

Abstract
Purpose of Review
Gonorrhea is a major global health concern, caused by the bacterium Neisseria gonorrhoeae. The main clinical feature of acute gonorrhea is neutrophilic influx that is unable to clear infection. Women of reproductive age are predominantly at risk for serious sequelae of gonorrhea, including pelvic inflammatory disease, ectopic pregnancy, and infertility. This review will highlight how neutrophils are recruited to the female reproductive tract (FRT) in response to N. gonorrhoeae, how N. gonorrhoeae resists killing by neutrophils, and the connection between neutrophilic inflammation and cellular damage.
Recent Findings
Epithelial cells and immune cells of the FRT recognize and respond to N. gonorrhoeae lipid A and heptose bisphosphate of lipooligosaccharide, porin, lipoproteins, and peptidoglycan fragments. N. gonorrhoeae skews the resulting immune response towards an neutrophilic, Th17-like response. N. gonorrhoeae has multiple, non-redundant mechanisms to survive inside neutrophils and in neutrophil extracellular traps. Infection that ascends to the upper FRT induces the further release of inflammatory cytokines and matrix metalloproteinases, which cause epithelial damage.
Summary
N. gonorrhoeae is remarkable in its ability to recruit neutrophils, yet survive in their midst. New models being developed for FRT infection with N. gonorrhoeae will be useful to reveal the mechanisms underlying these observations.
Keywords: neutrophil, Neisseria gonorrhoeae, female reproductive tract, inflammation, infectious diseases
INTRODUCTION
The Gram-negative diplococcus Neisseria gonorrhoeae (the gonococcus or Gc) causes the sexually transmitted infection gonorrhea, with an estimated 78 million cases worldwide each year [1]. Increasing rates of infection, the emergence of multidrug-resistant strains, and the lack of a protective vaccine have prompted the CDC and WHO to classify Gc as a top infectious threat [1]. Women are particularly at risk for negative outcomes associated with gonorrhea, including pelvic inflammatory disease (PID), ectopic pregnancy, and infertility [2].
Infection elicits a robust inflammatory response featuring the influx of neutrophils. Despite this potent immune response, secretions from infected individuals contain viable bacteria, many of which are associated with neutrophils [3]. Sustained neutrophil influx in other infectious conditions has been linked to epithelial cell damage and pathology associated with disease [4,5,6]. We posit that the ability of Gc to elicit neutrophilic inflammation in the female reproductive tract (FRT) is central to its ability to persist within its obligate human host and be transmitted to new hosts, while contributing to the negative outcomes in women. This review will highlight recent contributions to our understanding of the neutrophilic inflammatory response to Gc in the FRT, the infection models underlying these discoveries, and opportunities for the field moving forward.
GC STIMULATES A NON-PROTECTIVE NEUTROPHILIC INFLAMMATORY RESPONSE IN THE FRT
The initial site of Gc infection in women is the endocervix, the transition from the lower to upper FRT (Figure 1). Despite neutrophilic cervicitis, the majority of women with cervical gonorrhea do not report symptoms [reviewed in 7,15]. Although genital mucosal secretions contain cationic antimicrobial peptides (CAMPs) and other bactericidal components, Gc survives in their midst (Figure 1). The MisR-MisS two-component regulatory system confers inducible resistance in Gc to CAMPs by directing the expression of genes important for envelope integrity [12*], which enhances Gc colonization and extends infection duration in a mouse model of lower FRT gonorrhea [13*].
Figure 1. Gc initially infects the endocervix, a transition from the lower FRT to the upper FRT.
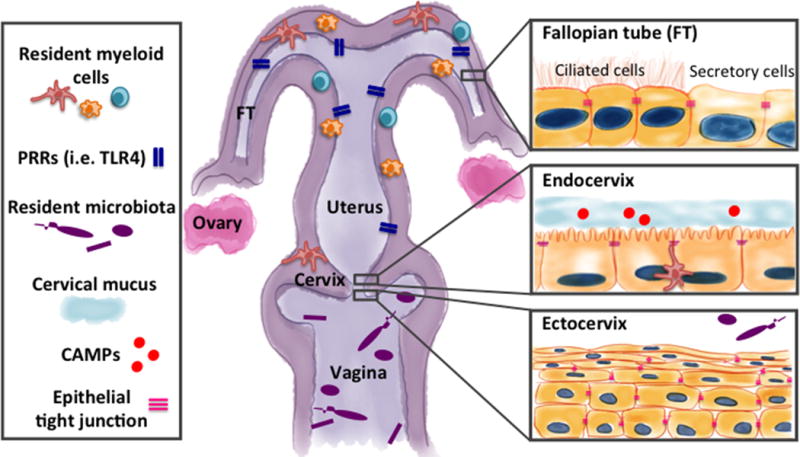
The endocervix marks a transition from multilayered squamous epithelium lining the lower FRT to single columnar epithelium lining the upper reproductive tract. Epithelial cells provide a number of barriers to infection including maintaining epithelial tight junctions and barrier integrity, producing thick cervical mucus, and producing and secreting cationic antimicrobial peptides (CAMPs) that accumulate in cervical mucus [reviewed in 8]. Gc factors contributing to survival from CAMPs in the genital tract include expression of the MtrCDE efflux pump, modification of lipid A with phosphoethanolamine (see Table 1 for more information) [9,10,11], and the MisR-MisS two-component regulatory system [12*,13*]. The transition from the lower to the upper FRT is also marked by changes in microbiota, pattern-recognition receptor (PRR) expression, and myeloid cell frequency [reviewed by 14]. While the lower FRT has a resident microbiota, low PRR expression, and low myeloid cell frequency, the upper FRT is considered sterile and has increased PRR expression and myeloid cell frequencies.
The recruitment and activation of neutrophils in response to Gc is coordinated by cellular and soluble factors. In the FRT, Gc interacts with epithelial and immune cells, including macrophages, dendritic cells, T cells, and neutrophils, to elicit the local production of inflammatory mediators and activation of a Th17-type response (Figure 2) [27,28,29,30*]. Moreover, Gc infection was recently shown to activate non-muscle myosin II in human cervical tissue, leading to epithelial junctional disruption, exfoliation of endocervical cells, and bacterial subepithelial penetration [31*].
Figure 2. Epithelial and resident immune cells initiate the immune response to Gc.
