

Abstract
Purpose
Non-small cell lung cancer is the leading cause of cancer related deaths globally.
Considering the side effects and diminishing chemosensitivity to chemotherapy, novel treatment approaches are sought. Hence, we aim to develop a liposomal co-delivery system of pDNA expressing shRNA against PFKFB3 (pshPFKFB3) and docetaxel (DTX).
Methods
Cationic DTX liposomes complexed with pshPFKFB3 (PSH-DL) were developed. In vitro cell line studies were performed to evaluate transfection, PFKFB3 mRNA silencing, cytotoxicity, pGP inhibition, and protein markers expression. In vivo efficacy study was performed in A549 xeno-graft nude mice model.
Results
Cytotoxicity studies showed significantly enhanced anticancer activity of PSH-DL against individual treatment alone confirming the chemoenhancing effect of pshPFKFB3 on DTX activity. Fluorescence microscopy and RT-PCR showed effective transfection and RNAi by pshPFKFB3. pGP inhibition assay and western blotting revealed that PFKFB3 downregulation caused diminution of pGP activity leading to changes in cell cycle (Cdk2), survival (survivin), apoptosis (Bcl2 and cleaved caspase 3) and stress (p-JNK and p-p38) markers so that induces apoptosis by PSH-DL in NSCLC cells. PSH-DL also showed ~3.8-fold reduction in tumor volume in A549 xenograft model which was significantly higher than individual treatments alone.
Conclusion
Targeting PFKFB3 through shRNA based RNAi is a promising approach for potentiating activity of DTX in NSCLC.
Keywords: docetaxel liposome, gene delivery, lung cancer, PFKFB, shRNA
INTRODUCTION
Lung cancer and particularly non-small cell lung cancer (NSCLC) is the foremost cancer killer for both men and women in the United States (1). The survival rate with lung cancer is approximately 15% which is significantly less than other prevalent cancers, such as breast and prostate cancer. First line of treatment for lung cancer has been chemotherapy for decades and only little success has been achieved; the therapy usually is accompanied with serious dose-limiting side effects (2). Hence, delivery approaches for effective treatment with limited toxicity are sought.
Cancer cells maintain an unusually high glycolytic rate even in the presence of oxygen, a phenomenon first described by Otto Warburg over 75 years ago (3). The glycolytic flux is 2 to 17-folds higher in tumor cells compared to normal cells (4). This is because of the high expression of all the glycolytic enzymes such as phosphofructokinase (6-Phosphofructo-2-Kinase/Fructose-2, 6-Biphosphatase-3, PFKFB3), hexokinase, pyruvate kinase, etc. and transporters induced by several oncogenes and the hypoxia-inducible factor-1 (HIF-1) (5). This leads to the excretion of large amounts of lactate even under aerobic conditions, a phenomenon known as the “Warburg effect”. This high glycolytic rate contributes to the ATP supply for cellular work and provides numerous intermediaries which are precursors for the synthesis of macromolecules, such as polysaccharides, nucleic acids, triglycerides and proteins required for cell proliferation (6). Thus, such high glycolytic flux extrapolates well to tumor progression, invasiveness, metastatic tendency and patient mortality and morbidity (7,8). Hence, glycolytic pathway is a suitable target in which over-expressed enzymes can be exploited for drug delivery for the treatment of cancer.
The type of isoenzyme expression is another important parameter besides the higher glycolytic rate exhibited by tumor cells (9). Among the enzymes playing role in glycolysis, four allosteric PFKFB enzymes 1–4 expressed by four independent PFKFB genes, catalyze the rate-limiting phosphorylation of fructose-6-phosphate to fructose-1, 6-bisphosphate, using ATP as the energy source in the glycolysis pathway. Among these four allosteric enzymes, PFKFB3 enzyme retains the highest Kinase/Biphosphatase activity ratio and is expressed by PFKFB3 gene which has been demonstrated to be highly expressed in leukemic cells and in solid tumors (10). Moreover, mitogenic, hypoxic and inflammatory conditions have an inductive effect on the expression of PFKFB3. Hence upregulation of PFKFB genes specific to cancer cells compared to their normal counterparts (from the same patients) with more robust over-expression in breast and lung cancer make it a more appropriate target (8).
