

Abstract
The overarching goal of modern drug development is to optimize therapeutic benefits while minimizing adverse effects. However, inadequate efficacy and safety concerns remain to be the major causes of drug attrition in clinical development. For the past 80 years, toxicity testing has consisted of evaluating the adverse effects of drugs in animals to predict human health risks. The U.S. Environmental Protection Agency recognized the need to develop innovative toxicity testing strategies and asked the National Research Council to develop a long-range vision and strategy for toxicity testing in the 21st century. The vision aims to reduce the use of animals and drug development costs through the integration of computational modeling and in vitro experimental methods that evaluates the perturbation of toxicity-related pathways. Towards this vision, collaborative quantitative systems pharmacology and toxicology modeling endeavors (QSP/QST) have been initiated amongst numerous organizations worldwide. In this article, we discuss how quantitative structure-activity relationship (QSAR), network-based, and pharmacokinetic/pharmacodynamic modeling approaches can be integrated into the framework of QST models. Additionally, we review the application of QST models to predict cardiotoxicity and hepatotoxicity of drugs throughout their development. Cell and organ specific QST models are likely to become an essential component of modern toxicity testing, and provides a solid foundation towards determining individualized therapeutic windows to improve patient safety.
1. Introduction
While the origin of systems toxicology lies in studying the cumulative effects of various environmental exposures on human health, there has been a tremendous increase in the application of this approach in the field of medicine. The Food, Drug, and Cosmetic Act (FDCA) passed by Congress in 1938, in response to the 1937 sulfanilamide tragedy where over 100 people died from nephrotoxicity, has set the precedence for the current toxicity testing strategy, which assesses the effects of a drug on animals prior to administration in humans. However, toxicity testing performed in animals is not always translatable to the clinic. For example, the teratogenic effects of thalidomide, which led to over 10,000 cases of birth defects, was not identified in rat toxicity studies [1]. In response to this event, the Kefauver-Harris amendment was made to the FDCA, requiring proof of drug effectiveness and safety. Thus, inaccuracies in preclinical-to-clinical translatability, significant worldwide resource cost, and the sacrifice of millions of animals for toxicity testing, warrants a novel toxicity testing strategy that moves away from traditional animal toxicity testing. In fact, a decade ago, the National Academy of Sciences published a report titled, ‘Toxicity Testing in the 21st Century’, which advocated the development of a systems approach to replace current toxicity testing. Accordingly, organ/disease specific quantitative systems toxicology models, which integrate in vitro human cell toxicity assays with multi-scale in silico modeling of drug exposures, could serve as an efficient tool to assess and predict human toxicity of drug molecules.
Quantitative systems pharmacology (QSP) has been defined as, “an approach to translational medicine that combines computational and experimental methods to elucidate, validate, and apply new pharmacological concepts to the development and use of small molecule and biologic drugs.” [2] Here we provide aworking definition for quantitative systems toxicology (QST) as an approach to quantitatively understand the toxic effects of a chemical on a living organism, from molecular alterations to phenotypical observations, through the integration of computational and experimental methods. A quantitative understanding of holistic drug effects will allow the distinction between three forms of toxicity, on-target/on-pathway, on-target/off-pathway, and off-target. Although QST may be considered to be a part of QSP modeling by many, we believe QST will likely find its own niche in the development of organ specific toxicity platforms. With collaborations between academic/nonprofit institutions, pharmaceutical industries, and regulatory agencies, current toxicity testing could begin to be replaced with in silico modeling, which would be of best interest to all parties. The Comprehensive in Vitro Pro-Arrhythmia (CIPA) and Drug Induced Liver Injury (DILI)-sim initiatives are two such collaborative efforts that aim to improve patient safety, decrease resource expenditure in drug development, and reduce the need for animal toxicity testing through the development of cardiac and hepatic QST models. Although in this chapter we have covered QST modeling in the context of its applications in pharmaceutical sciences, it should be noted that this type of modeling would also be of interest to other fields such as environmental sciences and ecotoxicology. Here, we have discussed the foundation and application of QST models in drug development, along with a discussion of the different mathematical modeling approaches that could be incorporated into QST model development.
2. Modeling approaches in systems toxicology
Several systems toxicology-modeling approaches have been developed to predict the adverse effects of drugs on human health. Here we briefly review QSAR/ADMET, network-based, and PK/PD modeling approaches, since these three are integral in the development of QST models.
2.1. Quantitative structure-activity relationship (QSAR) and ADMET modeling
The history of quantifying toxicity based upon similarities in chemical structure dates back to 1863, where Cross identified that the toxicity of primary aliphatic alcohols to mammals increased as its water solubility decreased [3]. At the end of the 19th century, Meyer and Overton separately showed that the anesthetic potency of narcotics is correlated with their olive oil/water partition coefficient, reflective of increased membrane permeability due to greater lipophilicity [4,5]. In 1937, Hammett formulated the first quantitative relationship between molecular structure and activity to describe electronic effects of organic reactions. The foundation of modern day QSAR has been attributed to Hansch and Fujita. They integrated Hammett’s constant (σ) with oil-water partition coefficients, later defined as a hydrophobicity parameter (π), in order to relate the physicochemical properties of phenoxyacetic acids with their plant growth activity [6]. The major advancement in QSAR occurred when it was shown that the concentration required to induce a biological response could correlate with the linear sum of different physicochemical parameters. The ability to make accurate in silico predictions of biological, pharmacological, and toxicological activity/properties of a compound, based upon molecular descriptors and physicochemical properties, is the underlying goal of QSAR modeling.