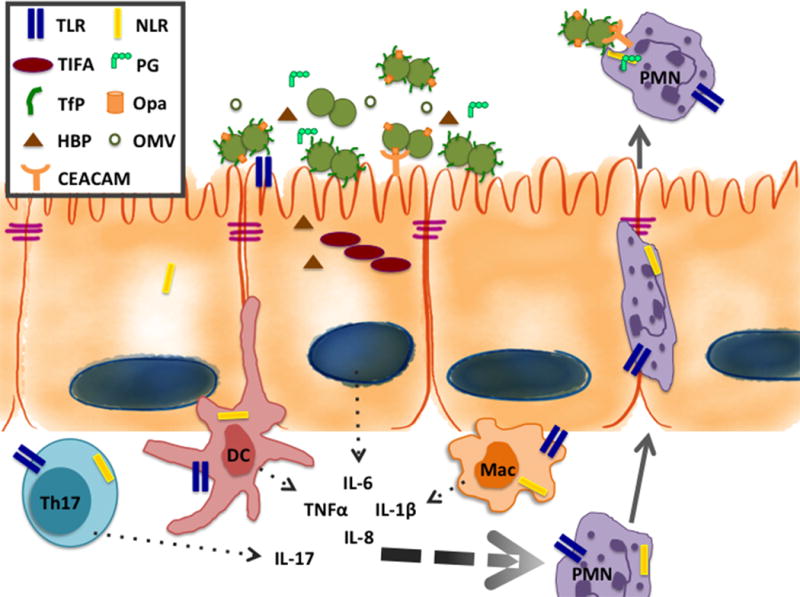
Gc uses adhesins such as type IV pili and opacity-associated (Opa) proteins to interact with the epithelial cells lining the human FRT [reviewed in 7]. The roles of several important Gc surface structures, including opacity associated (Opa) proteins, the type IV pilus (TfP), lipooligosaccharide (LOS), and porin, have been well defined in mediating adherence, invasion, and host cell signaling. During infection, there is a mixture of Gc with varied expression of Opa proteins and the TfP. Opa proteins interact with CEACAMs on host epithelial and immune cells to influence adherence, internalization, and signaling. PRRs on epithelial and resident immune cells recognize PAMPs presented on the surface of Gc and/or released from the bacterium freely or as components of outer membrane vesicles (OMVs). In addition to well-known PAMPs such as lipooligosaccharide (LOS) and PG, the Gc cell envelope and outer membrane vesicles (OMVs) may harbor additional factors that affect the host response to infection [16]. Cervical epithelial cells respond to Gc through stimulation of TLR2 but not TLR4, while resident immune and upper FRT cells, including fallopian tube epithelial cells, are particularly poised to respond to LOS via TLR4 [17,18]. Gc porin and other lipoproteins stimulate TLR2 to activate NFkB-driven inflammatory cytokine production [19,20]. Similarly, TLR4 is potently activated by the lipid A portion of LOS from Gc and its close relative, Neisseria meningitidis [reviewed in 21]. While surface-expressed TLRs recognize Gc LOS, porin, and other lipoproteins, PG is primarily recognized by intracellular receptors. The best-described intracellular receptors for PG are NOD1, which recognizes gamma-glutamyl-diaminopimelic acid, and NOD2, which recognizes muramyl dipeptide [reviewed in 22]. These receptors are expressed in epithelial cells as well as sentinel innate immune cells. Human NOD1 and NOD2, and mouse NOD2, recognize and mount an NF-kB-driven innate immune response to PG from Gc, in the context of whole bacteria or to PG fragments in conditioned media [23]. An additional mechanism of intracellular detection of gram-negative bacteria is the TIFA-dependent detection of heptose-1,7-bisphosphate (HBP), an intermediate in LPS/LOS production [24**,25,26*]. As a result, epithelial and resident myeloid cells secrete pro-inflammatory cytokines, including TNFα, IL-1β, IL-6, and IL-8 and sentinel Th17 cells release IL-17. The combination of these cytokines creates a cytokine gradient that serves to recruit neutrophils from the bloodstream and activate them at the site of infection.
PRR activation by Gc PAMPs
During infection, cytokine production is driven by signaling via epithelial and resident immune cell pattern recognition receptors (PRRs) that recognize Gc pathogen-associated molecular patterns (PAMPs) (Figure 2). Levels of PRRs, particularly TLR4, which recognizes the lipid A moiety of Gc lipooligosaccharide (LOS), and numbers of myeloid cells both increase from the lower to upper FRT, such that an immune response is mounted only to pathogens that ascend into the upper FRT and not to the resident microbiota of the lower FRT (Figure 1). PRRs include membrane-associated Toll-like receptors (TLRs) and cytoplasmic NOD-like receptors (NLRs) that recognize a variety of Gc PAMPs (Figures 1 & 2) [22,32,33]. Recent work from Gray-Owen and colleagues has identified a new PAMP from Gc, the LOS biosynthesis pathway intermediate heptose-1,7-bisphosphate (HBP) [24**,25,26*]. Activation of NFkB signaling by HBP occurs via phosphorylation-dependent oligomerization of TRAF-interacting protein with forkhead-associated domain (TIFA) [24**]. Recognition of HBP represents a novel mechanism to detect and mount an immune response to Gram-negative bacterial pathogens [26*]; this may be particularly important for signaling in sentinel cells that lack TLR4 expression, such as cervical epithelial cells.
Gc modulates innate immune cell responses
There is strong evidence to suggest that Gc skews innate immune cell recognition and response as an element of its survival strategy. Addition of phosphoethanolamine (PEA) to lipid A by the enzyme LptA, which is found in the pathogenic Neisseria, not only enhances TLR4 recognition to stimulate NFkB-driven cytokine production, but also aids the bacteria in defense against CAMPs found in mucosal secretions and neutrophils [10,34,35*]. Therefore, the potentially damaging effects of lipid A-mediated activation of TLR4 and subsequent inflammation are mitigated by the intrinsic defense against killing that is conferred by modifying lipid A with PEA. Another example was recently elucidated by Golenbock and colleagues, who showed that exposure to Gc leads to STING-dependent activation of the intracellular DNA sensor cyclic GMP-AMP synthase (cGAS) in monocytes and macrophages [36**]. In combination with TLR4 activation, this stimulates the production of IFN-β. Rather than helping to control infection, IFN-β increases the availability of intracellular iron in macrophages and neutrophils, which is correlated with enhanced survival of Gc [36**]. Gc may have additional mechanisms to survive and replicate in association with macrophages [37*], including through the expression of surface-exposed factors such as macrophage infectivity potentiator-like protein (MIP) [38,39*].
Not all features of Gc are immunostimulatory. Gc has been reported to polarize macrophages to an immune-regulatory, M2 phenotype, which would downregulate the antimicrobial activity and proinflammatory cytokine production of macrophages [40*]. Moreover, while the peptidoglycan (PG) fragments released by Gc are toxic to Fallopian tube cells, they are poorly recognized by innate immune receptors. Specifically, Duncan and Dillard and colleagues recently found that the PG monomers released by Gc, via the activities of the lytic transglycosylases LtgA and LtgD, are poor activators of mouse NOD2 and TLR2, compared with multimeric PG. This is due to the anhydro moiety on the terminus of Ltg-cleaved fragments, rather than a free reducing (hydroxyl) end, as would be found following digestion by lysozyme [41**]. These studies suggest that the large amounts of PG fragments released by Gc during normal growth may serve as decoys, to limit PRR activation in the lower FRT and enhance overall Gc survival.
Gc manipulates the adaptive immune response, skewing the inflammatory environment to attract and activate neutrophils
Despite the proinflammatory nature of Gc PAMPs, the subsequent host immune response is not sufficient to clear infection. Gc has a remarkable ability to evade host antibody-mediated immunity, due to extensive antigenic and phase variation of its immunogenic surface structures as well as by expression of Rmp, which limits the generation of bactericidal antibodies [42,43]. Gc also manipulates cellular immune responses to limit adaptive immune cell activation and direct the immune response towards a neutrophilic, non-protective presentation, as described below.
CEACAM1-mediated limitation of immune cell activation
Most members of the family of opacity-associated (Opa) outer-membrane proteins of Gc interact with human CEACAM1, which is expressed on T cells, B cells, dendritic cells, and epithelial cells. CEACAM1 has an immunoreceptor tyrosine-based inhibition motif on its cytoplasmic tail, which recruits the SHP phosphatase to block signaling in trans from activating receptors [44]. Engagement of CEACAM1 by Opa+ Gc also drives bacterial internalization into epithelial cells, within which Gc can survive while avoiding exposure to CAMPs, antibodies, complement factors, and other bactericidal components. The importance of the Opa-CEACAM1 interaction to the pathogenesis of gonorrhea is reflected in the strong selection for CEACAM1-binding Gc in vivo [45**]. However, the opa genes phase-vary at high frequency and diversify by recombination and mutation, changing their ability to engage CEACAM1 [46].
TH17 response
Gc infection induces the local production of TGF-β and IL-10 to skew the resulting adaptive response away from Th1 and Th2-driven immunity and towards a Th17 response, which in the context of gonorrhea enhances inflammation and prevents the development of protective immunity [47,48,49,15]. Th17 cells produce IL-17, which is important for neutrophil recruitment in the human and mouse lower FRT in response to Gc [30*,50]. The Russell group has proposed reorientation of the T cell response to Gc away from Th17 and towards Th1 as a therapeutic intervention to ameliorate neutrophilic inflammation. In support of this possibility, female mice immunized with Gc antigens in combination with IL-12, a Th1-activating cytokine, clear Gc more rapidly when first infected and are protected against reinfection [51**].
