

Abstract
The Myh11-CreERT2 mouse line (Cre+) has gained increasing application due to its high lineage specificity relative to other Cre drivers targeting smooth muscle cells (SMCs). This Cre allele, however, was initially inserted into the Y chromosome (X/YCre+), which excluded its application in female mice. Our group established a Cre+ colony from male ancestors. Surprisingly, genotype screening identified female carriers that stably transmitted the Cre allele to the following generations. Crossbreeding experiments revealed a pattern of X-linked inheritance for the transgene (k>1000), indicating that these female carries acquired the Cre allele through a mechanism of Y to X chromosome translocation. Further characterization demonstrated that in hemizygous X/XCre+ mice Cre activity was restricted to a subset arterial SMCs, with Cre expression in arteries decreased by 50% compared to X/YCre+ mice. This mosaicism, however, diminished in homozygous XCre+/XCre+ mice. In a model of aortic aneurysm induced by a SMC-specific Tgfbr1 deletion, the homozygous XCre+/XCre+ Cre driver unmasked the aortic phenotype that is otherwise subclinical when driven by the hemizygous X/XCre+ Cre line. In conclusion, the Cre allele carried by this female mouse line is located on the X chromosome and subjected to X-inactivation. The homozygous XCre+/XCre+ mice produce uniform Cre activity in arterial SMCs.
Keywords: Genetics, Process, Vasculature, Tissue, Mammal, Organism, Mutagenesis, Process
INTRODUCTION
Smooth muscle cells (SMCs) are key players in the pathogenesis of vascular diseases such as hypertension, atherosclerosis, and restenosis of stented arteries and vein bypass grafts. Studies in the early 1980s suggested that these cells acquire a proliferative/synthetic phenotype and migrate from the tunica media to the intima, driving hyperplastic neointimal thickening(Hoofnagle et al., 2006). The lack of reliable approaches to identify the SMC-derived cells in vascular lesions creates a challenge in obtaining direct evidence to support this hypothesis(Nguyen et al., 2013). Precursor lineages that populate SMCs in the vasculature during embryonic development leave little footprints that otherwise can be used as a marker for SMC identity. Although SMCs produce a panel of contractile (e.g., α-actin, SM myosin heavy chain, and calponin) and cytoskeleton (e.g., SM22 and smoothelin) proteins that distinguish themselves as a unique cell lineage, none of them can serve as a definitive lineage marker due to the production of one or more of these proteins by other cell groups such as endothelial cells, fibroblasts, and myeloid derived cells (Gomez et al., 2012). The task of fate mapping of SMCs was further complicated by their phenotypic plasticity. By undergoing a phenotypic switch, SMCs can present various morphologies and acquire distinct molecular signatures in different type (e.g. atherosclerosis vs. restenosis) and at different stages (e.g., early vs. advanced) of vascular diseases (Owens et al., 2004). Tools that enable researchers to track SMCs and selectively modify gene expression in SMCs were anxiously waited in the field of vascular biology.
Initial efforts in mapping the fate of SMCs focused on the expression of genes coding SMC-differentiation markers. Mouse lines carrying a reporter gene driven by the promoter of Acta2 (Wu et al., 2007), Tagln (Li et al., 1996), or Myh11 (Regan et al., 2000) were created and evaluated for their application. Although all these reporter lines are able to label medial SMCs in the mature vasculature, a major issue that limits their application is the “off-target” Cre activity in cells other than SMCs due to episodic expression of these genes in various cell types during embryonic development, pathogenesis of vascular diseases, and generation of gametes (Frutkin et al., 2006; Regan et al., 2000). The development of inducible Cre/LoxP technology offered an opportunity to modify gene expression in a cell-type specific and timely controlled manner. This technology not only allows selective attachment of a permanent “tag” to mature SMCs but also enables modification of the expression of SMC-specific genes during the postnatal life (Feil et al., 2009; Nguyen et al., 2013). Several lines of inducible Cre drivers have been made available to target SMCs by placing the CreERT2-expressing cassette under the promoter of Acta2 (Wendling et al., 2009), Tagln (Kuhbandner et al., 2000), or Myh11 (Wirth et al., 2008). Among these various Cre lines, the Myh11-CreERT2 strain has gained an increasing application due to its ability to induce recombination of the floxed genes in SMCs at a high specificity and efficiency (Hu et al., 2015; Li et al., 2014; Schmit et al., 2015; Shankman et al., 2015). We have previously reported that the Cre activity of this mouse line is restricted to medial SMCs, and it drives successful recombination in aortas and peripheral muscular arteries at efficiency rate greater than 90% (Yang et al., 2016a; Yang et al., 2016b). A limitation of this mouse line, however, is that the Cre allele was initially inserted into the Y-chromosome, which excludes its application in female mice. Gender has been identified as a risk factor for multiple vascular diseases such as aortic aneurysm and coronary heart disease (Mosca et al., 2011). Female mice carrying the same Myh11-CreERT2 allele will provide a useful tool to dissect mechanisms underlying vascular diseases with sexually dimorphic phenotypic presentations.
Our group has used the Myh11-CreER+ mouse line to study the role of SMCs in the pathogenesis of neointimal hyperplasia and aortic aneurysm development (Schmit et al., 2015; Yang et al., 2016a; Yang et al., 2016b). During the genotype screening process, we identified female carriers of the Cre allele and observed a stable inheritance of the Cre allele by both male and female offspring. This inheritance pattern indicated that the Cre allele was translocated from the Y chromosome to the X chromosome or even an autosome. Results obtained from the subsequent cross-breeding experiments demonstrated that the Cre allele was passed to the next generation in an X-linked fashion, thereby indicating translocation of the Cre allele from the Y to the X chromosome. In view of this finding, we further characterized this X-linked Myh11-CreERT2 mouse line with a focus being placed on vascular SMCs.
RESULTS
The Myh11-CreERT2 allele can translocate from the Y to an X chromosome
Our lab has used Tgfbr1f/f;R26R+;Myh11-CreERT2+ mice for a SMC-specific Tgfbr1 deletion (Schmit et al., 2015; Yang et al., 2016b). These mice received the Myh11-CreERT2 allele from a male founder mouse of which the transgene was integrated into the Y chromosome (Wirth et al., 2008). During the genotype screening process, we identified female Myh11-CreERT2 carriers in the offspring. It is because of this unexpected finding, that the Tgfbr1f/f;R26R+;Myh11-CreERT2+ colony were separated into two subcolonies. The 1st subcolony passed the Myh11-CreERT2 allele only to male progenies. The 2nd subcolony passed the Myh11-CreERT2 allele to both male and female littermates. For the stake of simplicity, the symbols Cre+ and Cre− are utilized to represent Tgfbr1f/f;R26R+mice carrying or not carrying the Myh11-CreERT2 allele, respectively; while location of the Myh11-CreERT2 allele on X- or Y-chromosome is indicated with the symbols XCre+ and YCre+, respectively.
