using both genetic and pharmacologic methods, we show that its effects are mediated through the EP1 receptor. We also show that in the developing human brain, COX2 is expressed by microglia, endothelial cells and maturing astrocytes. In human neonatal white matter pathology, reactive astrocytes with an “A2” phenotype strongly induce COX2, and treatment with a COX2-specific inhibitor is protective in a mouse model of inflammation-induced NWMI with preserved myelination and attenuated cognitive impairment. Taken together, our findings support a model (Fig. 7) in which systemic inflammation and perinatal insults can induce “A2” reactive astrocytes to produce PGE2 that directly impairs OPC maturation and myelination.
Figure 7. Model of COX2-PGE2 signaling pathway in human neonatal white matter injury and oligodendrocyte progenitor cell maturation arrest.
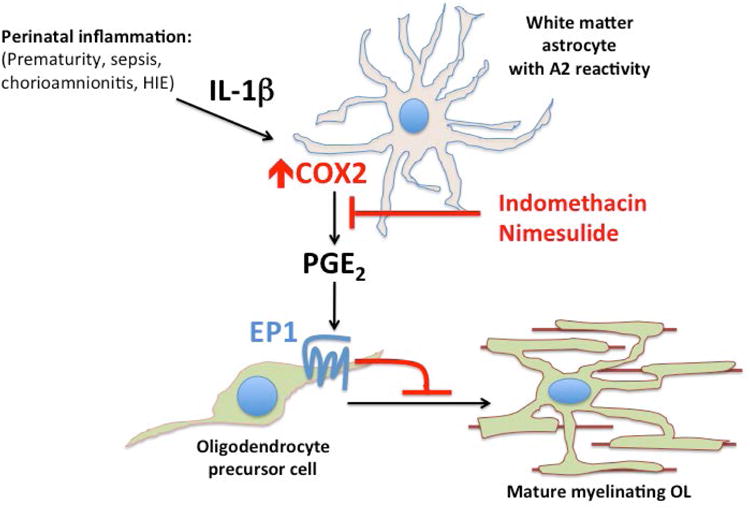
Systemic inflammation from perinatal insults can induce COX2 in reactive glia such as “A2” reactive astrocytes. PGE2 production from COX2 leads to EP1-receptor mediated maturation arrest of OPCs. Indomethacin or COX2-specific inhibitors such as Nimesulide may provide neuroprotection through inhibition of PGE2 production.
There are four receptors for PGE2, and differential expression patterns for these receptors and specific effects of these have been reported across species and injury models (Legler et al., 2010). Using transgenic EP1-/- mice, we purified OPCs and demonstrated that PGE2 directly inhibits OPC maturation in an EP1-dependant manner. Pharmacologic blockade with EP1-specific inhibitors (ONO-8711 or SC-51089) also attenuated effects of PGE2 to inhibit OPC maturation. What is downstream of EP1 signaling in OPCs? Interestingly, EP2 specific activation by PGE2 has been reported to modulate cellular differentiation through the activation of Wnt pathway signaling (Castellone et al., 2005; Goessling et al., 2009), which is capable of causing OPC maturation arrest (Fancy et al., 2009; 2011; Guo et al., 2015). However, we did not find any evidence for Wnt pathway activation. Also, a survey of multiple kinase pathways did not reveal significant changes with PGE2 treatment in OPCs. Thus, further work is needed to identify potential downstream pathways of EP1 in OPCs.