GC THWARTS NEUTROPHIL FUNCTIONS TO ENHANCE ITS SURVIVAL AND PROMOTE CONTINUED INFLAMMATION
The predominant clinical feature of acute gonorrhea is the presence of viable Gc in association with neutrophils in mucosal secretions. While neutrophils have a robust antimicrobial arsenal (Figure 3), our laboratory and others have identified mechanisms Gc uses to evade killing by neutrophils (Figure 3 & Table 1). Antimicrobial components are a crucial part of neutrophils’ functionality, but they have the potential to damage host cells. Reactive oxygen species (ROS) oxidize lipids, proteins, and DNA, and also serves as a second messenger to enhance inflammation and neutrophil recruitment [71]. Proteases, including neutrophil-derived matrix metalloproteases, degrade tissue extracellular matrix and contribute to sustained neutrophil recruitment [reviewed in 5]. The CAMP LL-37 interacts with host cell receptors including TLR4 and contributes to their continued activation during infection and inflammation [72]. Histones, released from neutrophils in neutrophil extracellular traps (NETs), induce direct epithelial cell damage [73]. NETs are also thought to contribute to the pathology associated with autoimmune diseases by providing self-antigen (such as in Systemic Lupus Erythematosus), and with cardiovascular disease by enhancing formation of atherosclerotic plaques [74,75]. NETs are formed in response to Gc, but the bacteria have multiple mechanisms to survive in association with NETs, including expression of a nuclease (Nuc) that degrades NET DNA (Figure 3 & Table 1) [59*,60*,61*]. Although the contribution of Gc-induced NETs to cellular damage remains to be elucidated, it is likely that Nuc could release CAMPs and histones from NETs, to directly damage epithelial cells and stimulate continued inflammation.
Figure 3. Neutrophils possess an arsenal of extracellular and intracellular killing mechanisms that Gc can evade, leading to off-target host cell damage.
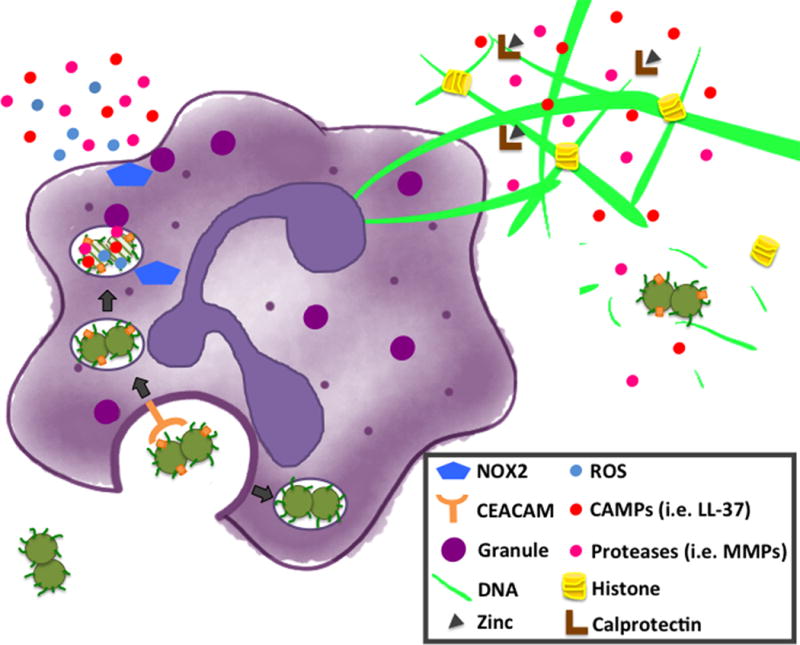
Neutrophil killing mechanisms include phagocytosis, production of reactive oxygen species (ROS), degranulation, and neutrophil extracellular trap (NET) formation. Granule components include cationic antimicrobial proteins (CAMPs; i.e. LL-37) and proteases (i.e. MMPs). Gc has evolved elegant mechanisms to resist neutrophil killing [see 3, 27 for overviews]. An important example is variation in Opa protein expression, which can affect neutrophil activation and Gc survival. In women, Gc expressing Opa proteins are predominantly recovered during the follicular phase of the menstrual cycle, while Opa-negative Gc predominate in the luteal phase and in upper FRT infection [7]. Opa-expressing Gc are phagocytosed in a CEACAM-dependent manner and trafficked to mature phagosomes while Opa-deficient Gc survive longer in immature phagosomes. Opa+ Gc that interact with CEACAM3 stimulate opsonin-independent phagocytosis, Syk and PI3 kinase dependent degranulation, ROS production, and fusion of granules to the phagosome, resulting in bacterial intracellular killing [52,53,54,55*,56]. In contrast, Opa-negative Gc is also internalized by adherent human neutrophils in an opsonin-independent manner, but suppresses ROS production and limits phagosome-granule fusion [57,58,54]. Our lab and others have recently uncovered additional mechanisms of resisting neutrophil killing including resistance to antimicrobial compounds contained in granules released at the plasma and phagosomal membranes and as components of NETs formed in response to Gc [35,59*,60*,61*,62*]. We and others have found that Gc stimulates NET formation [59*,61*]. However Gc encodes a thermonuclease (Nuc) that is released extracellularly and is necessary and sufficient for Gc to degrade the DNA backbone of NETs [59*]. Additionally, Gc uses the TonB-dependent transporter TdfH to obtain Zn from calprotectin, a protein that is abundant in the neutrophil cytosol and NETs [60*]. Gc stimulation of NET formation does not require an oxidative burst or ROS [61*]. Nuc and TdfH, along with the LOS-modifying enzyme LptA, enhance Gc survival in association with NETs [59*,60*,35*]. Further resistance to graunule components is conferred by phosphoethanolamine (PEA) addition to the lipid A moiety of LOS by LptA [35*] and envelope integrity maintained via PG modification by LtgA and LtgD [62**]. Since Gc can evade neutrophil killing, reactive products generated by neutrophils are instead able to induce host cell damage.
Table 1.
Gc proteins that confer resistance to neutrophils.
| Protein Name | Mechanisms of Resistance to Neutrophils | References |
|---|---|---|
| Mpg/NGO1686 | PG peptidase enhances resistance to CAMPs and ROS via presentation of type IV pili | 63, 64, 65 |
| RecN | Unclear | 64 |
| LtgA, LtgD | PG lytic transglycosylases together confer envelope integrity to defend Gc from lysozyme and neutrophil elastase; limit granule exocytosis and phagosome-granule fusion | 62** |
| TdfH | TonB-dependent transporter extracts Zn from calprotectin and enhances Gc survival in NETs | 60* |
| Nuc | Thermonuclease released extracellularly cleaves NETs | 59* |
| LptA | Phosphoethanolamine transferase for lipid A enhances Gc resistance to neutrophil CAMPs and proteases; limits phagosome-granule fusion | 35* |
| MIP | Peptidyl-prolyl isomerase protects by unknown mechanism | 39* |
| LdhA, LdhD | D-lactate dehydrogenases enhance Gc survival in neutrophils, potentially by obtaining lactate as a carbon source | 66 |
| Pili | Type IV pili protect against CAMPs and ROS | 63 |
| Lst | Sialylation of Gc N-lactotetraose-containing LOS inhibits phagocytosis by neutrophils in suspension | 67,68 |
| PorB | Essential porin inhibits the neutrophil oxidative burst | 69,70 |
Gc defenses against neutrophils also modulate neutrophil activation
Some of the defenses used by Gc against neutrophil antimicrobial activities influence neutrophil activation and extracellular release of antimicrobial components (Figure 3). These components may contribute to the cellular damage associated with gonorrhea in the FRT.