This 1st subcolony was maintained by mating Cre+ male mice with Cre− female breeders. Table 1 lists the genotyping results of littermates produced by the most recent four breeding pairs. In an agreement with the Y-chromosome location of the Cre allele(Wirth et al., 2008), which is designated as X/YCre+ throughout this study, the results showed that all male (17/49) but none of the female pups (32/49) received the Cre allele carried by their fathers. This inheritance pattern favored a linkage to the Y chromosome over an autosome or X chromosome (Bayes factor K>1000), indicating that the Cre allele carried by the 1st subcolony remained located on the Y-chromosome.
Table 1.
Littermates produced by four Cre+ male and Cre− female breeding pairs
| Genotype | Female | Male | Total |
|---|---|---|---|
| Cre− | 32 | 0 | 32 |
| Cre+ | 0 | 17 | 17 |
|
| |||
| Total | 32 | 17 | 49 |
The 2nd subcolony that passed the Cre allele to both male and female littermates was maintained by crossing Cre+ female with Cre− male breeders. The inheritance pattern displayed by this subcolony excluded the possibility of Y-chromosome location of the Cre allele. The question raised was whether it was relocated onto an X chromosome or an autosome. To answer this question, we initiated cross-breeding experiments. The strategy was to follow the inheritance of the Cre allele of a male breeder that receives the transgene maternally. Therefore, the first breeding experiment was aimed at producing Cre+ male breeders. Two Cre+ female littermates (i.e., taken from a litter of the 2nd subcolony) were mated with a Cre− male produced in this subcolony. Table 2 shows the genotyping results of their F1 offspring pups. As expected, both male and female Cre+ littermates were identified, and Bayes factor analysis indicated an equal probability for offspring pups of either gender to receive the Cre allele (k=1 when comparing autosomes vs. X chromosomes). Then, an F1 Cre+ male was mated with two Cre− female breeders. Table 3 displays the genotyping results of their F2 offspring pups. All F2 female but none of the F2 male mice received the Cre allele from their father. These results favored an X-linked inheritance pattern (Bayes Factor k>1000), indicating that the F1 male carried the Cre allele on its X-chromosome (a genotype designated as X/XCre+ throughout this study). Since this subcolony was derived from a male mouse in which the Cre allele was located on the Y chromosome, the results obtained from these crossbreeding experiments suggest that the X/YCre+ mouse line passed its Cre allele to the 2nd colony through Y-to-X chromosome translocation (t(X;Y)) during the breeding process.
Table 2.
F1 littermates produced by crossing a Cre− male with two Cre+ female breeders
| Genotype | Female | Male | Total |
|---|---|---|---|
| Cre− | 11 | 12 | 23 |
| Cre+ | 4 | 10 | 14 |
|
| |||
| Total | 15 | 22 | 37 |
Table 3.
F2 littermates produced by crossing an F1 Cre+ male mouse with two Cre− female breeders
| Genotype | Female | Male | Total |
|---|---|---|---|
| Cre− | 0 | 12 | 12 |
| Cre+ | 20 | 0 | 20 |
|
| |||
| Total | 20 | 12 | 32 |
Expression of the Cre transgene at the level of mRNA and protein is attenuated in X/XCre+ mice
The t(X;Y) of the Myh11-CreERT2 allele resulted in a novel X/XCre+ mouse line that can potentially be utilized to drive SMC-specific gene modification in female mice. Therefore, we performed experiments to evaluate its application. An emphasis was placed on muscular arteries including the aorta, common carotid artery (CCA), and femoral artery (FA) due to the ever-increasing application of the Myh11-CreERT2 driver in the field of vascular biology. First, we measured the baseline levels of Cre expression in aortas of X/YCre+ and X/XCre+ mice not treated with tamoxifen (n=5 per group). The results showed that it took approximately 1.0 more cycles for the aortas of X/XCre+ mice to reach the threshold level than did the aortas of X/YCre+ mice irrespective of the anatomic location of the samples from either the ascending (ATA, P=0.020) or suprarenal (SRA, P=0.008) aortic regions (Figure 1a). This 1.0 cycle difference indicates that levels of the Cre mRNA expressed by X/XCre+ mice is only about 50% of that transcribed by X/YCre+ mice. In agreement with the reduced mRNA expression, the abundance of the Cre protein was also significantly lower in the aortas of X/XCre+ mice than in X/YCre+ mice when quantified with Western blotting assays (P<0.001, Figure 1b).
Figure 1.
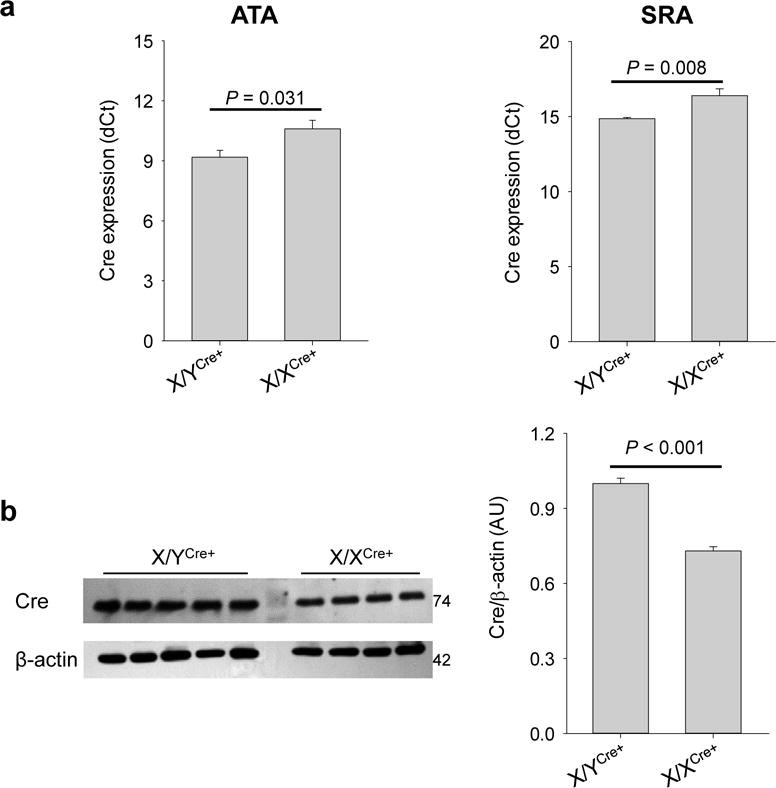
The X/XCre+ mouse line produces lower levels of Cre recombinase than the X/YCre+ mouse line prior to tamoxifen induction. (a) Expression of the Cre allele in ascending aortas (ATA) and suprarenal aortas (SRA) of mice not treated with tamoxifen. Data were dCt values calculated from the qRT-PCR assays. (b) Production of the Cre recombinase estimated with Western blotting assays. The intensity of the Cre blots (left panel) were normalized to β-actin expression and quantified in an arbitrary unit (AU). Data were analyzed using unpaired t-test.