Reactive astrogliosis is a pathological hallmark of human NWMI (Khwaja and Volpe, 2007) but its role in the maturation arrest of OPCs in the neonatal brain is unclear. Reactive astrocytes subtypes “A1” and “A2” (Liddelow et al., 2017) have been suggested to demarcate neurotoxic vs. regenerative forms (Sofroniew, 2015). While reactive astrocytes expressing “A1” markers are found in multiple adult human neurodegenerative conditions and are thought to confer neurotoxic effects, however, not much is known about the downstream effects of “A2” reactive astrocytes. In our study examining neonatal tissue from human white matter, we find that astrocytes predominantly express the “A2” marker S100A10 with COX2. These findings are consistent with the association of “A2” reactive astrocytes with the middle cerebral artery occlusion injury (Zamanian et al., 2012), a model of human neonatal HIE in early postnatal rodents. Our control cases included infants with diaphragmatic hernia, who may have been exposed to some milder degree of hypoxia that did not induce gliosis or inflammatory infiltration. We also find that astrocytes respond to systemic IL-1β with upregulation of “A2” markers, which is in agreement with in vitro findings that IL-1β can promote “A2” astrocyte reactivity associated with COX2 (Ptgs2) upregulation (Liddelow et al., 2017). Our findings indicate that COX2 is not only a marker of “A2” reactivity, but may also function to promote OPC maturation arrest through PGE2 production. Future studies may define whether “A1” or “A2” subtypes of reactive astrocytes are also associated with other astrocytic factors known to modulate OPC maturation, such as hyaluronan (Back et al., 2005), endothelin-1 (Hammond et al., 2014), BMP (Wang et al., 2011), or tenascin C (Nash et al., 2011).
Our study is in general agreement with observations that blocking PGE2 production prevents systemic IL-1β from exacerbating the extent and distribution of lesions in white matter injury (Favrais et al., 2007). Inhibition of PGE2 signaling also attenuates an in vitro model of excitotoxic OPC death (Carlson et al., 2015). Thus, in variable neurologic insults, PGE2 likely contributes to neuroglial damage through intrinsic and extrinsic pathways, and might exhibit detrimental effects on cell survival (Palumbo et al., 2011). However, the neuropathology in preterm infants exposed to systemic inflammation leads to hypomyelination, OPC maturation arrest, and typically occurs without increased cell death (Billiards et al., 2008; Favrais et al., 2011; Verney et al., 2012). As such, this role for PGE2 as a modulator rather than a toxic mediator in leading to OPC maturation arrest may be more consistent with today's predominant form of neonatal brain injury with diffuse NWMI.
Previous studies using nonspecific COX inhibitors, such as indomethacin or ibuprofen, to promote patent ductus arteriosus (PDA) closure in preterm infants showed benefit for the prevention of severe intraventricular hemorrhage (Ment et al., 1994; Schmidt et al., 2001) but they were not powered or designed to evaluate NWMI. A recent meta-analysis correlates maternal use of indomethacin as a tocolytic with poor neonatal outcomes (Hammers et al., 2015), but postnatal use of indomethacin have not demonstrated worse neurologic outcomes. On the contrary, a retrospective analysis PreMRI clinical trial data in preterm infants exposed to prolonged (less than three, but greater than seven days) courses of indomethacin showed decreased evidence of NWMI (Gano et al., 2014) suggesting a similar neuroprotective effect to what we report here. Indeed, our findings suggest a mechanism for white matter neuroprotection through indomethacin's anti-inflammatory inhibition of PGE2 production by reactive glia (Fig. 7).
In conclusion, this study identifies that COX2 mediated neuroinflammatory PGE2 production can impair the maturation of OPCs through engagement of EP1 receptor. We were able to demonstrate this association in vivo and prevent inflammation induced NMWI with the COX2 inhibitor nimesulide, and provide evidence for the expression of COX2 in human “A2” reactive astrocytes. This is an important mechanistic and proof-of-concept therapeutic support that targeting PGE2 production might be a viable therapeutic strategy in humans at risk for NWMI.
Supplementary Material
Main Points.
PGE2 generated by COX2 directly inhibits OPC maturation in an EP1 receptor-dependent manner. In human NWMI, astrocytes develop “A2” reactivity and induce COX2. Using an inflammation-induced model of NWMI, systemic COX2 inhibition protected myelination and preserved motor function.