Variations in Opa protein expression contribute to neutrophil activation and the ability of Gc to survive exposure to neutrophils. A subset of Gc express Opa proteins that interact with CEACAM3, which unlike CEACAM1 has an immunoreceptor tyrosine-based activation motif (ITAM) in its cytoplasmic tail. Interaction with CEACAM3 on neutrophils leads to increased ROS production and granule release, and decreased Gc survival (Figure 3) [52,53,54,55*,56]. Engagement of CEACAM3 also serves to further recruit and activate neutrophils at the site of infection in vivo [55*,56]. The same study that found a selection for an Opa protein repertoire that binds CEACAM1 also found a selection against CEACAM3-binding Opa proteins, underscoring the importance for Gc of avoiding recognition by this receptor [45**].
Some Gc defenses also limit the degree of granule fusion with phagosomes and the plasma membrane. We recently reported that addition of PEA to LOS by LptA is important for survival of Gc from neutrophils, not only by defending against CAMPs, but also by limiting the extent of phagolysosome formation [35*]. This was surprising since PEA-modified lipid A has a higher affinity for TLR4 [34], suggesting the activation of neutrophils by Gc may be TLR4-independent. As another example, we found the PG modifying enzymes LtgA and LtgD defend Gc from neutrophils, particularly from killing by lysozyme, and limit granule fusion with phagosomes and the plasma membrane [62**]. This observation is in agreement with recent findings that PG fragments produced by LtgA and LtgD are nonstimulatory for NLRs and TLRs [41**].
Challenges in modeling ascending infection with Gc and the consequences of neutrophil influx
A direct role for neutrophils in inducing epithelial cell damage in gonorrhea has not yet been described, owing to challenges of studying Gc infection and neutrophilic inflammation in the FRT. Ex vivo systems such as cell lines and tissue explants do not generally incorporate neutrophils, although basal-to-apical transepithelial migration in response to other mucosal pathogens has been modeled with monolayers of polarized epithelial cells [76]. A mouse model of lower FRT infection has been established by the Jerse group and used to probe the bacterial and host factors that are important for colonization and early neutrophil recruitment [77]. Subsequently, Gc infection models using mice transgenic for human receptors and other human-specific components have been developed. In particular, in mice that are transgenic for human CEACAMs, the Gray-Owen group has reported that Gc robustly infects the lower FRT and triggers a strong influx of CEACAM-expressing neutrophils that interact with the bacteria [56]. While mouse models have increased our understanding of Gc colonization and early inflammatory responses, at this time they do not reproduce features of Gc infection seen in women, including ascending infection, sustained neutrophil influx, and epithelial damage.
NEUTROPHILIC INFLAMMATION AND EPITHELIAL DAMAGE IN RESPONSE TO GC IN THE UPPER FRT
If treatment of Gc infection does not occur or is ineffective, Gc can ascend to the upper FRT, where the neutrophilic inflammatory response is more potent; this is correlated with increased epithelial expression of pattern recognition receptors (PRRs) and greater numbers of resident myeloid cells that are poised to detect foreign antigens (Figure 1) [14]. The interactions of Gc with epithelial cells during ascent from the lower to the upper FRT have recently been simulated using human Hec-1-A cells cultured in a bioreactor as three-dimensional organoids [78**]. Gc infection, but not colonization with commensal Lactobacillus crispatus or Gardnerella vaginalis, stimulates production of proinflammatory mediators (IL-1β, IL-8, and TNF-α) and alterations to host cells, including microvillus remodeling [78**].
A hallmark of upper FRT infection with Gc is the death of ciliated cells lining the Fallopian tube. Reduced motility in the Fallopian tube, along with tubal scarring, result in infertility and ectopic pregnancy. Human Fallopian tube explants have been instrumental in demonstrating that release of LOS and PG fragments by Gc stimulate the production of inflammatory cytokines and second messengers, including TNF-α and nitric oxide [79,80,81]. Unlike most bacteria, including other Neisseria species, Gc poorly recycles its PG during cell wall turnover [82]. Instead, PG fragments are released extracellularly and are responsible for ciliated cell death [83**,22]. A new contributor to Fallopian tube damage was recently reported by Velazquez, Christodoulides and colleagues [84**]. Gc-infected Fallopian tube epithelial cells increase production of matrix metalloproteinases (MMPs) and extracellular release of MMP-9 [84**], which may amplify tissue destruction by degrading extracellular matrix and interfering with tissue repair. Further, MMP-9 generates chemokine mimetics and extracellular matrix fragments, which further stimulate inflammation and neutrophil recruitment. Neutrophils themselves are a significant source of MMP-9, setting up a vicious cycle of inflammation and epithelial cell damage [5].
Much remains to be learned about the mechanisms underlying Gc-induced neutrophilic inflammation and damage in the upper FRT. It is unethical to conduct human challenge studies on women due to the risk of serious complications. Fallopian tube explants and endometrial organoids are useful models, especially if primary human neutrophils are introduced. A new female mouse model of upper FRT infection with Gc provides an in vivo platform for studying neutrophilic inflammation and its consequences [85**]. In this model, transcervical inoculation of Gc leads to infection of the uterine horns and corpus of the upper FRT, resulting in robust and rapid neutrophilic infiltration, edema, and other signs of PID [85**]. The inflammatory process is exaggerated in mice in diestrus; similarly, PID most commonly presents after the onset of menses in women with gonorrhea [85**]. Introduction of transgenic or knockout mice into this model may enable longer-term infection with Gc, to model the chronic inflammation and consequent damage associated with PID.
CONCLUSION
Gc induces potent neutrophilic inflammatory responses, yet survives in their midst. Sustained infection and neutrophilic inflammation likely underlie the pathology associated with gonorrhea in women. Recently developed models of infection provide new platforms for studying neutrophil influx, measuring consequent epithelial cell damage, and testing novel therapeutics to thwart the non-productive, sustained neutrophil response to Gc. Genetic manipulation of mouse and human cells will facilitate mechanistic studies of the pathways driving neutrophil influx and host damage. These advances will enable a better understanding of how sustained neutrophilic inflammation in response to Gc drives epithelial damage and serious clinical sequelae in women.
KEY POINTS.
N. gonorrhoeae infection stimulates a robust inflammatory response featuring the recruitment and activation of neutrophils.
N. gonorrhoeae has mechanisms to resist and thwart neutrophil antimicrobial activities, while promoting neutrophil production and release of proinflammatory products.
Sustained neutrophil influx in response to N. gonorrhoeae is linked to host epithelial damage and serious sequelae in women with gonorrhea.
Acknowledgments
None.
Financial support and sponsorship: This work was supported by R01 AI097312 and R21 AI110889 to AKC from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, and by the Robert R. Wagner Fellowship to JSS.
Funding: This work was supported by the National Institutes of Health.
Footnotes
Conflicts of interest: None.