Since the mice used in this study harbor floxed Tgfbr1 alleles, treatment with tamoxifen will delete SMC-specific Tgfbr1, leading to acute loss of TGFβ signaling in SMCs. It has been shown that abrogation of SMC TGFβ signaling inhibits Myh11 expression (Li et al., 2014; Yang et al., 2016b). We therefore examined the expression of the Cre recombinase at both the mRNA and protein levels in aortas over the period of tamoxifen induction. By the time of completion of tamoxifen injection (i.e. d5), expression of the Cre transgene in aortas remained at a difference of 1.0 cycles between X/XCre+ and X/YCre+ mice (n=5 per genotype, P=0.017, Figure 2a). Accordingly, X/XCre+ females produced about 50% Cre protein compared to X/YCre+ male mice (n=4–5 per genotype, P=0.003, Figure 2b). In agreement with the unchanged relative levels of Cre mRNA and protein between X/XCre+ and X/YCre+ mice on d5, no significant changes in the abundance of Cre recombinase was detected over the period of tamoxifen induction in aortas of mice on either genotype (n=4–5, Figure 2, c–d).
Figure 2.
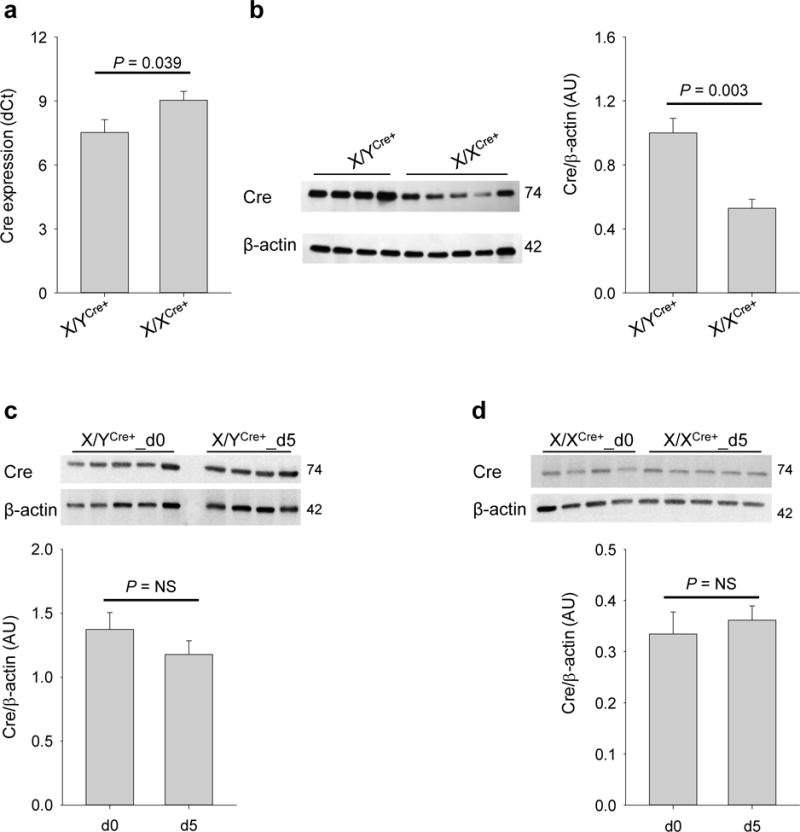
X/XCre+ mice produce lower levels of Cre recombinase than X/YCre+ mice after five doses of tamoxifen induction. (a) mRNA expression of the Cre allele in aortas of mice with the indicated genotype. Tissue samples were collected on the day following the last dose of tamoxifen (i.e., d5). (b) Protein abundance of the Cre recombinase in aortas of X/YCre+ and X/XCre+ mice on d5. (c-d) Changes of Cre abundance over the period of tamoxifen induction (from d0 to d5). Data were analyzed using unpaired t-test.
The Cre-driven recombination events in arteries of hemizygous X/XCre+ mice are mosaic but can be enhanced by prolonged duration of tamoxifen induction
The reduced expression and protein production of the Cre transgene in X/XCre+ mice raised a question concerning the recombination efficiency driven by this mouse line. To address this issue, we evaluated the recombination efficiency in arteries of X/XCre+ mice harboring an R26R reporter gene (n=4) and compared it to that of X/YCre+ mice (n=5). Since our previous studies demonstrate that sufficient recombination can be achieved in X/YCre+ mice after five doses of tamoxifen induction (Schmit et al., 2015; Yang et al., 2016a), the same protocol was followed in this experiment. Samples were collected on the day following the last dose of tamoxifen (i.e., d5). Consistent with our previous reports(Schmit et al., 2015; Yang et al., 2016a; Yang et al., 2016b), the recombination was confined to medial SMCs, as indicated by X-gal staining. The ATAs, CCAs, and FAs of X/XCre+ mice, however, displayed much more mosaic X-gal staining than did X/YCre+ mice (Figure 3a). As described in the methods section, X-gal staining prevented the counterstaining of X-gal positive but not X-gal negative nucleus by DAPI (4′,6-diamidino-2-phenylindole). An example is provided in Figure 3b (left panel), in which X-gal staining appeared in dark black and DAPI staining was assigned a pseudo color in pink. This differential response to DAPI counterstaining allowed us to count X-gal negative cells in the medial layer of the arteries. The total nuclei of the medial layer were obtained from the adjacent section. Specimens were stained with DAPI and imaged to record autofluorescence (green) and DAPI counterstain (pink) (Figure 3b, right panel). With these nuclear counts, the calculated recombination efficiency in ATAs, CCAs, and FAs was respectively 90%, 86%, and 83% for X/YCre+ mice and 56%, 44%, and 61% for X/XCre+ mice (Figure 3c, upper panel). Statistical analysis using two-way ANOVA revealed that the X/YCre+ mouse line induced significantly more recombination events than did the X/XCre+ strain in the examined arteries (P<0.001, Figure 3C, upper panel).
Figure 3.