Acknowledgments
We would like to thank Hui-Hsin Tsai and Eric J. Huang at the UCSF Pediatric Neuropathology brain bank, and Vien Nguyen, Sandra Chang, and Khalida Sabeur for technical assistance. L.R.S is an NICHD/NIH fellow of the Pediatric Scientist Development Program (K12HD000850) and received support from NICHD T32 training grant (32HD071860). L.S. was supported by a postdoctoral fellowship from the German Research Foundation (DFG, SCHI 1330/1-1). This study was supported by grants (to B.F, P.G) from Inserm, Université Paris Diderot, Université Sorbonne-Paris-Cité, Investissement d' (ANR-11-INBS-0011, NeurATRIS), ERA-NET Neuron (Micromet), DHU PROTECT, PremUP, Fondation de France, Institut Servier, Roger de Spoelberch Foundation, Grace de Monaco Foundation, Leducq Foundation, Cerebral Palsy Alliance Research Foundation Australia and funding (to D.H.R.) from the National Institute of Neurological Disease and Stroke (P01NS083513) and Howard Hughes Medical Institute. The supporting bodies played no role in any aspect of study design, analysis, interpretation or decision to publish these data.
Footnotes
Conflict of interest statement: L.S. filed a patent for the detection of antibodies against KIR4.1 in a subpopulation of patients with multiple sclerosis. Other authors declared that there are no conflicts of interest.
Author contributions: LRS, GF, LS, ALS, JNW, HN, SC, and CA performed the experiments. LRS, GF, LS, ALS, SC, CA, BF, PG, DHR were involved in the design of the experiments and the interpretation of the data. LRS, GF, BF, PG, DHR drafted the manuscript.
References
- Back SA, Miller SP. Brain injury in premature neonates: A primary cerebral dysmaturation disorder? Ann Neurol. 2014;75:469–486. doi: 10.1002/ana.24132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Rosenberg PA. Pathophysiology of glia in perinatal white matter injury. Glia. 2014;62:1790–1815. doi: 10.1002/glia.22658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Tuohy TMF, Chen H, Wallingford N, Craig A, Struve J, Luo NL, Banine F, Liu Y, Chang A, Trapp BD, Bebo BF, Rao MS, Sherman LS. Hyaluronan accumulates in demyelinated lesions and inhibits oligodendrocyte progenitor maturation. Nature Medicine. 2005;11:966–972. doi: 10.1038/nm1279. [DOI] [PubMed] [Google Scholar]
- Back SA. Perinatal white matter injury: The changing spectrum of pathology and emerging insights into pathogenetic mechanisms. Ment Retard Dev Disabil Res Rev. 2006;12:129–140. doi: 10.1002/mrdd.20107. [DOI] [PubMed] [Google Scholar]
- Billiards SS, Haynes RL, Folkerth RD, Borenstein NS, Trachtenberg FL, Rowitch DH, Ligon KL, Volpe JJ, Kinney HC. RESEARCH ARTICLE: Myelin Abnormalities without Oligodendrocyte Loss in Periventricular Leukomalacia. Brain Pathology. 2008;18:153–163. doi: 10.1111/j.1750-3639.2007.00107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björk L, Leifsdottir K, Saha S, Herlenius E. PGE-metabolite levels in CSF correlate to HIE score and outcome after perinatal asphyxia. Acta Paediatr. 2013;102:1041–1047. doi: 10.1111/apa.12361. [DOI] [PubMed] [Google Scholar]
- Boyle CA, Boulet S, Schieve LA, Cohen RA, Blumberg SJ, Yeargin-Allsopp M, Visser S, Kogan MD. Trends in the Prevalence of Developmental Disabilities in US Children, 1997-2008. Pediatrics. 2011;127:1034–1042. doi: 10.1542/peds.2010-2989. [DOI] [PubMed] [Google Scholar]