QSAR modeling has served as a useful tool throughout the drug discovery and development process. QSAR modeling has facilitated the discovery and development of new drugs through the ability to screen compounds for activity and favorable drug properties, complementing high throughput screening approaches [7]. QSAR offers the ability to design out unwanted drug properties, such as hERG inhibition and CYP450 modification, which is a powerful application. In terms of QSP/QST modeling, QSAR can be utilized to make initial predictions of parameters when no experimental information is available. QSAR predictions of parameters that relate to the absorption, distribution, metabolism, excretion, and toxicity of a drug is referred to as ADMET modeling. One of the most notable examples, although not quantitative, is Lipinski’s rule of five [8]. Due the ability to provide predictions of model parameters in the absence of experimental data, QSAR modeling can provide a bridge backwards for the utilization of QST models in the earliest stages of drug discovery and development.
2.2. Network-based modeling
The study of biology in the context of a system can be traced back to generalized systems theory [9]. The application of network theory in the study of biological systems has gained popularity within the past couple of decades due to the transition from a reductionist viewpoint of biological research back to one that is holistic [10,11]. A holistic viewpoint is one that aims to understand how the integration of molecular events give rise to biological processes across different scales of organization. The surge of interest in systems biology/pharmacology is met with advances in computational methods and software, curated databases, and analytical techniques. Genomics, proteomics, and metabolomics has enabled the generation of large quantities of data, which can be utilized in order to gain a systems-level understanding of biological phenomena through the complex dynamics of subcellular components.
Network models of biological systems, derived from the mathematical formalism of graph theory, aim to describe the complex qualitative relationships between biological components. There are different types of biological networks, which are dependent upon how nodes and edges are defined. Vertices, or nodes, typically represent genes or gene products, such as DNA, RNA, or proteins. Edges between nodes indicates a regulatory interaction. Analysis of network topology through applying measures of connectivity, centrality, and clustering, provides insight about important components/motifs and robustness. In order to observe how the expression of network components evolves over time, interaction networks need to be developed into dynamic networks. Dynamic networks can be built using ordinary/partial differential equations, Boolean algebra, or Bayesian inferences that range from continuous to discrete and deterministic to stochastic models. Network simulations, attractor analyses, and other dynamic network analysis techniques can be applied to study how the system evolves under various pharmacological/toxicological perturbations. Pathway analysis tools, such as ingenuity pathway analysis and DAVID/KEGG can be used to identify differentially expressed genes and pathways of importance. Inclusion of network-based approaches into QST models could improve predictions, elucidate unknown mechanisms of toxicity, and identify therapeutic targets to prevent toxicity.
2.3. Pharmacokinetic and pharmacodynamic modeling
Pharmacokinetic and pharmacodynamic (PK/PD) models aim to link drug concentrations at the site-of-action to pharmacological/toxicological responses. PKPD models are derived from ODEs and follow a law of parsimony. In addition, they are data-driven, compartmental, and empirical in nature. The foundation of modern day pharmacokinetics is attributed to Theorell, who published two seminal articles in 1937 [12]. Classic PK models include one/two-compartment models which describe the change in plasma drug concentration over time. More advanced PK models have been developed, which include target-mediated drug disposition (TMDD) to capture the effects of target binding on the disposition of drugs as well as physiologically-based pharmacokinetic models that compartmentalize human anatomy and include physiological parameters to obtain organ specific drug exposures [13]. In 1966, Levy was the first to link drug pharmacokinetics to a pharmacodynamic effect [14]. There are several types of classic pharmacodynamics models, such as direct/indirect effects, transduction, and tolerance models [15]. Over the past few decades, semi-mechanistic pharmacodynamic models have been developed and tailored to specific needs. However, recently there has been a shift towards mechanism-based systems, or enhanced, pharmacodynamic models [16,17].
The application of PK/PD principles in relation to chemical toxicity is referred to as toxicokinetics and toxicodynamics (TK/TD). There have been considerable efforts to incorporate TK/TD modeling framework into both ecotoxicology and drug development [18,19]. In drug development, TK/TD models aim to provide a quantitative relationship between drug concentration and toxicity in order to prevent unwanted adverse side effects and optimize dosing. A complete understanding of the dose–toxicity profile is especially important for drugs with a narrow therapeutic index. The integration of physiologically based pharmacokinetic/toxicokinetics (PBPK/PBTK) modeling into QST models is a necessary component to ensure accurate concentrations of drug at the site of toxicity and toxicodynamic predictions.
2.4. Quantitative systems pharmacology and toxicology modeling
QSP and QST are hybrid scientific disciplines where principles from PK/PD and TK/TD have merged with systems biology to gain a quantitative understanding of how drugs modulate cellular systems at the molecular level, and how therapeutic/toxic effects integrate across multiple layers of biological complexity to impact human pathophysiology. While QSTand QSP may seem similar, one distinguishable feature between the two is the development of organ specific QST models that aim to link drug exposure to clinical adverse drug reactions. The development of QST models requires the integration of several types of models and modeling techniques. QST models should include drug pharmacokinetics, a cell/organ systems physiology model, mechanism of toxicity, toxicodynamic biomarkers, and a projection of an adverse drug reaction (see Fig. 1). A PBPK model that simulates clinically relevant drug concentrations at the site-of -toxicity is ideal as input into a cell/organ systems physiology model. Cellular/organ models should include physiological processes and transduction pathways related to the mechanisms of toxicity of interest. For example, inclusion of ion channels, ion fluxes, and calcium signaling for a QST model of cardiac arrhythmias would be important since they directly relate to current and the generation of action potentials. Cellular systems physiology models canbe expanded to include additional potential mechanisms of toxicity through the inclusion of network models. The physiological model could also be enhanced with patient specific information to improve and individualize risk assessment. Incorporating QSAR modeling offers the ability to predict parameter values when no information is available, which is particularly important for investigational drugs. Cellular models could be extrapolated to organ level models or directly used to make predictions of a toxicodynamic biomarker. Lastly, a projection for the likelihood of a clinical adverse drug reaction should be made. Although the development of QST models is time and resource intensive, they are powerful tools and have a broad range of applicability throughout the drug development process.