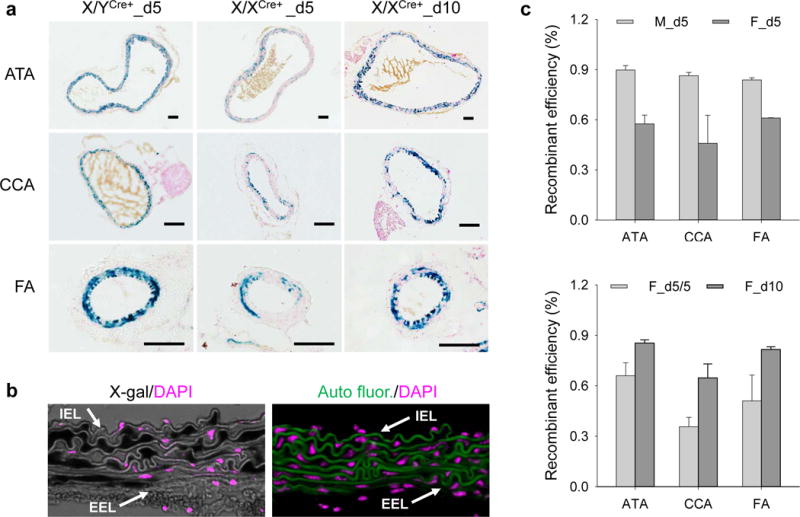
The X/XCre+ mouse line is less efficient than the X/YCre+ mouse line, but its efficiency can be enhanced by prolonged tamoxifen induction. (a) X-gal staining of arteries with indicated anatomic location, genotype, and duration of tamoxifen induction. Blue: positive X-gal staining; red: nuclear fast red counterstain; scale bars: 50 μm. (b) An example of imaging techniques utilized for assessing the recombinant efficiency. Left panel: X-gal staining (dark black) with DAPI counterstain (pink) of an ATA sample. Note the resistance of X-gal positive cells to DAPI counterstain. Right panel: DAPI counterstain of the section adjacent to that assigned to X-gal staining. Green: autofluorescence; pink: DAPI counterstain. Arrows point to internal and external elastic laminae (IEL and EEL, respectively). (c) Recombinant efficiency in muscular arteries. M and F represent X/YCre+ and X/XCre+ mice, respectively. Symbols d5, d10, and d5/5 indicate respectively 5, 10, and 5 tamoxifen doses for a duration of 5, 10, and 5 plus 5 days respectively. Data were analyzed using two-way ANOVA. M_d5 vs. F_d5: P<0.001; F_d5/5 vs. F_d10: P<0.001.
Since the efficiency of Cre-mediated recombination may depend on the duration of tamoxifen induction (Feil et al., 2009), we wondered whether a prolonged duration of tamoxifen administration would reduce the mosaicism of Cre activity in X/XCre+ mice. To address this issue, we treated a group of X/XCre+ mice (n=4) with ten tamoxifen doses and compared it to those receiving five administrations (n=3). Samples were collected ten days after the first injection (i.e., d10). Interestingly, the mosaicism of X-gal staining was reduced remarkably among medial SMCs of the treated arteries of X/XCre+ mice when the tamoxifen induction was increased from five to ten doses (Figure 3A). Compared with those receiving five doses of tamoxifen, mice treated with ten doses of tamoxifen displayed a significantly higher recombination efficiency (P<0.001, two-way ANOVA) in ATAs (85% vs. 66%), CCAs (65% vs. 36%), and FAs (82% vs. 51%). The difference of the recombination efficiency in ATAs and CCAs also reached the level of statistical significance (P=0.006, two-way ANOVA) (Figure 3c, lower panel).
The homozygous XCre+/XCre+ mouse line drives nearly homogeneous recombination in arterial SMCs
Although the recombination efficiency of the hemizygous X/XCre+ mouse line was improved through increasing the tamoxifen doses, it remained lower than that of the X/YCre+ counterparts. When receiving regular doses of tamoxifen induction, the hemizygous X/XCre+ mouse line achieved an inducibility of only 50% of the X/YCre+ strain at the levels of mRNA expression, protein production, and the resultant recombination. These results had led us hypothesize that the Cre allele carried by the X/XCre+ mouse line is subjected to random X chromosome inactivation. This hypothesis predicates that breeding the Cre allele into homozygosity would assure an active Cre expression in each individual SMCs. Due to the unknown insertion point of the Cre allele on the X chromosome, we performed qPCR to measure the copy number of this transgene in the genome. The gene Agtr1b, a homozygous allele located on the chromosome 3, was used as an internal reference for having two identical copies in the genome. Validation of the qPCR system was completed by varying the gDNA input by a serial of 2-fold reductions (data not shown). Using this validated quantitative system, we measured the content of the Cre transgene in Cre carriers with a known genotype of XCre+/Y (n=3) or X/XCre+ (n=3). Interestingly, the transgene consistently reached the threshold ahead of the Agtr1b by 0.60 ± 0.10 and 0.55 ± 0.08 cycles for XCre+/Y and X/XCre+ mice, respectively, with the differences between genotypes being statistically insignificant (P>0.05). As the reference gene Agtr1b has two copies, the 0.6 cycles of Ct differential corresponds to a copy number of 3, suggesting that the hemizygous mice carry 3 copies of the Cre gene on the X chromosome. The results obtained with the hemizygous mice established the copy number of 6 as the criteria for a Cre carrier being deemed homozygous. In screening of 29 offspring produced by a pair of XCre+/Y and X/XCre+ breeders, we identified 3 homozygotes in 9 female mice. All homozygous mice were grossly healthy. Figure 4a illustrates the genotyping data obtained for a hemizygous and a homozygous female mouse. The average dCt (CtCre-CtAgtr1b) value for the hemizygotes and homozygote is −0.6 and −1.5, which corresponds to copy numbers of 3 and 6, respectively. As aforementioned, the mice enrolled in the current study also carried an R26R reporter and were homozygous for the floxed Tgfbr1 allele. We have previously shown that acute loss of SMC Tgfbr1 causes thoracic aortic aneurysms and dissections (Schmit et al., 2015; Yang et al., 2016b). These genetic modifications allowed us to evaluate the Cre activity in arterial SMCs. We treated homozygous (n=3) and heterozygous (n=4) female mice with five consecutive doses of tamoxifen and collected the aortas 13 days after the first does of tamoxifen injection. Evans Blue was injected intravenously 30 min prior to tissue collection to assist visualization of early stage aortopathy(Schmit et al., 2015; Yang et al., 2016b). Consistent with the evaluation at d5 and d5/5, X-gal staining of X/XCre+ ATAs remained mosaic (53%) in medial SMCs at d13. In contrast, XCre+/XCre+ ATAs displayed nearly homogenous (91%) X-gal staining (Figure 4b). Gross examination under a dissection microscope detected only scarcely scattered blue spots on the luminal side of X/XCre+ ATAs, which corresponds with our previous report (Schmit et al., 2015). Evans Blue extravasation and elastic fiber breaks could be caught only occasionally (Figure 4, a–c, left panels). In agreement with the homogeneous Cre activity in the SMCs (Figure 4b), XCre+/XCre+ ATAs displayed grossly evident aortopathy featuring intramural hematoma, intimal/medial tears, and densely spreading Evans Blue staining on the luminal side. Early stage pathologies such as small intimal tears and elastic fiber breaks were frequently registered on cross sections (Figure 5, a–c, right panels).
Figure 4.