- Buser JR, Maire J, Riddle A, Gong X, Nguyen T, Nelson K, Luo NL, Ren J, Struve J, Sherman LS, Miller SP, Chau V, Hendson G, Ballabh P, Grafe MR, Back SA. Arrested preoligodendrocyte maturation contributes to myelination failure in premature infants. Ann Neurol. 2012;71:93–109. doi: 10.1002/ana.22627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson NG, Bellamkonda S, Schmidt L, Redd J, Huecksteadt T, Weber LM, Davis E, Wood B, Maruyama T, Rose JW. The role of the prostaglandin E2 receptors in vulnerability of oligodendrocyte precursor cells to death. J Neuroinflammation. 2015;12:33157–25. doi: 10.1186/s12974-015-0323-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellone MD, Teramoto H, Williams BO, Druey KM, Gutkind JS. Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-beta-catenin signaling axis. Science. 2005;310:1504–1510. doi: 10.1126/science.1116221. [DOI] [PubMed] [Google Scholar]
- Chew LJ, Coley W, Cheng Y, Gallo V. Mechanisms of Regulation of Oligodendrocyte Development by p38 Mitogen-Activated Protein Kinase. Journal of Neuroscience. 2010;30:11011–11027. doi: 10.1523/JNEUROSCI.2546-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Counsell SJ, Allsop JM, Harrison MC, Larkman DJ, Kennea NL, Kapellou O, Cowan FM, Hajnal JV, Edwards AD, Rutherford MA. Diffusion-weighted imaging of the brain in preterm infants with focal and diffuse white matter abnormality. Pediatrics. 2003;112:1–7. doi: 10.1542/peds.112.1.1. [DOI] [PubMed] [Google Scholar]
- Dammann O, Leviton A. Maternal intrauterine infection, cytokines, and brain damage in the preterm newborn. Pediatr Res. 1997;42:1–8. doi: 10.1203/00006450-199707000-00001. [DOI] [PubMed] [Google Scholar]
- Deng W. Neurobiology of injury to the developing brain. Nat Rev Neurol. 2010;6:328–336. doi: 10.1038/nrneurol.2010.53. [DOI] [PubMed] [Google Scholar]
- Deng Y, Xie D, Fang M, Zhu G, Chen C, Zeng H, Lu J, Charanjit K. Astrocyte-Derived Proinflammatory Cytokines Induce Hypomyelination in the Periventricular White Matter in the Hypoxic Neonatal Brain. PLoS ONE. 2014;9:e87420. doi: 10.1371/journal.pone.0087420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery B, Dugas JC. Purification of oligodendrocyte lineage cells from mouse cortices by immunopanning. Cold Spring Harbor Protocols. 2013;2013:854–868. doi: 10.1101/pdb.prot073973. [DOI] [PubMed] [Google Scholar]
- Fancy SPJ, Baranzini SE, Zhao C, Yuk DI, Irvine KA, Kaing S, Sanai N, Franklin RJM, Rowitch DH. Dysregulation of the Wnt pathway inhibits timely myelination and remyelination in the mammalian CNS. Genes & Development. 2009;23:1571–1585. doi: 10.1101/gad.1806309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fancy SPJ, Harrington EP, Yuen TJ, Silbereis JC, Zhao C, Baranzini SE, Bruce CC, Otero JJ, Huang EJ, Nusse R, Franklin RJM, Rowitch DH. Axin2 as regulatory and therapeutic target in newborn brain injury and remyelination. Nature Publishing Group. 2011;14:1009–1016. doi: 10.1038/nn.2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favrais G, Schwendimann L, Gressens P, Lelièvre V. Cyclooxygenase-2 mediates the sensitizing effects of systemic IL-1-beta on excitotoxic brain lesions in newborn mice. Neurobiology of Disease. 2007;25:496–505. doi: 10.1016/j.nbd.2006.10.012. [DOI] [PubMed] [Google Scholar]