Fig. 1.
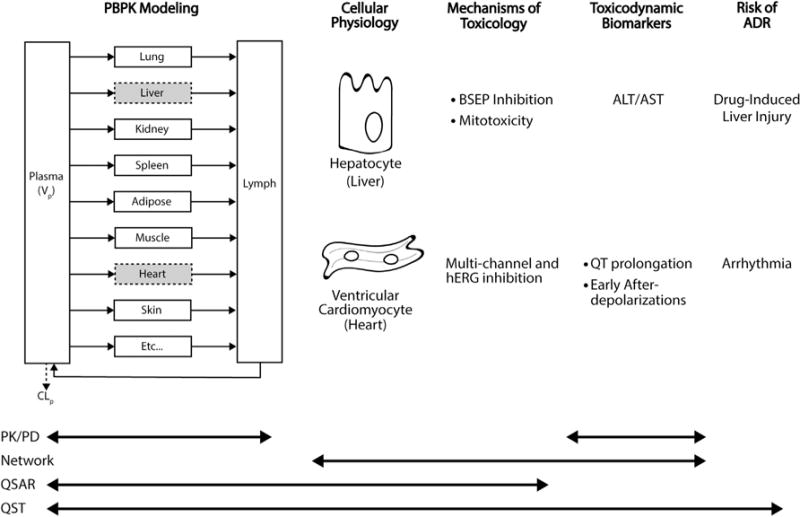
Overview of quantitative systems toxicology (QST) model structure. QST models contain the characterization of pharmacokinetics (PBPK modeling), a quantitative understanding of cellular physiological processes, mechanisms of toxicity, toxicodynamic biomarkers, and projected risk of an adverse drug reaction (ADR). For hepatotoxicity, drug concentrations at hepatocytes drive cellular pathophysiological changes and liver toxicodynamic biomarkers, aspartate transaminase (AST) and alanine transaminase (ALT). Liver enzyme dynamics can be used to predict the risk of DILI. For cardiotoxicity, drug concentrations at cardiomyocytes modulate ion channels, potentially resulting in QT prolongation and EADs. These toxicodynamic biomarkers can be used as surrogate markers to predict arrhythmias. Multiscale mechanism-based QST models include a vast range of pharmacological and physiological components, which enables a broad applicability. Whereas the scope of other modeling approaches, PK/PD, network-based, and QSAR, are often limited due to their empirical nature.
3. Application of quantitative systems toxicology in drug development
Extensive modeling efforts have been undertaken to predict cardiac and hepatic toxicity of drugs throughout preclinical and clinical phases of drug development, since these are the two main toxicities responsible for drug attrition/withdrawal due to safety concerns. Here we provide a few examples where quantitative systems toxicology modeling has been useful for making clinical projections of adverse drug reaction risk.
3.1. Cardiovascular safety
Cardiovascular safety concerns are the leading cause of drugs withdrawn from the US market and a large reason for attrition in drug development [20]. Drug-induced cardiac toxicities include QT prolongation, myocyte damage, blood pressure changes, thrombosis, and arrhythmias. QT prolongation is one of the most investigated cardiovascular safety concerns since it is known to be associated with Torsade de pointes (TdP), a life threatening ventricular tachyarrhythmia [21]. Initially only antiarrhythmic drugs were known to cause QT prolongation. However, in the 1980s–1990s, regulatory concerns about non-antiarrhythmic drug-induced QT prolongation began to rise with the withdrawal of prenylamine, lidoflazine, and terodiline from the EU market, as well as the approval of halofantrine and cisapride. In 1997, the Committee for Proprietary Medicinal Products recommended in vitro electrophysiology studies of non-cardiovascular drugs prior to clinical trials. The turning point in the history of cardiovascular safety monitoring occurred in 2005 when the International Conference of Harmonization (ICH) organized a committee that implemented two guidelines, S7B and E14. ICH-S7B suggests investigating hERG potassium current blockade in vitro and QT prolongation in an in vivo animal model. IHC-E14 involves a clinical investigation to assess the relationship between drug concentration and QT/QTc (TQT) interval change. Currently, we are in the midst of a paradigm shift from in vivo animal testing towards focusing on integrating in vitro drug effects on multiple cardiac ion channels with mechanistic in silico electrophysiology modeling to predict proarrhythmic risk. This shift towards the development of a quantitative systems toxicology models could prevent premature attrition due to QT prolongation or hERG inhibition, replace costly TQT studies, and improve patient safety through the development of safer drugs and enhancing predictions of severe cardiac events.