REFERENCES AND RECOMMENDED READING
- 1.Wi T, Lahra MM, Ndowa F, Bala M, Dillon JR, Ramon-Pardo P, et al. Antimicrobial resistance in Neisseria gonorrhoeae: Global surveillance and a call for international collaborative action. PLoS Med. 2017;14(7):e1002344. doi: 10.1371/journal.pmed.1002344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mayor MT, Roett MA, Uduhiri KA. Diagnosis and management of gonococcal infections. Am Fam Physician. 2012;86(10):931–8. [PubMed] [Google Scholar]
- 3.Johnson MB, Criss AK. Resistance of Neisseria gonorrhoeae to neutrophils. Front Microbiol. 2011;2:77. doi: 10.3389/fmicb.2011.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Williams IR, Parkos CA. Colonic neutrophils in inflammatory bowel disease: double-edged swords of the innate immune system with protective and destructive capacity. Gastroenterology. 2007;133(6):2049–52. doi: 10.1053/j.gastro.2007.10.031. [DOI] [PubMed] [Google Scholar]
- 5.Kruger P, Saffarzadeh M, Weber AN, Rieber N, Radsak M, von Bernuth H, et al. Neutrophils: Between host defence, immune modulation, and tissue injury. PLoS Pathog. 2015;11(3):e1004651. doi: 10.1371/journal.ppat.1004651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Porto BN, Stein RT. Neutrophil Extracellular Traps in Pulmonary Diseases: Too Much of a Good Thing? Front Immunol. 2016;7:311. doi: 10.3389/fimmu.2016.00311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Edwards JL, Butler EK. The Pathobiology of Neisseria gonorrhoeae Lower Female Genital Tract Infection. Front Microbiol. 2011;2:102. doi: 10.3389/fmicb.2011.00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quayle AJ. The innate and early immune response to pathogen challenge in the female genital tract and the pivotal role of epithelial cells. J Reprod Immunol. 2002;57(1–2):61–79. doi: 10.1016/s0165-0378(02)00019-0. [DOI] [PubMed] [Google Scholar]
- 9.Jerse AE, Sharma ND, Simms AN, Crow ET, Snyder LA, Shafer WM. A gonococcal efflux pump system enhances bacterial survival in a female mouse model of genital tract infection. Infection and immunity. 2003;71(10):5576–82. doi: 10.1128/IAI.71.10.5576-5582.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hobbs MM, Anderson JE, Balthazar JT, Kandler JL, Carlson RW, Ganguly J, et al. Lipid A’s structure mediates Neisseria gonorrhoeae fitness during experimental infection of mice and men. mBio. 2013;4(6):e00892–13. doi: 10.1128/mBio.00892-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kandler JL, Joseph SJ, Balthazar JT, Dhulipala V, Read TD, Jerse AE, et al. Phase-variable expression of lptA modulates the resistance of Neisseria gonorrhoeae to cationic antimicrobial peptides. Antimicrob Agents Chemother. 2014;58(7):4230–3. doi: 10.1128/AAC.03108-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12*.Kandler JL, Holley CL, Reimche JL, Dhulipala V, Balthazar JT, Muszynski A, et al. The MisR Response Regulator Is Necessary for Intrinsic Cationic Antimicrobial Peptide and Aminoglycoside Resistance in Neisseria gonorrhoeae. Antimicrob Agents Chemother. 2016;60(8):4690–700. doi: 10.1128/AAC.00823-16. Gc encounters high concentrations of CAMPs in cervical mucus and in neutrophils during infection. This study reports that Gc MisR influences the expression of genes important for membrane integrity, helping Gc to resist the antimicrobial activity of CAMPs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13*.Gangaiah D, Raterman E, Wu H, Fortney KR, Gao H, Liu Y, et al. Both MisR (CpxR) and MisS (CpxA) are Required for Neisseria gonorrhoeae Infection in a Murine Model of Lower Genital Tract Infection. Infection and immunity. 2017 doi: 10.1128/IAI.00307-17. Two component regulatory systems are often important during infection. This study demonstrated that the two-component system MisRS is important for Gc survival in an in vivo mouse model, increaseing colonization and allowing Gc to infect for up to seven days. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wira CR, Fahey JV, Sentman CL, Pioli PA, Shen L. Innate and adaptive immunity in female genital tract: cellular responses and interactions. Immunol Rev. 2005;206:306–35. doi: 10.1111/j.0105-2896.2005.00287.x. [DOI] [PubMed] [Google Scholar]
- 15.Jerse AE, Deal CD. Vaccine research for gonococcal infections: where are we? Sex Transm Infect. 2013;89(Suppl 4):iv63–8. doi: 10.1136/sextrans-2013-051225. [DOI] [PubMed] [Google Scholar]
- 16.Zielke RA, Wierzbicki IH, Weber JV, Gafken PR, Sikora AE. Quantitative proteomics of the Neisseria gonorrhoeae cell envelope and membrane vesicles for the discovery of potential therapeutic targets. Mol Cell Proteomics. 2014;13(5):1299–317. doi: 10.1074/mcp.M113.029538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fichorova RN, Cronin AO, Lien E, Anderson DJ, Ingalls RR. Response to Neisseria gonorrhoeae by cervicovaginal epithelial cells occurs in the absence of toll-like receptor 4-mediated signaling. J Immunol. 2002;168(5):2424–32. doi: 10.4049/jimmunol.168.5.2424. [DOI] [PubMed] [Google Scholar]
- 18.Fazeli A, Bruce C, Anumba DO. Characterization of Toll-like receptors in the female reproductive tract in humans. Hum Reprod. 2005;20(5):1372–8. doi: 10.1093/humrep/deh775. [DOI] [PubMed] [Google Scholar]
- 19.Massari P, Henneke P, Ho Y, Latz E, Golenbock DT, Wetzler LM. Cutting edge: Immune stimulation by neisserial porins is toll-like receptor 2 and MyD88 dependent. J Immunol. 2002;168(4):1533–7. doi: 10.4049/jimmunol.168.4.1533. [DOI] [PubMed] [Google Scholar]
- 20.Fisette PL, Ram S, Andersen JM, Guo W, Ingalls RR. The Lip lipoprotein from Neisseria gonorrhoeae stimulates cytokine release and NF-kappaB activation in epithelial cells in a Toll-like receptor 2-dependent manner. J Biol Chem. 2003;278(47):46252–60. doi: 10.1074/jbc.M306587200. [DOI] [PubMed] [Google Scholar]
- 21.John CM, Phillips NJ, Stein DC, Jarvis GA. Innate immune response to lipooligosaccharide: pivotal regulator of the pathobiology of invasive Neisseria meningitidis infections. Pathog Dis. 2017;75(3) doi: 10.1093/femspd/ftx030. [DOI] [PubMed] [Google Scholar]
- 22.Chan JM, Dillard JP. Attention seeker: Production, modification, and release of inflammatory peptidoglycan fragments in Neisseria. J Bacteriol. 2017 doi: 10.1128/JB.00354-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mavrogiorgos N, Mekasha S, Yang Y, Kelliher MA, Ingalls RR. Activation of NOD receptors by Neisseria gonorrhoeae modulates the innate immune response. Innate Immun. 2014;20(4):377–89. doi: 10.1177/1753425913493453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24**.Gaudet RG, Sintsova A, Buckwalter CM, Leung N, Cochrane A, Li J, et al. INNATE IMMUNITY. Cytosolic detection of the bacterial metabolite HBP activates TIFA-dependent innate immunity. Science. 2015;348(6240):1251–5. doi: 10.1126/science.aaa4921. This study is the first to describe the monosaccharide heptose-1,7-bisphosphate (HBP) as a novel intracellular PAMP, which activates a potent innate immune response via TIFA. HBP is an intermediate of the LPS synthesis pathway in gram-negative bacteria, so it represents a previously unappreciated way that the immune system can recognize and mount a response to gram-negative bacteria in the host cell cytosol. [DOI] [PubMed] [Google Scholar]
- 25.Gaudet RG, Gray-Owen SD. Heptose Sounds the Alarm: Innate Sensing of a Bacterial Sugar Stimulates Immunity. PLoS Pathog. 2016;12(9):e1005807. doi: 10.1371/journal.ppat.1005807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26**.Gaudet RG, Guo CX, Molinaro R, Kottwitz H, Rohde JR, Dangeard AS, et al. Innate Recognition of Intracellular Bacterial Growth Is Driven by the TIFA-Dependent Cytosolic Surveillance Pathway. Cell Rep. 2017;19(7):1418–30. doi: 10.1016/j.celrep.2017.04.063. Gc and other bacteria often invade host cells, helping them to evade the immune response. This study demonstrated an important role of intracellular TIFA-dependent recognition of HBP during innate immune recognition of gram-negative bacteria attempting to establish a replicative niche in the host cell cytosol. [DOI] [PubMed] [Google Scholar]