There are few reports of potent and selective PFKFB3 inhibitors. Briefly, 3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one (3PO) is a PFKFB3 inhibitor and has shown cytostatic activity in leukemic and other cancer cells (11). Recently, a novel derivative, PFK158, which is a potent and selective inhibitor of PFKFB3 is being investigated in phase I study in patients with advanced solid malignancies (12). According to Boyd et al., 3PO and PFK158 are inactive in the PFKFB3 kinase assay due to absence of binding sites in these molecules. Other inhibitors, such as pyridazinone inhibitors are also being investigated which have been reported to have an IC50 greater that 1 mM (13). Thus, we propose to use a gene delivery system which will utilize plasmid for shRNA against PFKFB3. The approach is novel in its being a plasmid which will provide constant shRNA expression for RNAi (RNA interference) and provide better therapeutic action by removing the target mRNA and hence the protein, unlike the inhibitors which will only block the function of protein. This approach is also advantageous over siRNA based RNAi by having low off-target effects, low immunological reactivity and toxicity, high in vivo stability and therapeutic potential (14).
Docetaxel (DTX) has been used as a primary agent for the treatment of solid tumors such as in lung, breast and pancreatic cancers (15–17). Currently available marketed formulations of taxanes (DTX and paclitaxel) face issues of resistance development and toxicities which are dose limiting (18). Abraxane, which is a nanoparticle albumin-bound paclitaxel, has been approved for metastatic pancreatic, lung and breast cancer. It is used in combination with carboplatin for the initial treatment of patients with locally advanced or metastatic NSCLC who are not candidates for curative surgery or radiation therapy (19). Taxotere and taxol, which are micellar formulations of DTX and paclitaxel, respectively, are used for intravenous therapy of breast, lung, prostate, gastric, head and neck, ovarian and pancreatic cancer. However, both the drugs have dose dependent side effects like neutropenia, alopecia and anemia. Hence, a formulation strategy is required that can overcome its toxicity while providing better alternative treatment approach in comparison to those available in the clinic. Liposomes are able to entrap hydrophobic drugs in the bilayer and provide protection from degradation, reduce toxicity, and possibly help in overcoming multidrug resistance (20). Furthermore, liposomes can be modified using appropriate selection of lipids to load large hydrophobic drugs such as DTX in the bilayer along with the use of cationic lipids to complex with therapeutic genes (21). Liposomal co-delivery of the drug with the shRNA plasmid against PFKFB3 would be of added advantage by providing a simultaneous delivery of drug and gene using the same nano-particulate carrier to the tumor site.
The objective of this study was to evaluate the therapeutic potential of plasmids to provide pool of shRNA in cells for silencing PFKFB3 and evaluate the efficacy of the co-delivery of a chemotherapeutic agent with such RNAi agent. The objective here was to develop a non-toxic liposomal system of DTX and plasmid which can express PFKFB3 shRNA for inhibiting a key enzyme in the glucose metabolism for parental delivery. The co-delivery liposomal system was evaluated in lung cancer cells in vitro for its therapeutic effects. Further, in vivo A549 xenograft model was used to evaluate the therapeutic potential of the system and proof-of-concept was established through mechanistic studies to characterize the involvement of specific pathways in the therapeutic effectiveness of the combination.