Mathematical models of cardiac electrophysiology have been developed to characterize the effect of drugs on ion channels and currents, in order to predict QT prolongation, action potential duration (APD), and proar-rhythmic potential. In 1960, Noble developed the first model of cardiac action potential based upon Hodgkin and Huxley’s equations [22]. This mathematical framework has been extended to quantitatively describe the generation of action potentials by specific membrane currents in cardiac ventricular myocytes [23]. The Luo-Rudy and, more recent, O’Hara-Rudy dynamic (ORd) models are amongst the most extensively used cardiac ventricular models. CIPA has recently adopted the ORd model as a base model that can be extended and applied to make important regulatory decisions regarding cardiovascular safety [24]. The ORd model describes cellular electrophysiology mechanisms in human ventricular myocytes, which consists of 4 compartments, 15 ion channels, 6 ionic fluxes, 5 buffers, and CaMK [25]. This model has great utility since all parameters, initial conditions, and scaling factors are known and model predictions have been rigorously validated against experimentally obtained data of ion current kinetics, APD rate dependence, APD restitution, and early afterdepolarizations (EAD). In order to broaden applicability and predictability, others have modified the original ORd model to incorporate the temperature dependence of hERG potassium currents, disease state, and gender differences [26–28]. Dose–response information for the modulation of specific ion channel conductance by a drug of interest can be incorporated into cardiac myocyte models in order to assess proarrhythmic risk. Mirams et al. (2014), in an attempt to predict the results of TGT studies, combined a conductance-block model with three different cardiac myocyte models and used experimentally obtained IC50’s for the inhibition of 5 channels by 34 compounds to drive predictions of APD [29]. The ORd model performed the best in comparison with the Tusscher-Noble-Noble-Panfilov (TNNP) and Grandi models [30,31]. Okada et al. (2015) developed a multi-scale finite element method-based model of a whole human heart, scaled from the ORd cardiac ventricular myocyte model, coupled to a human torso model [32]. This multi-scale heart model, UT-Heart, enabled realistic electrocardiogram (ECG) simulations based upon dose–response information for the inhibition of multiple ion channels, which was used to predict arrhythmogenic risk. Certara’s Cardiac Safety Simulator (CSS) is quantitative systems toxicology platform that integrates physiologically based pharmacokinetic modeling with a cardiomyocyte model to assess the proarrhythmic potential of drugs. The CSS was able to predict the effects for five of six antipsychotic drugs on QT prolongation as well as the QT prolongation due to the drug–drug interaction between domperidone and ketoconazole [33,34].
QST models for the in silico assessment of cardiovascular safety should include clinically relevant drug concentrations, accurate dose–response information for the inhibition of multiple ion channels, a calibrated cardiomyocyte model, output for the change in arrhythmia surrogate markers (APD, EAD, and QT prolongation), and statistical risk of arrhythmia. The ability to predict cardiotoxicity lays within the scope of the model, hence unknown molecular mechanisms of toxicity as well as inhibition of additional ion channels, not experimentally measured, could lead to discrepancies between in silico predicted and clinically observed toxicity. Therefore, the inclusion of a network-based systems pharmacology approach could provide utility for elucidating additional unknown mechanisms of toxicity [35]. Due to their multi-targeted nature, the off-target non-QT cardiotoxicity associated with tyrosine kinase inhibitors warrants a novel systems level method to identify mechanisms of toxicity, such as the multi-layer strategy suggested by Kariya et al. (2016) [36]. Amemiya et al. (2015), using a comparative systems toxicology approach, elucidated the molecular mechanisms responsible for sunitinib cardiotoxicity and identified a prophylactic intervention [37]. In the case where experimental data for the inhibition of ion channels by a drug is unavailable, QSAR modeling could be utilized to obtain approximations of initial parameter values [38]. Sensitivity analyses on multiscale systems toxicology models may provide insights into the importance of certain components on model outputs as well as help guide decisions on measurements to obtain experimentally. For example, if the complete inhibition of an ion channel results in a marginal change in APD or QT prolongation, in essence the clinical endpoint of interest is insensitive to changes in a particular ion channel, then spending resources to obtain accurate dose-inhibition measurements for that ion channel may not be necessary.
3.2. Hepatotoxicity
Hepatotoxicity, or drug induced liver injury (DILI), is also a leading cause of drugs withdrawn from the market and one of the main reasons for attrition during drug development [20]. DILI is defined as damage to liver cells due to the exposure of a drug, and it is of concern since it can lead to acute liver failure. DILI has been separated into two distinct classes of toxicity, intrinsic and idiosyncratic. Intrinsic hepatotoxicity is reproducible, dose-dependent, and easy to predict. Idiosyncratic hepatotoxicity occurs only in select individuals, exhibits a complicated dose–response profile, and is unpredictable. A complete mechanistic understanding of the complex physiological interactions between the administered drug, inflammatory processes, mitochondrial stress, metabolism, and other drugs, can enable better predictions of clinical DILI [39]. However, the complexities associated with predicting DILI, warrants a QST modeling approach.
There has been several in silico modeling efforts to predict DILI, which primary consist of (Q)SAR related approaches that aimed to relate structure-activity relationships between compounds to general hepatotoxicity as well as liver enzymes dynamics [40]. Cheng and Dixon (2003) were one of the first who attempted to predict hepatotoxicity using an in silico approach [41]. They built a QSAR-based decision tree model, which was able to predict the occurrence of dose-dependent hepatotoxicity for 44 out of 54 drugs. QSAR based approaches have been able to predict DILI with accuracy ranging from 60 to 90%, however analyses with few external validation compounds have biased these predictions towards one that is more favorable [42].