- 27.Criss AK, Seifert HS. A bacterial siren song: intimate interactions between Neisseria and neutrophils. Nat Rev Microbiol. 2012;10(3):178–90. doi: 10.1038/nrmicro2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Masson L, Mlisana K, Little F, Werner L, Mkhize NN, Ronacher K, et al. Defining genital tract cytokine signatures of sexually transmitted infections and bacterial vaginosis in women at high risk of HIV infection: a cross-sectional study. Sex Transm Infect. 2014;90(8):580–7. doi: 10.1136/sextrans-2014-051601. [DOI] [PubMed] [Google Scholar]
- 29.Liu Y, Feinen B, Russell MW. New concepts in immunity to Neisseria gonorrhoeae: innate responses and suppression of adaptive immunity favor the pathogen, not the host. Front Microbiol. 2011;2:52. doi: 10.3389/fmicb.2011.00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30*.Masson L, Salkinder AL, Olivier AJ, McKinnon LR, Gamieldien H, Mlisana K, et al. Relationship between female genital tract infections, mucosal interleukin-17 production and local T helper type 17 cells. Immunology. 2015;146(4):557–67. doi: 10.1111/imm.12527. This study measured IL-17 concentrations and Th17 cell frequencies in cervicovaginal lavages from women with FRT infections. Importantly, the authors report increased IL-17 concentrations in women infected with Gc and chlamydia. This study also highlights the asymptomatic nature of gonorrhea in women, despite the presence of Gc and increased genital cytokines. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31*.Wang LC, Yu Q, Edwards V, Lin B, Qiu J, Turner JR, et al. Neisseria gonorrhoeae infects the human endocervix by activating non-muscle myosin II-mediated epithelial exfoliation. PLoS Pathog. 2017;13(4):e1006269. doi: 10.1371/journal.ppat.1006269. Gc infection of the endocervix leads to attachment and invasion of epithelial cells. This study showed that Gc induces epithelial apical junction disassembly and shedding of columnar epithelial cells by activating non-muscle myosin II at the apical surface. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McClure R, Massari P. TLR-Dependent Human Mucosal Epithelial Cell Responses to Microbial Pathogens. Front Immunol. 2014;5:386. doi: 10.3389/fimmu.2014.00386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Magalhaes JG, Sorbara MT, Girardin SE, Philpott DJ. What is new with Nods? Curr Opin Immunol. 2011;23(1):29–34. doi: 10.1016/j.coi.2010.12.003. [DOI] [PubMed] [Google Scholar]
- 34.Packiam M, Yedery RD, Begum AA, Carlson RW, Ganguly J, Sempowski GD, et al. Phosphoethanolamine decoration of Neisseria gonorrhoeae lipid A plays a dual immunostimulatory and protective role during experimental genital tract infection. Infection and immunity. 2014;82(6):2170–9. doi: 10.1128/IAI.01504-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35*.Handing JW, Criss AK. The lipooligosaccharide-modifying enzyme LptA enhances gonococcal defence against human neutrophils. Cellular microbiology. 2015;17(6):910–21. doi: 10.1111/cmi.12411. This study showed that PEA addition to Gc LOS by LptA helps Gc overcome neutrophil killing mechanisms by limiting phagolysosome maturation as well as helping Gc resist antimicrobial components of neutrophil granules delivered to the phagosome, released extracellularly, and contained in NETs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36**.Andrade WA, Agarwal S, Mo S, Shaffer SA, Dillard JP, Schmidt T, et al. Type I Interferon Induction by Neisseria gonorrhoeae: Dual Requirement of Cyclic GMP-AMP Synthase and Toll-like Receptor 4. Cell Rep. 2016;15(11):2438–48. doi: 10.1016/j.celrep.2016.05.030. This article showed that in addition to the known type I interferon (IFN) response to Gc LOS via surface TLR4/MD-2 recognition of extracellular bacteria, there is an additional, cytosolic response to Gc where bacterial DNA activates cyclic-GMP-AMP synthase (cGAS), leading to STING/TBK-1/IRF-3 mediated IFN expression. In concert, these pathways serve to increase IFN-β production in response to Gc. Importantly, this is an example of how Gc manipulates the host response to its survival advantage since IFN-β helps increase intracellular iron availability for Gc during infection of neutrophils and macrophages. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37*.Chateau A, Seifert HS. Neisseria gonorrhoeae survives within and modulates apoptosis and inflammatory cytokine production of human macrophages. Cellular microbiology. 2016;18(4):546–60. doi: 10.1111/cmi.12529. Modulation of the host immune response contributes to Gc survival. This article reports that Gc can survive and replicate inside two different in vitro macrophage models. Importantly, the results show that Gc induces inflammatory cytokine production in macrophages, which could contribute to the neutrophil rich inflammatory response seen during infection. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leuzzi R, Serino L, Scarselli M, Savino S, Fontana MR, Monaci E, et al. Ng-MIP, a surface-exposed lipoprotein of Neisseria gonorrhoeae, has a peptidyl-prolyl cis/trans isomerase (PPIase) activity and is involved in persistence in macrophages. Mol Microbiol. 2005;58(3):669–81. doi: 10.1111/j.1365-2958.2005.04859.x. [DOI] [PubMed] [Google Scholar]
- 39*.Reimer A, Seufert F, Weiwad M, Ebert J, Bzdyl NM, Kahler CM, et al. Inhibitors of macrophage infectivity potentiator-like PPIases affect neisserial and chlamydial pathogenicity. Int J Antimicrob Agents. 2016;48(4):401–8. doi: 10.1016/j.ijantimicag.2016.06.020. MIP is a PPIase activity found in Chlamydia and pathogenic Neisseria. Using two newly identified inhibitors of MIP, PipN3 and PipN4, the authors’ findings support a role for MIP during Gc survival in neutrophils. [DOI] [PubMed] [Google Scholar]
- 40*.Ortiz MC, Lefimil C, Rodas PI, Vernal R, Lopez M, Acuna-Castillo C, et al. Neisseria gonorrhoeae Modulates Immunity by Polarizing Human Macrophages to a M2 Profile. PLoS One. 2015;10(6):e0130713. doi: 10.1371/journal.pone.0130713. Gc is known to modulate the immune response by suppressing T and B cells, as well as antigen presenting cells (including DCs). This study showed that Gc interacts with macrophages to skew them to an M2 profile. Macrophages exposed to Gc displayed M2 surface markers, produced immunosuppressive cytokines, and stimulated less T-cell proliferaiton, demonstrating another mechanism by which Gc suppresses an adaptive immune response. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41**.Knilans KJ, Hackett KT, Anderson JE, Weng C, Dillard JP, Duncan JA. Neisseria gonorrhoeae Lytic Transglycosylases LtgA and LtgD Reduce Host Innate Immune Signaling through TLR2 and NOD2. ACS Infect Dis. 2017 doi: 10.1021/acsinfecdis.6b00088. While PG fragments are potent activators of innate immune receptors including TLR2 and NOD2, this very important study described how Gc LtgA and LtgD allow for extracellular release of monomeric instead of multimeric PG fragments. Compared to multimeric PG fragments, monomeric PG fragments lead to reduced activation of these receptors and decreased production of IL-1β and TNFα, dampening the immune response to Gc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Obergfell KP, Seifert HS. Mobile DNA in the Pathogenic Neisseria. Microbiol Spectr. 2015;3(1) doi: 10.1128/microbiolspec.MDNA3-0015-2014. [DOI] [PubMed] [Google Scholar]