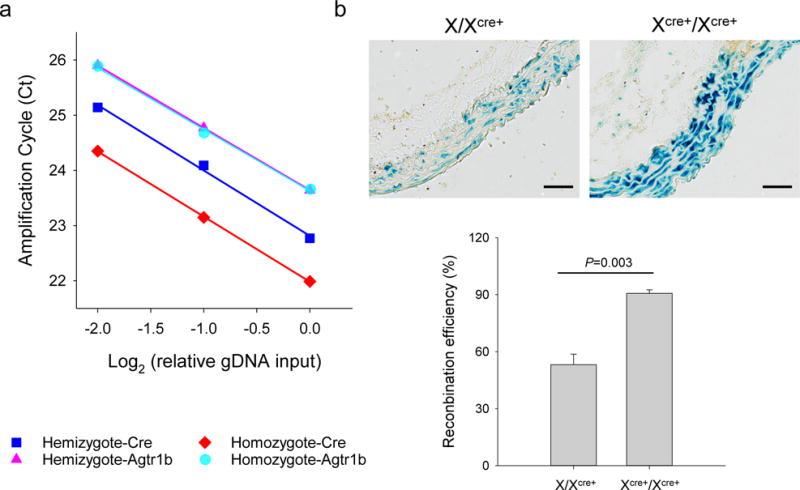
The homozygous XCre+/XCre+ mouse line is more efficient than the hemizygous X/XCre+ mouse line in activating expression of the R26R reporter gene in SMCs. (a) qPCR data obtained for a hemizygous X/XCre+ and a homozygous XCre+/XCre+ mouse. Note the one cycle difference of Ct values for the Cre transgene between these mice. (b) X-gal staining of ATAs. Blue: positive X-gal staining; scale bars: 50 μm. (c) Recombination efficiency in ATAs of female mice with the indicated genotype of the Cre transgene. Data were analyzed using unpaired t-test.
Figure 5.
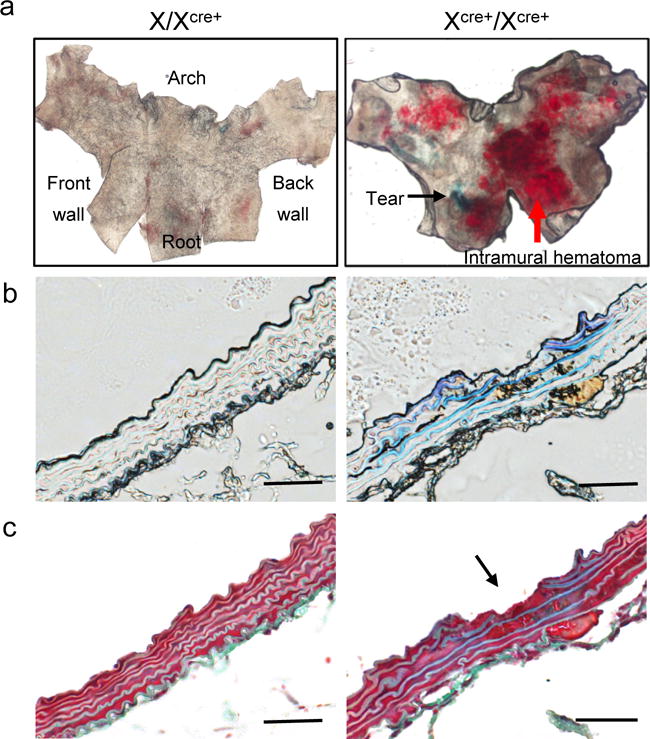
The homozygous XCre+/XCre+ mouse line unmasks the subclinical aortic phenotype driven by the hemizygous X/XCre+ strain in a model of SMC-specific Tgfbr1 deletion. (a) En face microscopy of the luminal surface of X/XCre+ and XCre+/XCre+ ATA segments. The black arrow points to an area of positive Evans Blue staining. The red arrow indicates intramural hematoma. (b) Bright field imaging of cross sections of ATAs with indicated genotype of the Cre transgene. Note the extravasation of Evans Blue in the XCre+/XCre+ ATA sample. (c) Masson’s staining of ATA specimens. The arrow points to an intimal/medial tear. Scale bars: 50 μm.
DISCUSSION
During the routine genotype-screening assays, we identified female carriers for the transgene Myh11-CreERT2 that was initially only carried by male mice on their Y chromosome (X/YCre+). With these carries, we established a new subcolony (named 2nd subcolony in this study) that stably passed the Cre allele to both male and female offspring. Subsequent cross-breeding experiments revealed that transmission of the Cre allele through a male breeder was restricted to female offspring. Therefore, the Cre allele carried by the 2nd subcolony must be located on the X chromosome (X/XCre+). Since this subcolony was established from a male mouse with the Cre allele on the Y chromosome, we concluded that the female Cre carriers received the Cre allele via Y to X chromosome-translocation. A limitation of the original X/YCre+ line is that female offspring could not receive the Cre allele, which has made it impossible for researchers to use this tool to decipher sexual dimorphism of the SMC biology in the pathogenesis of vascular diseases. To examine whether the X/XCre+ mouse line is a solution to this limitation, we characterized the function of the Cre allele under both hemizygous (X/XCre+) and homozygous (XCre+/XCre+) conditions. The results showed that expression of the Cre allele at the level of mRNA and protein as well as the Cre-driven recombination in the hemizygous X/XCre+ female mice were all reduced to a level of approximately 50% of that observed in X/YCre+ mice. Although this mosaicism of recombination was ameliorated through a prolonged duration of tamoxifen induction, it remained quite high compared to the homogeneous recombination in the hemizygous X/YCre+ male mice. We suspected that the mosaic Cre expression in X/XCre+ females is a consequence of the random X-chromosome inactivation. The results generated with homozygous XCre+/XCre+ female carriers supported this hypothesis. We found that the homozygous XCre+/XCre+ allele can activate the reporter gene almost uniformly in SMCs of ATAs. Furthermore, when evaluated with a model of aortic aneurysm formation induced by SMC-specific Tgfbr1 deletion, the XCre+/XCre+ female mice developed severe aortic aneurysmal degeneration, a phenotype that is otherwise rarely observed in hemizygous X/XCre+ carriers. Therefore, we have generated a novel inducible Cre line that allows sufficient Myh11-driven Cre expression in female mice.
The exchange in genetic materials between the X and Y chromosomes occurs primarily in a region with sequence identity, namely pseudoautosomal regions (PARs). During meiosis, genes located in the PARs of the X and Y chromosomes behaves like autosomal genes. As a result, a gene residing in PARs of a father’s Y chromosome will be passed to his daughters at a probability of 50%(White et al., 2012). For the X/YCre+ colony, the breeding results showed a stable transmission of the Cre allele exclusively to the male offspring. Although the exact location of the Cre allele was not defined by the original provider, the male-only transmission indicates that the Cre allele is unlikely to be located in one of the PARs. The t(Y;X) observed for the Cre allele, therefore, occurred most likely between the loci outside the PARs. In humans, t(Y;X) often result in an abnormal phenotype (McElreavey et al., 2001). Mice carrying the Cre allele on the X chromosome, however, are physically normal. The X/XCre+ breeders were reproductively fertile and passed the Cre allele to the offspring at the expected Mendelian frequency (Table 2). These results suggest that the t(Y;X) of the Cre allele resulted in only modest, if any, interruption to function of the reproductive system.