Similar to the endeavors to improve cardiovascular safety, collaborative efforts between academicians, pharmaceutical industries, and regulatory agencies have been taken in order to develop better computational and experimental methods in order to predict DILI. The DILI-sim Initiative is one of these collaborative efforts led by DILIsym Services, several pharmaceutical companies, and the FDA, in order to improve patient safety, reduce animal toxicity testing, and reduce resources required to develop new drugs. In order to facilitate this initiative, DILIsym Services has developed a QST platform (DILIsym™) to predict DILI. DILIsym™ has been used to predict hepatotoxicity in mice, rats, dogs, and humans based upon experimental data obtained in vitro, QSAR predictions for unknown parameter values, and in vitro-in vivo extrapolations. DILIsym™ was developed via a “middle-out” approach since modeling efforts began with the liver and bifurcated vertically to describe cellular/subcellular effects up to predictions of clinical biomarkers. DILIsym™ is broken up into a series of interconnected sub-models that include PBPK/metabolism, glutathione dynamics, mitochondrial dysfunction, ATP generation, NRF-2 pathway, bile salt homeostasis, hepatocyte life cycle, immune response, and biomarkers of DILI. The model has been used to predict species-specific toxicity of methapyrilene (MP), by using in vitro and physico-chemical characteristics of MP to estimate ADME properties, simulate clinically relevant pharmacokinetics, and predict plasma ALT concentrations [43]. However, the compound used in DILIsym™ development (acetaminophen) caused hepatotoxicity through the same mechanism as MP. Therefore, to ensure adequate predictability of other compounds, DILIsym™ has been extended to incorporate additional mechanisms of toxicity, such as the inclusion bile salt homeostasis through bile salt export pumps and efflux transporters [44]. Going forward, the combination of network/omics-based approaches could reveal novel mechanisms of drug-induced hepatotoxicity, which can be incorporated to extend the scope of the QST model. For example, an integrative cross-omics analysis revealed mechanisms of cyclosporine induced hepatotoxicity [45]. Components of the network generated in this study could be built into a QST model in order to better capture cyclosporine related DILI.
4. Future directions
Quantitative systems toxicology modeling maybe the solution to the long-range vision and strategy of the National Research Council (NRC) for the advancement of toxicity testing in the 21st century [46]. This vision was motivated by the need to expand our knowledge about the complex interactions of chemical exposures on human physiology and the impact on human health. To summarize this endeavor, a paradigm shift in toxicity testing is deemed necessary in order to satisfy four main objectives: (i) To provide broad coverage of chemicals, chemical mixtures, outcomes and life stages; (ii) To reduce the cost and time of testing; (iii) To use fewer animals and cause minimal suffering in the animals used; and (iv) To develop a more robust scientific basis for assessing health effects of environmental agents. These four objects, in essence, provide a guidance for the future of toxicity testing. Although these guidelines were prefaced for environmental sciences, we believe they are equally applicable to pharmaceutical sciences.
Compared to conventional approaches, NRCs vision of toxicity testing in the 21st century (TT21) proposes the use of modern technologies for elucidating mechanisms of toxicity, identifying and measuring toxicological biomarkers, and integrating computational methods to assess human health risks (Fig. 2). This vision proposes that toxicity testing has two components, toxicity-pathway assays and target testing, which serve as strategies to assess toxicological properties of new and existing compounds. Exposure to a toxic compound results in a perturbation of normal biology. Physiological adaptations at the cellular level are made to reverse harmful perturbations. However, repeated exposures may overwhelm the adaptive nature of this biological process, resulting in tissue injury and adverse health effects. The adaptive stress response can be enhanced with a pharmacological intervention in order to prevent toxicity and disease (Fig. 3). However, to accomplish this efficiently, the complex interactions between therapeutic drugs and the physiological system must be fully characterized.
Fig. 2.
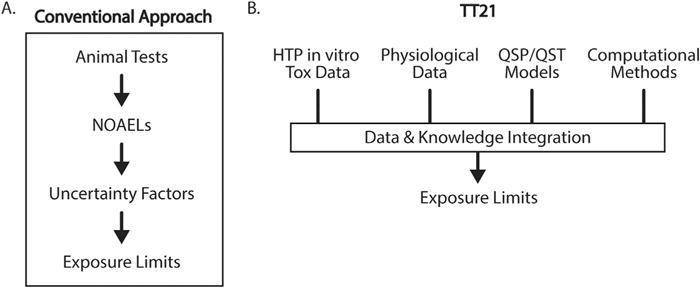
Comparison of conventional and TT21 approaches to the prediction of toxicity thresholds in humans. Historically, toxicity testing has relied on animal testing and experimental NOAELs. The NOAELs were used to determine exposure limits in humans via the application of multiple “rule of thumb” uncertainty factors, which were not informed by knowledge of toxicity mechanism or mathematical models of tissue dosimetry. By contrast, TT21 integrates knowledge and data from a variety of sources, including physiological and in vitro toxicity data. Extrapolation from in vitro systems to humans is performed using mathematical models, which allow in vitro data to be used in the context of cellular exposure. Computational methods are then applied to the model to account for physiological variability (inter- and intra-individual) and model or data uncertainty. The resulting exposure limit predictions are thus based on understanding of the underlying mechanism of toxicity, as well as knowledge of physiological variability in the target population.
Fig. 3.