- Favrais G, van de Looij Y, Fleiss B, Ramanantsoa N, Bonnin P, Stoltenburg-Didinger G, Lacaud A, Saliba E, Dammann O, Gallego J, Sizonenko S, Hagberg H, Lelièvre V, Gressens P. Systemic inflammation disrupts the developmental program of white matter. Ann Neurol. 2011;70:550–565. doi: 10.1002/ana.22489. [DOI] [PubMed] [Google Scholar]
- Fern RF, Matute C, Stys PK. White matter injury: Ischemic and nonischemic. Glia. 2014;62:1780–1789. doi: 10.1002/glia.22722. [DOI] [PubMed] [Google Scholar]
- Gano D, Andersen SK, Partridge JC, Bonifacio SL, Xu D, Glidden DV, Ferriero DM, Barkovich AJ, Glass HC. Diminished White Matter Injury over Time in a Cohort of Premature ewborns. J Pediatr. 2014 doi: 10.1016/j.jpeds.2014.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goessling W, North TE, Loewer S, Lord AM, Lee S, Stoick-Cooper CL, Weidinger G, Puder M, Daley GQ, Moon RT, Zon LI. Genetic Interaction of PGE2 and Wnt Signaling Regulates Developmental Specification of Stem Cells and Regeneration. Cell. 2009;136:1136–1147. doi: 10.1016/j.cell.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillen U, Weiss EM, Munson D, Maton P, Jefferies A, Norman M, Naulaers G, Mendes J, Justo da Silva L, Zoban P, Hansen TWR, Hallman M, Delivoria-Papadopoulos M, Hosono S, Albersheim SG, Williams C, Boyle E, Lui K, Darlow B, Kirpalani H. Guidelines for the Management of Extremely Premature Deliveries: A Systematic Review. Pediatrics. 2015;136:343–350. doi: 10.1542/peds.2015-0542. [DOI] [PubMed] [Google Scholar]
- Guo F, Lang J, Sohn J, Hammond E, Chang M, Pleasure D. Canonical Wnt signaling in the oligodendroglial lineage-puzzles remain. Glia. 2015:n/a–n/a. doi: 10.1002/glia.22813. [DOI] [PubMed] [Google Scholar]
- Hagberg H, Mallard C, Ferriero DM, Vannucci SJ, Levison SW, Vexler ZS, Gressens P. The role of inflammation in perinatal brain injury. Nature Publishing Group. 2015:1–17. doi: 10.1038/nrneurol.2015.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallinan EA, Hagen TJ, Husa RK, Tsymbalov S, Rao SN, vanHoeck JP, Rafferty MF, Stapelfeld A, Savage MA, Reichman M. N-substituted dibenzoxazepines as analgesic PGE2 antagonists. J Med Chem. 1993;36:3293–3299. doi: 10.1021/jm00074a010. [DOI] [PubMed] [Google Scholar]
- Hammers AL, Sanchez-Ramos L, Kaunitz AM. Antenatal exposure to indomethacin increases the risk of severe intraventricular hemorrhage, necrotizing enterocolitis, and periventricular leukomalacia: a systematic review with metaanalysis. Am J Obstet Gynecol. 2015;212:505.e1–13. doi: 10.1016/j.ajog.2014.10.1091. [DOI] [PubMed] [Google Scholar]
- Hammond TR, Gadea A, Dupree J, Kerninon C, Nait-Oumesmar B, Aguirre A, Gallo V. Astrocyte-Derived Endothelin-1 Inhibits Remyelination through Notch Activation. Neuron. 2014;81:588–602. doi: 10.1016/j.neuron.2013.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inder T, Neil J, Kroenke C, Dieni S, Yoder B, Rees S. Investigation of cerebral development and injury in the prematurely born primate by magnetic resonance imaging and histopathology. Dev Neurosci. 2005;27:100–111. doi: 10.1159/000085981. [DOI] [PubMed] [Google Scholar]
- Ji R, Chou CL, Xu W, Chen XB, Woodward DF, Regan JW. EP1 Prostanoid Receptor Coupling to Gi/o Up-Regulates the Expression of Hypoxia-Inducible Factor-1 through Activation of a Phosphoinositide-3 Kinase Signaling Pathway. Molecular Pharmacology. 2010;77:1025–1036. doi: 10.1124/mol.110.063933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuura G, Gottschall PE, Dahl RR, Arimura A. Interleukin-1 beta increases prostaglandin E2 in rat astrocyte cultures: modulatory effect of neuropeptides. Endocrinology. 1989;124:3125–3127. doi: 10.1210/endo-124-6-3125. [DOI] [PubMed] [Google Scholar]