- 43.Zhu W, Chen CJ, Thomas CE, Anderson JE, Jerse AE, Sparling PF. Vaccines for gonorrhea: can we rise to the challenge? Front Microbiol. 2011;2:124. doi: 10.3389/fmicb.2011.00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gray-Owen SD, Blumberg RS. CEACAM1: contact-dependent control of immunity. Nat Rev Immunol. 2006;6(6):433–46. doi: 10.1038/nri1864. [DOI] [PubMed] [Google Scholar]
- 45*.Sintsova A, Wong H, MacDonald KS, Kaul R, Virji M, Gray-Owen SD. Selection for a CEACAM receptor-specific binding phenotype during Neisseria gonorrhoeae infection of the human genital tract. Infection and immunity. 2015;83(4):1372–83. doi: 10.1128/IAI.03123-14. Gc Opa protein interaction with human CEACAM helps Gc attach and invade host cells, including epithelial cells, however engagement of the neutrophil-restricted CEACAM3 in particular reduces Gc survival in neutrophils. This very important study analyzed CEACAM-binding profiles of clinical-specimen-derived Gc isolates and found a selection for Opa proteins that bind CEACAM1 and CEACAM5, but not CEACAM3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wachter J, Hill S. Positive Selection Pressure Drives Variation on the Surface-Exposed Variable Proteins of the Pathogenic Neisseria. PLoS One. 2016;11(8):e0161348. doi: 10.1371/journal.pone.0161348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu Y, Russell MW. Diversion of the immune response to Neisseria gonorrhoeae from Th17 to Th1/Th2 by treatment with anti-transforming growth factor beta antibody generates immunological memory and protective immunity. mBio. 2011;2(3):e00095–11. doi: 10.1128/mBio.00095-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu Y, Islam EA, Jarvis GA, Gray-Owen SD, Russell MW. Neisseria gonorrhoeae selectively suppresses the development of Th1 and Th2 cells, and enhances Th17 cell responses, through TGF-beta-dependent mechanisms. Mucosal Immunol. 2012;5(3):320–31. doi: 10.1038/mi.2012.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu Y, Liu W, Russell MW. Suppression of host adaptive immune responses by Neisseria gonorrhoeae: role of interleukin 10 and type 1 regulatory T cells. Mucosal Immunol. 2014;7(1):165–76. doi: 10.1038/mi.2013.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Feinen B, Jerse AE, Gaffen SL, Russell MW. Critical role of Th17 responses in a murine model of Neisseria gonorrhoeae genital infection. Mucosal Immunol. 2010;3(3):312–21. doi: 10.1038/mi.2009.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51**.Liu Y, Hammer LA, Liu W, Hobbs MM, Zielke RA, Sikora AE, et al. Experimental vaccine induces Th1-driven immune responses and resistance to Neisseria gonorrhoeae infection in a murine model. Mucosal Immunol. 2017 doi: 10.1038/mi.2017.11. Gc infection does not elicit a protective adaptive immune response because Gc skews the response away from Th1 and Th2-driven immunity towards a non-productive Th17 response. This study demonstrated that female mice immunized with Gc OMVs with the addition of IL-12 cleared infection faster and mounted a robust adaptive immune response that protected against future challenge with antigenically different strains of Gc. This study may have important clinical applications for modulating the immue response to Gc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sarantis H, Gray-Owen SD. Defining the roles of human carcinoembryonic antigen-related cellular adhesion molecules during neutrophil responses to Neisseria gonorrhoeae. Infection and immunity. 2012;80(1):345–58. doi: 10.1128/IAI.05702-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sarantis H, Gray-Owen SD. The specific innate immune receptor CEACAM3 triggers neutrophil bactericidal activities via a Syk kinase-dependent pathway. Cellular microbiology. 2007;9(9):2167–80. doi: 10.1111/j.1462-5822.2007.00947.x. [DOI] [PubMed] [Google Scholar]
- 54.Smirnov A, Daily KP, Criss AK. Assembly of NADPH oxidase in human neutrophils is modulated by the opacity-associated protein expression State of Neisseria gonorrhoeae. Infection and immunity. 2014;82(3):1036–44. doi: 10.1128/IAI.00881-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55*.Johnson MB, Ball LM, Daily KP, Martin JN, Columbus L, Criss AK. Opa+ Neisseria gonorrhoeae exhibits reduced survival in human neutrophils via Src family kinase-mediated bacterial trafficking into mature phagolysosomes. Cellular microbiology. 2015;17(5):648–65. doi: 10.1111/cmi.12389. Opa protein expression is important for Gc attachment to and invasion of host epithelial cells, but has been shown to lead to decreased survival in neutrophils. This study reports that Opa interaction with the neutrophil-restricted CEACAM3 leads to Src-dependent neutrophil activation, phagosome maturation, and reduced Gc survival. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sintsova A, Sarantis H, Islam EA, Sun CX, Amin M, Chan CH, et al. Global analysis of neutrophil responses to Neisseria gonorrhoeae reveals a self-propagating inflammatory program. PLoS Pathog. 2014;10(9):e1004341. doi: 10.1371/journal.ppat.1004341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ball LM, Criss AK. Constitutively Opa-expressing and Opa-deficient neisseria gonorrhoeae strains differentially stimulate and survive exposure to human neutrophils. J Bacteriol. 2013;195(13):2982–90. doi: 10.1128/JB.00171-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Johnson MB, Criss AK. Neisseria gonorrhoeae phagosomes delay fusion with primary granules to enhance bacterial survival inside human neutrophils. Cellular microbiology. 2013;15(8):1323–40. doi: 10.1111/cmi.12117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59*.Juneau RA, Stevens JS, Apicella MA, Criss AK. A thermonuclease of Neisseria gonorrhoeae enhances bacterial escape from killing by neutrophil extracellular traps. J Infect Dis. 2015;212(2):316–24. doi: 10.1093/infdis/jiv031. NETs can help to trap and kill invading microbes, however this study describes a mechanism by which Gc uses a nuclease to degrade NET DNA. Imporantly, Gc induces NET formation but efficiently evades NET killing, which may contribute to the sustained an innefficient neutrophil response to Gc infection. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60*.Jean S, Juneau RA, Criss AK, Cornelissen CN. Neisseria gonorrhoeae Evades Calprotectin-Mediated Nutritional Immunity and Survives Neutrophil Extracellular Traps by Production of TdfH. Infection and immunity. 2016;84(10):2982–94. doi: 10.1128/IAI.00319-16. Nutritional immunity, the sequestration of essential nutrients including Zn, is an important host defense against pathogens. This study demonstrates that Gc uses TonB-dependent transporters to overcome sequestration of Zn by directly binding calprotectin, a host Zn-binding protein that is produced by neutrophils and contained in NETs. Expression of TdfH affords Gc a survival advantage when in association with NETs, in a Zn-dependent manner. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61*.Gunderson CW, Seifert HS. Neisseria gonorrhoeae elicits extracellular traps in primary neutrophil culture while suppressing the oxidative burst. mBio. 2015;6(1) doi: 10.1128/mBio.02452-14. This study evaluated NET formation in response to Gc, reporting that NETs form in the absence of an oxidative burst and ROS production and that Gc is not killed by NETs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62**.Ragland SA, Schaub RE, Hackett KT, Dillard JP, Criss AK. Two lytic transglycosylases in Neisseria gonorrhoeae impart resistance to killing by lysozyme and human neutrophils. Cellular microbiology. 