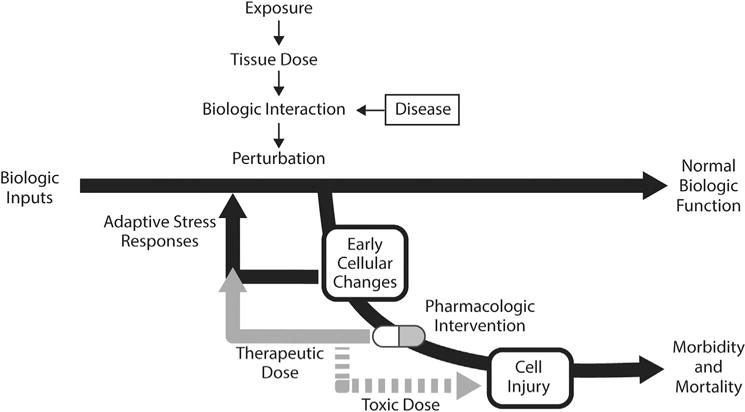
Interpretation of toxicity as a perturbation of normal biological function, and the effect of pharmacologic intervention. TT21 posits that exposure to toxic levels of chemicals induces changes in cellular biology. At low level, the cells’ innate adaptive stress responses can drive the cell back to normal biologic function. Similarly, disease can induce early changes in cellular function. Pharmacologic intervention at therapeutic doses can help return the cell to a state of normal biologic function. At toxic drug doses, however, the perturbation increase to a state of cell injury and potential morbidity or mortality.
Due to the complex interactions between pharmaceuticals and human physiology, the development of a quantitative computational framework like QST models is warranted. Information regarding drug pharmacokinetics, cellular physiology, toxicodynamics, interpatient characteristics, environmental exposures, and other potential descriptors/covariates relating to toxicity can be integrated into QST models. Technological advances that underpin TT21 also provide the foundation for what has become the Precision Medicine Initiative (PMI), or in other words personalized medicine. In particular, high throughput in vitro assays, such as Next-Generation Sequencing, combined with data repositories such as KEGG, the GO database, and the Protein Atlas, have provided the initial tools to understand toxicity mechanisms at the genomic scale. Therefore, QST models can be extended to incorporate patient-specific information in order to make individualized predictions of toxicity and risk assessments. Ultimately, as a means to satisfy and integrate the endeavors of TT21 and PMI, the development of quantitative systems pharmacology and toxicology models can allow for a complete inquisition of the personalized window between a therapeutic and toxic response.
In sum, immense collaboration and investment of resources amongst pharmaceutical industries, regulatory agencies, and non-profit/academic organizations, is required to fuel the development of disease/organ specific QST models. These models will serve as a platform where they can be applied in order to reduce resources in toxicity testing, improve patient safety, and advance our knowledge about pharmacological perturbations of physiological systems. There is much work needed to be done to enumerate the various mechanisms of toxicity for a plethora of chemicals, to elucidate and understand physiological responses across multiple scales of biological organization, and to gather the necessary data required to develop computational frameworks for toxicological risk assessments. Fortunately, we are in the era where technological/computational advancements in toxicity testing have begun to converge and the future of toxicity testing is just over the horizon (Fig. 4). One might be tempted, in fact, to think of this evolution in toxicological analysis as “precision toxicity,” or perhaps “individualized toxicity.”
Fig. 4.
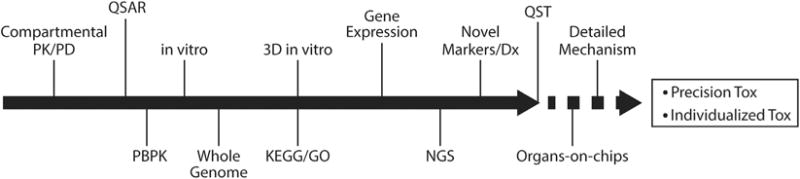
A vision for the convergence of technologies toward the future of QST. Starting with classical compartmental PK/PD, and continuing through the introduction of advanced in vitro test systems and genomic, high-volume data, toxicity testing has evolved greatly over the last few decades. At present, sophisticated models are being used to further elucidate the understanding the variety of toxicity mechanisms which lead to failed drugs and environmental risk. As the acquisition of patient-specific genomic and specialized diagnostics become more available, QST models will enable precise, individualized predictions of toxicity.
Acknowledgments
Funding
DKS is funded by NIH grant GM114179.