- Khwaja O, Volpe JJ. Pathogenesis of cerebral white matter injury of prematurity. Archives of Disease in Childhood - Fetal and Neonatal Edition. 2007;93:F153–F161. doi: 10.1136/adc.2006.108837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legler DF, Bruckner M, Uetz-von Allmen E, Krause P. Prostaglandin E2 at new glance: novel insights in functional diversity offer therapeutic chances. Int J Biochem Cell Biol. 2010;42:198–201. doi: 10.1016/j.biocel.2009.09.015. [DOI] [PubMed] [Google Scholar]
- Leviton A, Allred EN, Fichorova RN, Kuban KCK, Michael O'Shea T, Dammann O ELGAN study investigators. Systemic inflammation on postnatal days 21 and 28 and indicators of brain dysfunction 2years later among children born before the 28th week of gestation. Early Human Development. 2016;93:25–32. doi: 10.1016/j.earlhumdev.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung WS, Peterson TC, Wilton DK, Frouin A, Napier BA, Panicker N, Kumar M, Buckwalter MS, Rowitch DH, Dawson VL, Dawson TM, Stevens B, Barres BA. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541:481–487. doi: 10.1038/nature21029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo F, Burke K, Kantor C, Miller RH, Yang Y. Cyclin-dependent kinase 5 mediates adult OPC maturation and myelin repair through modulation of Akt and GsK-3β signaling. J Neurosci. 2014;34:10415–10429. doi: 10.1523/JNEUROSCI.0710-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaeb S, Dammann O. Fetal inflammatory response and brain injury in the preterm newborn. Journal of Child Neurology. 2009;24:1119–1126. doi: 10.1177/0883073809338066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy KD, de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. The Journal of Cell Biology. 1980;85:890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ment LR, Oh W, Ehrenkranz RA, Philip AG, Vohr B, Allan W, Duncan CC, Scott DT, Taylor KJ, Katz KH. Low-dose indomethacin and prevention of intraventricular hemorrhage: a multicenter randomized trial. Pediatrics. 1994;93:543–550. [PubMed] [Google Scholar]
- Molina-Holgado E, Ortiz S, Molina-Holgado F, Guaza C. Induction of COX-2 and PGE(2) biosynthesis by IL-1beta is mediated by PKC and mitogen-activated protein kinases in murine astrocytes. British Journal of Pharmacology. 2000;131:152–159. doi: 10.1038/sj.bjp.0703557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore T, Hennessy EM, Myles J, Johnson SJ, Draper ES, Costeloe KL, Marlow N. Neurological and developmental outcome in extremely preterm children born in England in 1995 and 2006: the EPICure studies. BMJ. 2012;345:e7961–e7961. doi: 10.1136/bmj.e7961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nash B, Thomson CE, Linington C, Arthur AT, McClure JD, McBride MW, Barnett SC. Functional duality of astrocytes in myelination. J Neurosci. 2011;31:13028–13038. doi: 10.1523/JNEUROSCI.1449-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobuta H, Ghiani CA, Paez PM, Spreuer V, Dong H, Korsak RA, Manukyan A, Li J, Vinters HV, Huang EJ, Rowitch DH, Sofroniew MV, Campagnoni AT, de Vellis J, Waschek JA. STAT3-Mediated astrogliosis protects myelin development in neonatal brain injury. Ann Neurol. 2012;72:750–765. doi: 10.1002/ana.23670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northam GB, Liégeois F, Chong WK, S Wyatt J, Baldeweg T. Total brain white matter is a major determinant of IQ in adolescents born preterm. Ann Neurol. 2011;69:702–711. doi: 10.1002/ana.22263. [DOI] [PubMed] [Google Scholar]