MATERIALS AND METHODS
Materials
Docetaxel (DTX) (LC Laboratories, Woburn, MA) was dissolved in dimethyl sulfoxide (DMSO) to prepare 1 mM stock solution and aliquots were stored at −20°C. Stock solutions were diluted to the desired final concentrations with medium just before use. 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), N- (2,3-Dioleoyloxy-1-propyl) trimethylammonium bromide (DOTAB) and 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-methoxy (polyethyleneglycol) (DSPE-PEG-2000) were obtained from Lipoid LLC, Germany. Dimethyldioctadecyl-ammonium bromide (DDAB) was purchased from Sigma–Aldrich (St. Louis, Missouri). All the chemicals were used without any further purification. The mouse antibodies against β- actin were purchased from Santa Cruz Biotechnology (Dallas, TX). The rabbit antibodies against Cdk2, Survivin, Bcl2, Cleaved Caspase 3, JNK, pJNK, p38 and p-p38 were purchased from Cell Signaling Technology (Danvers, MA). Secondary goat anti-rabbit or anti-mouse antibodies conjugated with horseradish peroxidase was bought from Santa Cruz Biotechnology (Dallas, TX). iScript™ Advanced cDNA Synthesis Kit and SsoAdvanced™ Universal SYBR® Green Supermixwere purchased from Bio rad (California, USA). Q-PCR primers for mouse PFKFB3 were purchased from Origene (Rockville, MD). PFKFB3 plasmid (pshPFKFB) was obtained from Origene Technologies (Rockville, MD).
Cell Lines
The cell lines, A549 (CCL- 185), H460 (HTB- 177) and HEK293 (CRL- 1573), were obtained from American Type Culture Collection (ATCC). All cell lines were maintained in Dulbecco’s Modified Eagle’s medium (DMEM; Sigma Aldrich, St Louis, MO) nutrient mixture supplemented with 10% fetal bovine serum (FBS) from Invitrogen (Grand Island, NY) and antibiotic-antimycotic mixture comprising penicillin (5000 U/mL), streptomycin (0.1 mg/mL) and neomycin (0.2 mg/mL) from Sigma Aldrich (St Louis, MO, USA). The cell lines were sub-cultured when they were approximately 80% confluent with 0.25% trypsin-EDTA (Invitrogen, Grand Island, NY).
Methods
Preparation and Characterization of co-Loaded Liposomes
Preparation of Different Liposomal Formulations
DTX liposomes (DL) were prepared by ether injection method. Briefly, DTX, DOPC, DOPE, DOTAP, DDAB and DSPE-mPEG2000 were dissolved in 1.5 mL diethyl ether. The solution was then injected into 10 ml of distilled water at 55°C. The suspension formed was kept at the same temperature for 60 min under constant stirring to form multi-lamellar vesicles (MLV). The small unilamellar vesicles were obtained from the MLV suspension by ultra-sonication (Vibra Cell™, 130w, 20 kHz, 4 min). DL were optimized using DoE study using Box-Behnken design (Design Expert 7.0, Sate Ease, MA) using entrapment efficiency and particle size. Optimized liposomes were used for complexation with pshPFKFB3 to obtain co-loaded liposomes (PSH-DL). DTX liposomes complexed with negative control pDNA (NC-pDNA) (NC-pDNA-DL) were used for in vitro and in vivo studies. For this, DL were incubated with pshPFKFB3 or NC-pDNA at room temperature at increasing w/w ratio of pshPFKFB3 to cationic lipids and were analyzed for complexation efficiency. Similarly, liposomes without DTX complexed with pshPFKFB3 (PSH-L) were also prepared using the same lipid composition for in vitro studies.
Physicochemical Characterization of Liposome
Particle size and polydispersity of the liposomes were determined by photon correlation spectroscopy (PCS) using Zeta sizer (PSS Systems, USA). Zeta potential of liposomes was determined by electrophoretic mobility determination using Zeta sizer (PSS systems, USA). The liposome samples were analyzed after appropriate dilution with deionized water.