References
- 1.Schardein JL, Schwetz BA, Kenel MF. Species sensitivities and prediction of teratogenic potential. Environ Health Perspect. 1985;61:55–67. doi: 10.1289/ehp.856155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sorger PK, et al. An NIH white paper by the QSP workshop group. NIH; Bethesda: [Google Scholar]
- 3.Cros A. Action de l’alcool amylique sur l’organisme. 1863 [Google Scholar]
- 4.Meyer H. Zur theorie der alkoholnarkose. Naunyn-Schmiedeberg’s Arch Pharmacol. 1899;42:109–118. [Google Scholar]
- 5.Overton CE. Studien über die Narkose zugleich ein Beitrag zur allgemeinen Pharmakologie. Fischer; 1901. [Google Scholar]
- 6.Hansch C, Maloney PP, Fujita T, Muir RM. Correlation of biological activity of phenoxyacetic acids with Hammett substituent constants and partition coefficients. Nature. 1962;194:178–180. [Google Scholar]
- 7.Bajorath J. Integration of virtual and high-throughput screening. Nat Rev Drug Discov. 2002;1:882–894. doi: 10.1038/nrd941. http://dx.doi.org/10.1038/nrd941. [DOI] [PubMed] [Google Scholar]
- 8.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 1997;23:3–25. doi: 10.1016/s0169-409x(00)00129-0. http://dx.doi.org/10.1016/S0169-409x(96)00423-1. [DOI] [PubMed] [Google Scholar]
- 9.Von Bertalanffy L. General system theory. 1968:40. New York 41973. [Google Scholar]
- 10.Kitano H. Computational systems biology. Nature. 2002;420:206–210. doi: 10.1038/nature01254. http://dx.doi.org/10.1038/nature01254. [DOI] [PubMed] [Google Scholar]
- 11.Kitano H. Systems biology: a brief overview. Science. 2002;295:1662–1664. doi: 10.1126/science.1069492. http://dx.doi.org/10.1126/science.1069492. [DOI] [PubMed] [Google Scholar]
- 12.Teorell T. Kinetics of distribution of substances administered to the body, I: the extravascular modes of administration. Arch Int Pharmacodyn Ther. 1937;57:205–225. [Google Scholar]
- 13.Mager DE, Jusko WJ. General pharmacokinetic model for drugs exhibiting target-mediated drug disposition. J Pharmacokinet Pharmacodyn. 2001;28:507–532. doi: 10.1023/a:1014414520282. [DOI] [PubMed] [Google Scholar]
- 14.Levy G. Kinetics of pharmacologic effects. Clin Pharmacol Ther. 1966;7:362–372. doi: 10.1002/cpt196673362. [DOI] [PubMed] [Google Scholar]
- 15.Mager DE, Wyska E, Jusko WJ. Diversity of mechanism-based pharmacodynamic models. Drug Metab Dispos. 2003;31:510–518. doi: 10.1124/dmd.31.5.510. [DOI] [PubMed] [Google Scholar]
- 16.Iyengar R, Zhao S, Chung SW, Mager DE, Gallo JM. Merging systems biology with pharmacodynamics. Sci Transl Med. 2012;4:126ps127. doi: 10.1126/scitranslmed.3003563. http://dx.doi.org/10.1126/scitranslmed.3003563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jusko WJ. Moving from basic toward systems pharmacodynamic models. J Pharm Sci. 2013;102:2930–2940. doi: 10.1002/jps.23590. http://dx.doi.org/10.1002/jps.23590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jager T, Albert C, Preuss TG, Ashauer R. General unified threshold model of survival–a toxicokinetic-toxicodynamic framework for ecotoxicology. Environ Sci Technol. 2011;45:2529–2540. doi: 10.1021/es103092a. http://dx.doi.org/10.1021/es103092a. [DOI] [PubMed] [Google Scholar]
- 19.Peck CC, et al. Opportunities for integration of pharmacokinetics, pharmacodynamics, and toxicokinetics in rational drug development. J Clin Pharmacol. 1994;34:111–119. doi: 10.1002/j.1552-4604.1994.tb03974.x. [DOI] [PubMed] [Google Scholar]
- 20.Piccini JP, et al. Current challenges in the evaluation of cardiac safety during drug development: translational medicine meets the Critical Path Initiative. Am Heart J. 2009;158:317–326. doi: 10.1016/j.ahj.2009.06.007. http://dx.doi.org/10.1016/j.ahj.2009.06.007. [DOI] [PubMed] [Google Scholar]
- 21.Yap YG, Camm AJ. Drug induced QT prolongation and torsades de pointes. Heart. 2003;89:1363–1372. doi: 10.1136/heart.89.11.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Noble D. Cardiac action and pacemaker potentials based on the Hodgkin-Huxley equations. Nature. 1960;188:495–497. doi: 10.1038/188495b0. [DOI] [PubMed] [Google Scholar]
- 23.Beeler GW, Reuter H. Reconstruction of the action potential of ventricular myocardial fibres. J Physiol. 1977;268:177–210. doi: 10.1113/jphysiol.1977.sp011853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Colatsky T, et al. The Comprehensive in Vitro Proarrhythmia Assay (CiPA) initiative – update on progress. J Pharmacol Toxicol Methods. 2016;81:15–20. doi: 10.1016/j.vascn.2016.06.002. http://dx.doi.org/10.1016/j.vascn.2016.06.002. [DOI] [PubMed] [Google Scholar]