- Palumbo S, Toscano CD, Parente L, Weigert R, Bosetti F. The cyclooxygenase-2 pathway via the PGE2 EP2 receptor contributes to oligodendrocytes apoptosis in cuprizone-induced demyelination. Journal of Neurochemistry. 2011;121:418–427. doi: 10.1111/j.1471-4159.2011.07363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekny M, Nilsson M. Astrocyte activation and reactive gliosis. Glia. 2005;50:427–434. doi: 10.1002/glia.20207. [DOI] [PubMed] [Google Scholar]
- Pierson CR, Folkerth RD, Billiards SS, Trachtenberg FL, Drinkwater ME, Volpe JJ, Kinney HC. Gray matter injury associated with periventricular leukomalacia in the premature infant. Acta Neuropathol. 2007;114:619–631. doi: 10.1007/s00401-007-0295-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Y, Cai J, Wu Y, Wu R, Lee J, Fu H, Rao M, Sussel L, Rubenstein J, Qiu M. Control of oligodendrocyte differentiation by the Nk×2.2 homeodomain transcription factor. Development. 2001;128:2723–2733. doi: 10.1242/dev.128.14.2723. [DOI] [PubMed] [Google Scholar]
- Riddle A, Dean J, Buser JR, Gong X, Maire J, Chen K, Ahmad T, Cai V, Nguyen T, Kroenke CD, Hohimer AR, Back SA. Histopathological correlates of magnetic resonance imaging-defined chronic perinatal white matter injury. Ann Neurol. 2011;70:493–507. doi: 10.1002/ana.22501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schang AL, Van Steenwinckel J, Chevenne D, Alkmark M, Hagberg H, Gressens P, Fleiss B. Failure of thyroid hormone treatment to prevent inflammation-induced white matter injury in the immature brain. Brain Behavior and Immunity. 2014;37:95–102. doi: 10.1016/j.bbi.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schildge S, Bohrer C, Beck K, Schachtrup C. Isolation and culture of mouse cortical astrocytes. JoVE. 2013:e50079–e50079. doi: 10.3791/50079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt B, Davis P, Moddemann D, Ohlsson A, Roberts RS, Saigal S, Solimano A, Vincer M, Wright LL Trial of Indomethacin Prophylaxis in Preterms Investigators. Long-term effects of indomethacin prophylaxis in extremely-low-birth-weight infants. N Engl J Med. 2001;344:1966–1972. doi: 10.1056/NEJM200106283442602. [DOI] [PubMed] [Google Scholar]
- Serenius F, Källén K, Blennow M, Ewald U, Fellman V, Holmström G, Lindberg E, Lundqvist P, Maršál K, Norman M, Olhager E, Stigson L, Stjernqvist K, Vollmer B, Strömberg B EXPRESS Group. Neurodevelopmental outcome in extremely preterm infants at 2.5 years after active perinatal care in Sweden. JAMA. 2013;309:1810–1820. doi: 10.1001/jama.2013.3786. [DOI] [PubMed] [Google Scholar]
- Sharma K, Schmitt S, Bergner CG, Tyanova S, Kannaiyan N, Manrique-Hoyos N, Kongi K, Cantuti L, Hanisch UK, Philips MA, Rossner MJ, Mann M, Simons M. Cell type– and brain region–resolved mouse brain proteome. Nat Neurosci. 2015:1–16. doi: 10.1038/nn.4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Johansson J, Woodling NS, Wang Q, Montine TJ, Andreasson K. The prostaglandin E2 E-prostanoid 4 receptor exerts anti-inflammatory effects in brain innate immunity. The Journal of Immunology. 