The evolutionary gene decay in the Y chromosome has led to a differential content of the X-linked genes between X/X and X/Y mammals. To balance the gene dosage, one of the two X chromosomes is silenced by X inactivation in X/X individuals (Bachtrog 2013). During murine embryonic development, although the paternal X chromosome always gets imprinted initially, it is reactivated in the blastocyst and reassumes the transcription thereafter at an equal probability to the maternal X chromosome (Berletch et al., 2011). As a result, cells in a given tissue or organ are mosaic in the parental origin of the active X-linked genes. This X inactivation, while balancing the dosage of endogenous genes, can disrupt the function of exogenous transgenes (Wutz 2011). For example, the hemizygous female mice carrying a CAG-EGFP reporter gene on the X chromosome produced EGFP proteins only in a subset of somatic cells (Hadjantonakis et al., 2001). In the current study, the F2 X/XCre+ mice received the Cre allele from a Y/XCre+ father. In concurrence with those previous reports, our results demonstrated a reduction in the expression, production, and function of the Cre transgene, with all the levels around 50% of those presented by Y/XCre+ males, indicating that the Cre allele was randomly silenced by X inactivation in X/XCre+ mice. It is estimated that only 3% of the native genes can escape from X inactivation in mice(Berletch et al., 2011). Whether a transgene can escape from X inactivation appears to depend on the promoter initially assembled to the expression cassette. For example, the Cre transgene is actively transcribed in almost all target cells of hemizygous females when driven by a Ggt1 promoter (Dworniczak et al., 2007). Although a full characterization of the in vivo response of the Myh11 promoter assembled to the Cre transgene is beyond the scope of the current study, we made a few interesting findings. First, a prolonged duration of tamoxifen induction turned >50% arterial SMCs of hemizygous X/XCre+ mice into X-gal positive. Such a dramatic improvement potentially conflicts with the theory of X inactivation. Although the duration of tamoxifen induction is a known factor for the recombination efficiency of Cre drivers (Agarwal et al., 2012; Feil et al., 2009), theoretically, the recombination efficiency achieved by X/XCre+ drivers should be capped at 50%. We suspect that this potential conflict was caused by the semi-quantitative nature of the X-gal staining or possibly reactivation of the inactivated Cre allele. In addition, we demonstrated a trend of reduction in the Cre production in SMCs following Tgfbr1 deletion. TGFβ is a stimulator of the endogenous Myh11 promoter in SMCs. Whether our results point towards the lack of requisite sequences for the exogenous Myh11 promoter to exhibit a full spectrum of response to the changes of molecular cues remains to be determined.
The X/XCre+ mouse line, although exhibiting mosaicism in its Cre activity among individual SMCs, is a valuable tool for modification of gene expression in arterial SMCs of female mice. For example, we have previously shown that an X/XCre+-mediated deletion of SMC Tgfbr1 results in a subclinical phenotype of aortic aneurysm formation in female mice. This genetic predisposition makes the animals highly responsive to environmental risk factors. Using this model, we were able to demonstrate protective and detrimental effects for female sex hormones and hypertension, respectively, on aortic aneurysm development (Schmit et al., 2015). Additionally, we have demonstrated that the hemizygous X/XCre+ Cre line can be breed to produce viable homozygous XCre+/XCre+ mice. Due to the presence of the Cre transgene on both X chromosomes, the Cre activity in individual SMCs is no longer mosaic, as evidenced by the homogeneous X-gal staining in the tunica media of aortas and the similar capacity of driving SMC specific Tgfbr1 deletion to the originally created X/YCre+ male mouse line. Finally, the hemizygous strain (i.e., X/XCre+) together with the homozygous strain (i.e., XCre+/XCre+) comprises a system where the varied mosaicism of Cre activity may allow in vivo investigations of interactions between SMCs with and without modification of a gene of interest.
Sex chromosomes, either X or Y, are occasionally selected for knock-in of exogenous transgenes. In addition to the X inactivation and exclusion of female transmission of the Myh11-CreERT2 allele, the t(Y;X) phenomenon exhibited by this transgene suggests that inheritance of a transgene integrated into the sex chromosomes may not always follow the predicated pattern. Although other inducible Cre lines such as Acta2-CreERT2 and SM22-CreERT2 do not have these issues, the off-target effect of these Cre drivers in the myeloid-derived cells and other non-SMC lineages (Shankman et al., 2015; Shen et al., 2012) may confound data interpretation, particularly under conditions with preexisting vascular pathologies. The Myh11 gene is the most definitive marker identified to date for vascular SMCs (Shankman et al., 2015). The X/XCre+ and XCre+/XCre+ mouse lines, therefore, represent novel tools that are complementary to the previously created Cre lines.
MATERIALS AND METHODS
Animals
This study conforms to the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Institutional Animal Care and Use Committee of the University of Florida. The Myh11-CreERT2 strain (two male mice) was kindly provided by Dr. Stefan Offermanns (University of Heidelberg). The Tgfbr1-floxed (Tgfbr1f/f) mouse line was kindly provided by Dr. Paul S. Oh (University of Florida). The R26R strain was purchased from the Jackson Laboratory. All animals were backcrossed to C57BL/6J for at least five generations The Myh11-CreERT2 and R26R mice were crossed with the Tgfbr1f/f breeders. Offspring progenies were genotyped using PCR-based genotyping assays with the primers specified in Table 4. Mice with a genotype of Tgfbr1f/f;R26R+ were then crossed with Tgfbr1f/f;Myh11-CreERT2+ mice to establish the Tgfbr1f/f;R26R+;Myh11-CreERT2+ colony (denoted as Cre+ in this study). This colony was maintained by breeding Cre+ male mice with Cre− female mice. As reported previously (Schmit et al., 2015; Yang et al., 2016a), tamoxifen (2.5 mg/mouse, Sigma, T5648) was administered to mice at an age of 9–11 weeks via a daily intraperitoneal injection. All animals received five consecutive doses of tamoxifen unless otherwise specified.
Table 4.
Primers used for genotype screening of Cre and R26R carriers
| Gene | Primers | Band Size |
|---|---|---|
| Cre | P1: 5′-tgaccccatctcttcactcc-3′ P2: 5′-aactccacgaccacctcatc-3′ P3: 5′-agtccctcacatcctcaggtt-3 |
Wt: 225 bps Mutant: 225 bps + 287 bps |
| R26R | P1: 5′- gcgaagagtttgtcctcaacc-3′; P2: 5′- aaagtcgctctgagttgttat-3′ P3: 5′- ggagcgggagaaatggatatg-3 |
Wt: 650 bps Mutant: 300 bps |
Cross-breeding experiments
The first cross-breeding experiment was set up with breeding pairs consisting of two Cre+ female mice and two Cre− male mice. Following this step, an F1 Cre+ male mouse produced in the first cross-breeding experiment was mated with two Cre− female mice to generate F2 offspring. All F1 and F2 mice were genotyped for the Cre and R26R alleles.