2017;19(3) doi: 10.1111/cmi.12662. Gc LtgA and LtgD are important for PG release during Gc growth. This study showed that these enzymes also help Gc resist killing by neutrophils, in particular by resisting killing by lysozyme. Importantly, this study also demonstrated that LtgA and LtgD activity reduces neutrophil activation by reducing granule mobilization to the phagosomal and plasma membranes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stohl EA, Dale EM, Criss AK, Seifert HS. Neisseria gonorrhoeae metalloprotease NGO1686 is required for full piliation, and piliation is required for resistance to H2O2- and neutrophil-mediated killing. mBio. 2013;4(4) doi: 10.1128/mBio.00399-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stohl EA, Criss AK, Seifert HS. The transcriptome response of Neisseria gonorrhoeae to hydrogen peroxide reveals genes with previously uncharacterized roles in oxidative damage protection. Mol Microbiol. 2005;58(2):520–32. doi: 10.1111/j.1365-2958.2005.04839.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stohl EA, Chan YA, Hackett KT, Kohler PL, Dillard JP, Seifert HS. Neisseria gonorrhoeae virulence factor NG1686 is a bifunctional M23B family metallopeptidase that influences resistance to hydrogen peroxide and colony morphology. J Biol Chem. 2012;287(14):11222–33. doi: 10.1074/jbc.M111.338830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Atack JM, Ibranovic I, Ong CL, Djoko KY, Chen NH, Vanden Hoven R, et al. A role for lactate dehydrogenases in the survival of Neisseria gonorrhoeae in human polymorphonuclear leukocytes and cervical epithelial cells. J Infect Dis. 2014;210(8):1311–8. doi: 10.1093/infdis/jiu230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rest RF, Frangipane JV. Growth of Neisseria gonorrhoeae in CMP-N-acetylneuraminic acid inhibits nonopsonic (opacity-associated outer membrane protein-mediated) interactions with human neutrophils. Infection and immunity. 1992;60(3):989–97. doi: 10.1128/iai.60.3.989-997.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim JJ, Zhou D, Mandrell RE, Griffiss JM. Effect of exogenous sialylation of the lipooligosaccharide of Neisseria gonorrhoeae on opsonophagocytosis. Infection and immunity. 1992;60(10):4439–42. doi: 10.1128/iai.60.10.4439-4442.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lorenzen DR, Gunther D, Pandit J, Rudel T, Brandt E, Meyer TF. Neisseria gonorrhoeae porin modifies the oxidative burst of human professional phagocytes. Infection and immunity. 2000;68(11):6215–22. doi: 10.1128/iai.68.11.6215-6222.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen A, Seifert HS. Neisseria gonorrhoeae-mediated inhibition of apoptotic signalling in polymorphonuclear leukocytes. Infection and immunity. 2011;79(11):4447–58. doi: 10.1128/IAI.01267-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB. Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal. 2014;20(7):1126–67. doi: 10.1089/ars.2012.5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Verjans ET, Zels S, Luyten W, Landuyt B, Schoofs L. Molecular mechanisms of LL-37-induced receptor activation: An overview. Peptides. 2016;85:16–26. doi: 10.1016/j.peptides.2016.09.002. [DOI] [PubMed] [Google Scholar]
- 73.Saffarzadeh M, Juenemann C, Queisser MA, Lochnit G, Barreto G, Galuska SP, et al. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS One. 2012;7(2):e32366. doi: 10.1371/journal.pone.0032366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Papayannopoulos V, Zychlinsky A. NETs: a new strategy for using old weapons. Trends Immunol. 2009;30(11):513–21. doi: 10.1016/j.it.2009.07.011. [DOI] [PubMed] [Google Scholar]
- 75.Doring Y, Soehnlein O, Weber C. Neutrophil Extracellular Traps in Atherosclerosis and Atherothrombosis. Circ Res. 2017;120(4):736–43. doi: 10.1161/CIRCRESAHA.116.309692. [DOI] [PubMed] [Google Scholar]
- 76.Szabady RL, McCormick BA. Control of neutrophil inflammation at mucosal surfaces by secreted epithelial products. Front Immunol. 2013;4:220. doi: 10.3389/fimmu.2013.00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jerse AE, Wu H, Packiam M, Vonck RA, Begum AA, Garvin LE. Estradiol-Treated Female Mice as Surrogate Hosts for Neisseria gonorrhoeae Genital Tract Infections. Front Microbiol. 2011;2:107. doi: 10.3389/fmicb.2011.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78**.Laniewski P, Gomez A, Hire G, So M, Herbst-Kralovetz MM. Human Three-Dimensional Endometrial Epithelial Cell Model To Study Host Interactions with Vaginal Bacteria and Neisseria gonorrhoeae. Infection and immunity. 2017;85(3) doi: 10.1128/IAI.01049-16. Ascending Gc infection leads to significant risk of serious complications. This very important study established an in vitro model of ascending Gc infection, modeling the TLR response to Gc infection and comparing this to the response to two vaginal commensals, Lactobacillus crispatus and Gardnerella vaginalis. Importantly, only infection with Gc, but not commensal bacteria, stimulated the immune response. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Melly MA, Gregg CR, McGee ZA. Studies of toxicity of Neisseria gonorrhoeae for human fallopian tube mucosa. J Infect Dis. 1981;143(3):423–31. doi: 10.1093/infdis/143.3.423. [DOI] [PubMed] [Google Scholar]
- 80.Gregg CR, Melly MA, Hellerqvist CG, Coniglio JG, McGee ZA. Toxic activity of purified lipopolysaccharide of Neisseria gonorrhoeae for human fallopian tube mucosa. J Infect Dis. 1981;143(3):432–9. doi: 10.1093/infdis/143.3.432. [DOI] [PubMed] [Google Scholar]
- 81.Melly MA, McGee ZA, Rosenthal RS. Ability of monomeric peptidoglycan fragments from Neisseria gonorrhoeae to damage human fallopian-tube mucosa. J Infect Dis. 1984;149(3):378–86. doi: 10.1093/infdis/149.3.378. [DOI] [PubMed] [Google Scholar]
- 82.Woodhams KL, Chan JM, Lenz JD, Hackett KT, Dillard JP. Peptidoglycan fragment release from Neisseria meningitidis. Infection and immunity. 2013;81(9):3490–8. doi: 10.1128/IAI.00279-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83**.Chan JM, Dillard JP. Neisseria gonorrhoeae Crippled Its Peptidoglycan Fragment Permease To Facilitate Toxic Peptidoglycan Monomer Release. J Bacteriol. 2016;198(21):3029–40. doi: 10.1128/JB.00437-16. Peptidoglycan (PG) fragments are important PAMPs produced and released by gram-negative bacteria. This study showed that Gc releases significantly more PG fragments compared to N. meningiditis and nonpathogenic Neisseria due to the decreased PG recycling efficiency of its AmpG permease. This is an important finding, as the toxic PG monomers released by Gc stimulate pro-inflammatory signaling through host NOD and TLR receptors, contributing to the inflammatory response to Gc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84**.Rodas PI, Perez D, Jaffret C, Gonzalez Y, Carreno C, Tapia CV, et al. Modified profile of matrix metalloproteinase-2 and -9 production by human Fallopian tube epithelial cells following infection in vitro with Neisseria gonorrhoeae. J Infect Dis. 2016 doi: 10.1093/infdis/jiw568. This critical study demonstrated that human Fallopian tube epithelial cells secrete MMP9 and have increased cytosolic MMP2 upon Gc infection. This study is important because MMP9 could play a significant role in the epithelial scarring associated with ascending Gc infections, eading to complications including infertility. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85**.Islam EA, Shaik-Dasthagirisaheb Y, Kaushic C, Wetzler LM, Gray-Owen SD. The reproductive cycle is a pathogenic determinant during gonococcal pelvic inflammatory disease in mice. Mucosal Immunol. 2016;9(4):1051–64. doi: 10.1038/mi.2015.122. This very important article established a murine model of ascending Gc infection and PID. In this model, infection and the immune response, including neutrophil influx and signs of epithelial cell damage, are worse when mice are infected during diestrus. This models the common clinical presentation of PID immediately after the onset of menses and will be a very useful tool for the field moving forward. [DOI] [PMC free article] [PubMed] [Google Scholar]