2010;184:7207–7218. doi: 10.4049/jimmunol.0903487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siljehav V, Hofstetter AM, Leifsdottir K, Herlenius E. Prostaglandin E2 Mediates Cardiorespiratory Disturbances during Infection in Neonates. J Pediatr. 2015;167:1207–13.e3. doi: 10.1016/j.jpeds.2015.08.053. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV. Astrogliosis. Cold Spring Harbor Perspectives in Biology. 2015;7:a020420–17. doi: 10.1101/cshperspect.a020420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supramaniam V, Vontell R, Srinivasan L, Wyatt-Ashmead J, Hagberg H, Rutherford M. Microglia activation in the extremely preterm human brain. Pediatr Res. 2013;73:301–309. doi: 10.1038/pr.2012.186. [DOI] [PubMed] [Google Scholar]
- Vela J. Interleukin-1 Regulates Proliferation and Differentiation of Oligodendrocyte Progenitor Cells. Molecular and Cellular Neuroscience. 2002;20:489–502. doi: 10.1006/mcne.2002.1127. [DOI] [PubMed] [Google Scholar]
- Verney C, Pogledic I, Biran V, Adle-Biassette H, Fallet-Bianco C, Gressens P. Microglial reaction in axonal crossroads is a hallmark of noncystic periventricular white matter injury in very preterm infants. J Neuropathol Exp Neurol. 2012;71:251–264. doi: 10.1097/NEN.0b013e3182496429. [DOI] [PubMed] [Google Scholar]
- Wang Y, Cheng X, He Q, Zheng Y, Kim DH, Whittemore SR, Cao QL. Astrocytes from the contused spinal cord inhibit oligodendrocyte differentiation of adult oligodendrocyte precursor cells by increasing the expression of bone morphogenetic proteins. J Neurosci. 2011;31:6053–6058. doi: 10.1523/JNEUROSCI.5524-09.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K, Kawamori T, Nakatsugi S, Ohta T, Ohuchida S, Yamamoto H, Maruyama T, Kondo K, Ushikubi F, Narumiya S, Sugimura T, Wakabayashi K. Role of the prostaglandin E receptor subtype EP1 in colon carcinogenesis. Cancer Research. 1999;59:5093–5096. [PubMed] [Google Scholar]
- Williams A, Piaton G, Lubetzki C. Astrocytes—Friends or foes in multiple sclerosis? Glia. 2007;55:1300–1312. doi: 10.1002/glia.20546. [DOI] [PubMed] [Google Scholar]
- Woodward LJ, Anderson PJ, Austin NC, Howard K, Inder TE. Neonatal MRI to Predict Neurodevelopmental Outcomes in Preterm Infants. N Engl J Med. 2006;355:685–694. doi: 10.1056/NEJMoa053792. [DOI] [PubMed] [Google Scholar]
- Xia Q, Hu Q, Wang H, Yang H, Gao F, Ren H, Chen D, Fu C, Zheng L, Zhen X, Ying Z, Wang G. Induction of COX-2-PGE2 synthesis by activation of the MAPK/ERK pathway contributes to neuronal death triggered by TDP-43-depleted microglia. Cell Death and Disease. 2015;6:e1702. doi: 10.1038/cddis.2015.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Chalimoniuk M, Shu Y, Simonyi A, Sun AY, Gonzalez FA, Weisman GA, Wood WG, Sun GY. Prostaglandin E2 production in astrocytes: regulation by cytokines, extracellular ATP, and oxidative agents. Prostaglandins Leukotrienes and Essential Fatty Acids. 2003;69:437–448. doi: 10.1016/j.plefa.2003.08.016. [DOI] [PubMed] [Google Scholar]
- Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, Barres BA. Genomic Analysis of Reactive Astrogliosis. Journal of Neuroscience. 2012;32:6391–6410. doi: 10.1523/JNEUROSCI.6221-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, Barres BA, Wu JQ. An RNA-Sequencing Transcriptome and Splicing Database of Glia, Neurons, and Vascular Cells of the Cerebral Cortex. Journal of Neuroscience. 2014;34:11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.