Gene expression
Total RNA was isolated using the RNeasy® Plus micro kit (QIAGEN, 74004) and reverse transcribed to cDNA templates (cDNA reverse transcription Kit, AB applied Biosystems, 4368814). Quantitative (q)RT-PCR (TaqMan; Applied Biosystems, 4488593) was performed as we have described previously (Fu et al., 2012). The primer and probe set specific for codon-improved Cre recombinase (Cre) was purchased from Applied Biosystems (Mr00733719_cn). Expression of the Cre gene was normalized to 18s rRNA. Assays were carried out in triplicate with an ABI Prism 7500 system.
Western blot
Total proteins were extracted with a Tris·HCl lysis buffer containing a cocktail of protease inhibitor (Thermo Fisher, 78430). After determination of the protein concentration with BCA assays (Thermo Fisher, 23225), 10 μg total protein of each sample were separated with SDS PAGE gels. A rabbit mAb (1: 1000; Cell Signaling, D3U7F) was applied to detect the Cre-specific band. The immuno-blots were developed with chemiluminescent substrate (Lumigen, TMA-6) and quantified for their intensity using a Kodak imager. Data were expressed as intensity normalized to β-actin expression.
Zygosity determination
The copy number of the Cre transgene integrated into the genome of X/YCre, X/XCre, and XCre/XCre strains was determined using qPCR assays with genomic DNA templates. This method was adapted from a previous report (Sakurai et al., 2008). In brief, genomic DNA was extracted from tail snips (ZR Genomic DNA-Tissue MiniPrep Kit, ZymoResearch). The amplification efficiency of each primer and probe set was evaluated by measuring changes of the Ct values between reactions loaded with a 2-fold differential of gDNA input with the maximum loading amount being 40 ng. After testing a panel of primer and probe sets specific for different autosome genes, we selected Agtr1b as an internal reference for having two identical copies in the genome because of the similar efficiency of the primer and probe sets for the Agtr1b gene (Mm02620758_s1, Applied Biosystems) and Cre transgene (Mr00733719_cn, Applied Biosystems). The copy number of the Cre transgene integrated into a single X chromosome was determined with mice with a known genotype of XCre/Y or X/XCre. Using the validated protocol, mice doubling the copy number of the Cre transgene were deemed as homozygotes.
X-gal staining
Samples were fixed with 4% paraformaldehyde on ice for 30 min, incubated with 30% sucrose in PBS overnight, and embedded in OCT compound. Frozen sections (8.0 μm) were then cut from the OCT blocks, air-dried, and incubated with X-gal staining solution (Mirus, MIR 2600) at 37C° overnight. Three sets of adjacent sections were collected for each sample. The first set was stained with X-gal reagents, counterstained with nuclear fast red, and imaged using a color camera to record X-gal staining. The second set was also stained with X-gal reagent, but the counterstain was finished with DAPI instead of nuclear fast red at a concentration of 0.1 μM. At this concentration, we found that the nuclei of cells with positive X-gal staining displayed a very faint and often non-detectable DAPI signal, whereas those negative for X-gal staining presented an intense DAPI signal. The X-gal staining illuminated under a bright field and the DAPI fluoresce were recorded using a monochrome camera to separate channels and then merged into a single image. The merged image allowed us to define the region of the medial layer by tracing the internal and external elastic laminae as well as to identify and count X-gal negative cells located in the tunica media. Sections of the third set were stained with DAPI and illuminated to record autofluorescence and DAPI signal. Total nuclei in the medial layer were then obtained using the Interactive Measurement module of Zen lite (Zeiss), and the recombination efficiency was calculated as the percentage of X-gal positive cells in the tunica media. Two cropped images to illustrate this method are provided in Figure 3B.
Histology
This procedure was performed as we have previously described (Schmit et al., 2015; Yang et al., 2016b). In brief, Evans Blue (0.2 ml 5% in saline) was administrated to mice via the tail vein 30 minutes prior to opening the chest. After perfusion fixation with 10% neutral buffered formalin, the ATA segments were cut open longitudinally, mounted on slides with the luminal surface facing coverslip, and imaged using bright field illumination. Cross-sections (5 μm-thick) were then made and stained using Masson’s trichrome methods.
Statistics analysis
Data are expressed as mean ± SEM. Bayes factor analysis was performed using SPSS to determine the significance of differences in the frequency of female and male Cre carriers. Other quantitative data were analyzed using unpaired t test, one-way ANOVA, or two-way ANOVA as appropriate. Data sets failing the normality and/or equal variance tests were transformed using the functions provided by SigmaPlot (version 13) to meet these assumptions. A P value less than 0.05 was considered statistically significant.
Acknowledgments
Supported by NIH1R01HL105764 (Zhihua Jiang), 7JK05 (Zhihua Jiang), and China National Natural Science Foundation 81670429 (Zhisheng Jiang)
References
- Agarwal A, Dibaj P, Kassmann CM, Goebbels S, Nave KA, Schwab MH. In vivo imaging and noninvasive ablation of pyramidal neurons in adult NEX-CreERT2 mice. Cereb Cortex. 2012;22:1473–1486. doi: 10.1093/cercor/bhr214. [DOI] [PubMed] [Google Scholar]
- Bachtrog D. Y-chromosome evolution: emerging insights into processes of Y-chromosome degeneration. Nat Rev Genet. 2013;14:113–124. doi: 10.1038/nrg3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berletch JB, Yang F, Xu J, Carrel L, Disteche CM. Genes that escape from X inactivation. Hum Genet. 2011;130:237–245. doi: 10.1007/s00439-011-1011-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dworniczak B, Skryabin B, Tchinda J, Heuck S, Seesing FJ, Metzger D, Chambon P, Horst J, Pennekamp P. Inducible Cre/loxP recombination in the mouse proximal tubule. Nephron Exp Nephrol. 2007;106:e11–e20. doi: 10.1159/000100554. [DOI] [PubMed] [Google Scholar]
- Feil S, Valtcheva N, Feil R. Inducible Cre mice. Methods Mol Biol. 2009;530:343–363. doi: 10.1007/978-1-59745-471-1_18. [DOI] [PubMed] [Google Scholar]