- 25.O’Hara T, Virag L, Varro A, Rudy Y. Simulation of the undiseased human cardiac ventricular action potential: model formulation and experimental validation. PLoS Comput Biol. 2011;7:e1002061. doi: 10.1371/journal.pcbi.1002061. http://dx.doi.org/10.1371/journal.pcbi.1002061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Z, et al. A temperature-dependent in silico model of the human ether-a-go-go-related (hERG) gene channel. J Pharmacol Toxicol Methods. 2016;81:233–239. doi: 10.1016/j.vascn.2016.05.005. http://dx.doi.org/10.1016/j.vascn.2016.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gomez JF, Cardona K, Romero L, Ferrero JM, Jr, Trenor B. Electrophysiological and structural remodeling in heart failure modulate arrhythmogenesis. 1D simulation study. PLoS One. 2014;9:e106602. doi: 10.1371/journal.pone.0106602. http://dx.doi.org/10.1371/journal.pone.0106602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang PC, Clancy CE. In silico prediction of sex-based differences in human susceptibility to cardiac ventricular tachyarrhythmias. Front Physiol. 2012;3:360. doi: 10.3389/fphys.2012.00360. http://dx.doi.org/10.3389/fphys.2012.00360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mirams GR, et al. Prediction of thorough QT study results using action potential simulations based on ion channel screens. J Pharmacol Toxicol Methods. 2014;70:246–254. doi: 10.1016/j.vascn.2014.07.002. http://dx.doi.org/10.1016/j.vascn.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.ten Tusscher KH, Noble D, Noble PJ, Panfilov AV. A model for human ventricular tissue. Am J Physiol Heart Circ Physiol. 2004;286:H1573–H1589. doi: 10.1152/ajpheart.00794.2003. http://dx.doi.org/10.1152/ajpheart.00794.2003. [DOI] [PubMed] [Google Scholar]
- 31.Grandi E, Pasqualini FS, Bers DM. A novel computational model of the human ventricular action potential and Ca transient. J Mol Cell Cardiol. 2010;48:112–121. doi: 10.1016/j.yjmcc.2009.09.019. http://dx.doi.org/10.1016/j.yjmcc.2009.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Okada J, et al. Screening system for drug-induced arrhythmogenic risk combining a patch clamp and heart simulator. Sci Adv. 2015;1:e1400142. doi: 10.1126/sciadv.1400142. http://dx.doi.org/10.1126/sciadv.1400142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Glinka A, Polak S. The effects of six antipsychotic agents on QTc–an attempt to mimic clinical trial through simulation including variability in the population. Comput Biol Med. 2014;47:20–26. doi: 10.1016/j.compbiomed.2014.01.010. http://dx.doi.org/10.1016/j.compbiomed.2014.01.010. [DOI] [PubMed] [Google Scholar]
- 34.Mishra H, Polak S, Jamei M, Rostami-Hodjegan A. Interaction between domperidone and ketoconazole: toward prediction of consequent QTc prolongation using purely in vitro information. CPT Pharmacomet Syst Pharmacol. 2014;3:e130. doi: 10.1038/psp.2014.26. http://dx.doi.org/10.1038/psp.2014.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao S, Iyengar R. Systems pharmacology: network analysis to identify multiscale mechanisms of drug action. Annu Rev Pharmacol Toxicol. 2012;52:505–521. doi: 10.1146/annurev-pharmtox-010611-134520. http://dx.doi.org/10.1146/annurev-pharmtox-010611-134520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kariya Y, Honma M, Suzuki H. In: Systems pharmacology and pharmacodynamics. Mager Donald E, Kimko Holly HC, editors. Springer International Publishing; 2016. pp. 353–370. [Google Scholar]
- 37.Amemiya T, et al. Elucidation of the molecular mechanisms underlying adverse reactions associated with a kinase inhibitor using systems toxicology. NPJ Syst Biol Appl. 2015;1 doi: 10.1038/npjsba.2015.5. http://dx.doi.org/10.1038/npjsba.2015.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wisniowska B, Mendyk A, Szlek J, Kolaczkowski M, Polak S. Enhanced QSAR models for drug-triggered inhibition of the main cardiac ion currents. J Appl Toxicol. 2015;35:1030–1039. doi: 10.1002/jat.3095. http://dx.doi.org/10.1002/jat.3095. [DOI] [PubMed] [Google Scholar]
- 39.Russmann S, Kullak-Ublick GA, Grattagliano I. Current concepts of mechanisms in drug-induced hepatotoxicity. Curr Med Chem. 2009;16:3041–3053. doi: 10.2174/092986709788803097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Przybylak KR, Cronin MT. In silico models for drug-induced liver injury–current status. Expert Opin Drug Metab Toxicol. 2012;8:201–217. doi: 10.1517/17425255.2012.648613. http://dx.doi.org/10.1517/17425255.2012.648613. [DOI] [PubMed] [Google Scholar]
- 41.Cheng A, Dixon SL. In silico models for the prediction of dose-dependent human hepatotoxicity. J Comput Aided Mol Des. 2003;17:811–823. doi: 10.1023/b:jcam.0000021834.50768.c6. [DOI] [PubMed] [Google Scholar]
- 42.Chen M, et al. Toward predictive models for drug-induced liver injury in humans: are we there yet? Biomark Med. 2014;8:201–213. doi: 10.2217/bmm.13.146. http://dx.doi.org/10.2217/bmm.13.146. [DOI] [PubMed] [Google Scholar]
- 43.Howell BA, et al. In vitro to in vivo extrapolation and species response comparisons for drug-induced liver injury (DILI) using DILIsym: a mechanistic, mathematical model of DILI. J Pharmacokinet Pharmacodyn. 2012;39:527–541. doi: 10.1007/s10928-012-9266-0. http://dx.doi.org/10.1007/s10928-012-9266-0. [DOI] [PubMed] [Google Scholar]
- 44.Woodhead JL, et al. Mechanistic modeling reveals the critical knowledge gaps in bile acid-mediated DILI. CPT Pharmacomet Syst Pharmacol. 2014;3:e123. doi: 10.1038/psp.2014.21. http://dx.doi.org/10.1038/psp.2014.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Van den Hof WF, et al. Integrative cross-omics analysis in primary mouse hepatocytes unravels mechanisms of cyclosporin A-induced hepatotoxicity. Toxicology. 2014;324:18–26. doi: 10.1016/j.tox.2014.06.003. http://dx.doi.org/10.1016/j.tox.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 46.Council NR. Toxicity testing in the 21st century: a vision and a strategy. National Academies Press; 2007. [PubMed] [Google Scholar]