- Frutkin AD, Shi H, Otsuka G, Leveen P, Karlsson S, Dichek DA. A critical developmental role for tgfbr2 in myogenic cell lineages is revealed in mice expressing SM22-Cre, not SMMHC-Cre. J Mol Cell Cardiol. 2006;41:724–731. doi: 10.1016/j.yjmcc.2006.06.067. [DOI] [PubMed] [Google Scholar]
- Fu C, Yu P, Tao M, Gupta T, Moldawer LL, Berceli SA, Jiang Z. Monocyte chemoattractant protein-1/CCR2 axis promotes vein graft neointimal hyperplasia through its signaling in graft-extrinsic cell populations. Arterioscler Thromb Vasc Biol. 2012;32:2418–2426. doi: 10.1161/ATVBAHA.112.255786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez D, Owens GK. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc Res. 2012;95:156–164. doi: 10.1093/cvr/cvs115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjantonakis AK, Cox LL, Tam PP, Nagy A. An X-linked GFP transgene reveals unexpected paternal X-chromosome activity in trophoblastic giant cells of the mouse placenta. Genesis. 2001;29:133–140. doi: 10.1002/gene.1016. [DOI] [PubMed] [Google Scholar]
- Hoofnagle MH, Thomas JA, Wamhoff BR, Owens GK. Origin of neointimal smooth muscle: we’ve come full circle. Arterioscler Thromb Vasc Biol. 2006 Dec;26(12):2579–81. doi: 10.1161/01.ATV.0000249623.79871.bc. 2006. 26, 2579–2581. [DOI] [PubMed] [Google Scholar]
- Hu JH, Wei H, Jaffe M, Airhart N, Du L, Angelov SN, Yan J, Allen JK, Kang I, Wight TN, Fox K, Smith A, Enstrom R, Dichek DA. Postnatal Deletion of the Type II Transforming Growth Factor-beta Receptor in Smooth Muscle Cells Causes Severe Aortopathy in Mice. Arterioscler Thromb Vasc Biol. 2015;35:2647–2656. doi: 10.1161/ATVBAHA.115.306573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhbandner S, Brummer S, Metzger D, Chambon P, Hofmann F, Feil R. Temporally controlled somatic mutagenesis in smooth muscle. Genesis. 2000;28:15–22. doi: 10.1002/1526-968x(200009)28:1<15::aid-gene20>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Li L, Miano JM, Mercer B, Olson EN. Expression of the SM22alpha promoter in transgenic mice provides evidence for distinct transcriptional regulatory programs in vascular and visceral smooth muscle cells. J Cell Biol. 1996;132:849–859. doi: 10.1083/jcb.132.5.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Li Q, Jiao Y, Qin L, Ali R, Zhou J, Ferruzzi J, Kim RW, Geirsson A, Dietz HC, Offermanns S, Humphrey JD, Tellides G. Tgfbr2 disruption in postnatal smooth muscle impairs aortic wall homeostasis. J Clin Invest. 2014;124:755–767. doi: 10.1172/JCI69942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElreavey K, Cortes LS. X-Y translocations and sex differentiation. Semin Reprod Med. 2001;19:133–139. doi: 10.1055/s-2001-15393. [DOI] [PubMed] [Google Scholar]
- Mosca L, Barrett-Connor E, Wenger NK. Sex/gender differences in cardiovascular disease prevention: what a difference a decade makes. Circulation. 2011;124:2145–2154. doi: 10.1161/CIRCULATIONAHA.110.968792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen AT, Gomez D, Bell RD, Campbell JH, Clowes AW, Gabbiani G, Giachelli CM, Parmacek MS, Raines EW, Rusch NJ, Speer MY, Sturek M, Thyberg J, Towler DA, Weiser-Evans MC, Yan C, Miano JM, Owens GK. Smooth muscle cell plasticity: fact or fiction? Circ Res. 2013;112:17–22. doi: 10.1161/CIRCRESAHA.112.281048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- Regan CP, Manabe I, Owens GK. Development of a smooth muscle-targeted cre recombinase mouse reveals novel insights regarding smooth muscle myosin heavy chain promoter regulation. Circ Res. 2000;87:363–369. doi: 10.1161/01.res.87.5.363. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Kamiyoshi A, Watanabe S, Sato M, Shindo T. Rapid zygosity determination in mice by SYBR Green real-time genomic PCR of a crude DNA solution. Transgenic Res. 2008;17:149–155. doi: 10.1007/s11248-007-9134-7. [DOI] [PubMed] [Google Scholar]
- Schmit BM, Yang P, Fu C, DeSart K, Berceli SA, Jiang Z. Hypertension overrides the protective effect of female hormones on the development of aortic aneurysm secondary to Alk5 deficiency via ERK activation. Am J Physiol Heart Circ Physiol. 2015;308:H115–H125. doi: 10.1152/ajpheart.00521.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, Swiatlowska P, Newman AA, Greene ES, Straub AC, Isakson B, Randolph GJ, Owens GK. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med. 2015;21:628–637. doi: 10.1038/nm.3866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Z, Li C, Frieler RA, Gerasimova AS, Lee SJ, Wu J, Wang MM, Lumeng CN, Brosius FC, III, Duan SZ, Mortensen RM. Smooth muscle protein 22 alpha-Cre is expressed in myeloid cells in mice. Biochem Biophys Res Commun. 2012;422:639–642. doi: 10.1016/j.bbrc.2012.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendling O, Bornert JM, Chambon P, Metzger D. Efficient temporally-controlled targeted mutagenesis in smooth muscle cells of the adult mouse. Genesis. 2009;47:14–18. doi: 10.1002/dvg.20448. [DOI] [PubMed] [Google Scholar]
- White MA, Ikeda A, Payseur BA. A pronounced evolutionary shift of the pseudoautosomal region boundary in house mice. Mamm Genome. 2012;23:454–466. doi: 10.1007/s00335-012-9403-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth A, Benyo Z, Lukasova M, Leutgeb B, Wettschureck N, Gorbey S, Orsy P, Horvath B, Maser-Gluth C, Greiner E, Lemmer B, Schutz G, Gutkind JS, Offermanns S. G12-G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat Med. 2008;14:64–68. doi: 10.1038/nm1666. [DOI] [PubMed] [Google Scholar]
- Wu Z, Yang L, Cai L, Zhang M, Cheng X, Yang X, Xu J. Detection of epithelial to mesenchymal transition in airways of a bleomycin induced pulmonary fibrosis model derived from an alpha-smooth muscle actin-Cre transgenic mouse. Respir Res. 2007;8:1. doi: 10.1186/1465-9921-8-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wutz A. Gene silencing in X-chromosome inactivation: advances in understanding facultative heterochromatin formation. Nat Rev Genet. 2011;12:542–553. doi: 10.1038/nrg3035. [DOI] [PubMed] [Google Scholar]
- Yang P, Hong MS, Fu C, Schmit BM, Su Y, Berceli SA, Jiang Z. Preexisting smooth muscle cells contribute to neointimal cell repopulation at an incidence varying widely among individual lesions. Surgery. 2016a;159:602–612. doi: 10.1016/j.surg.2015.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang P, Schmit BM, Fu C, DeSart K, Oh SP, Berceli SA, Jiang Z. Smooth muscle cell-specific Tgfbr1 deficiency promotes aortic aneurysm formation by stimulating multiple signaling events. Sci Rep. 2016b;6:35444. doi: 10.1038/srep35444. [DOI] [PMC free article] [PubMed] [Google Scholar]