For the entrapment efficiency, the amount of DTX encapsulated in the liposomes was determined by HPLC. A reverse-phase HPLC column (Agilent Eclipse XDB-C18, 4.6 × 250 mm, 5 μm) was used. Briefly, formulation was centrifuged at 5000 rpm for 10 min for pelleting the unentrapped DTX. Supernatant containing DTX liposomes was mixed with methanol (1:4 v/v) for liposomal disruption and then diluted appropriately with mobile phase consisting of acetonitrile and deionized water (60:40, v/v). The flow rate of mobile phase was set to 1.0 mL/min. The run time was set at 15 min and the column temperature was ambient. Prior to the injection of the drug solution, the column was equilibrated for at least 0.5 h with the mobile phase flowing through the system. The column effluent was detected with a PDA detector at 227 nm. The calibration curve was linear in the range of 50–50,000 ng/ml with a regression coefficient R2 = 0.999. The drug encapsulation efficiency was defined as the percentage of the amount of DTX encapsulated in the liposomes to total amount of drug.
Complexation efficiency of pshPFKFB3 with the liposomes was evaluated by centrifugation and UV analysis was done using NanoDrop 1000 (ThermoScientific, USA). Briefly, PSH-L and PSH-DL were centrifuged at 18000 rpm for 2 h at 4°C (Beckman J2-HC Centrifuge, Beckman, US) to settle down the liposomes. Supernatant was analyzed spectrophotometrically and content of pDNA was calculated using the standard expression OD1 = 50 μg/mL of double stranded pDNA. Readings from formulations were compared to readings from standard dilution of naked pshPFKFB3. Complexation efficiency was calculated using following equation;
Where, ApshPFKFB3standard is absorbance of the standard naked pshPFKFB3, Asupernatant is absorbance of supernatant after centrifugation:
In Vitro Cell Cytotoxicity
Briefly, A549, H460 and HEK293 cells were plated at 10,000 cells/well in a 96-well plate. Cells were treated either with DTX solution or NC-pDNA-DL at different concentrations for 48 h to check the effect of the liposomes. Cells were transfected with either control plasmid or plasmid directed against PFKFB3 (pshPFKFB3) at 40 ng/well along with DTX liposomes with various concentrations for 48 h. Cells were washed with PBS and 100 μl of 0.5% (v/v) glutaraldehyde was added for fixing the plate and incubated for 30 min. Next 0.5% (w/v) crystal violet dye was added and the plate was incubated for another 20 min. The plate was washed with tap water and dried before adding 1% (w/v) sodium lauryl sulfate (SLS) and read at 570 nm.
Cellular Uptake and Transfection Efficiency
Briefly, A549 cells were grown to 60–70% confluency and incubated in serum-free medium with PSH-L for 4 h at 37°C and 5% CO2 in quantities required to deliver 20–100 ng of DNA per 10,000 cells. 100 ng of pshPFKFB3 was complexed with Lipofectamine 2000 using manufacturer’s protocol to use as positive reference control. The cells were incubated for 48 h with regular media. After incubation, the medium was removed and cells were washed twice with sterile PBS and GFP expression was visualized by fluorescence microscopy using an Olympus BX40 fluorescence microscope (22).
Quantitative RT-PCR
Total RNA from cells was extracted using Tiazol reagent and treated with RNase-free DNase to remove any residual genomic DNA. Single-stranded cDNAs were synthesized by incubating total RNA (5 μg) with 5X reaction mix, reverse transcriptase and Nuclease free water at 46°C for 20 min and 95°C for 1 min in a final volume of 20 μl. Quantitative real-time PCR (RT-PCR) was performed to compare expression levels of each of the total RNA sample. RT-PCR was performed on CFX96 Real-Time System (Bio rad, California, USA) in the presence of 10 μl SYBR Green Supermix, 1 μl cDNA, and H2O was added to a final volume of 20 μl. RT-PCR was performed with an initial denaturation step of 30 s at 95°C, followed by 40 cycles of 30 s at 95°C, 5 s at 65°C and annealing temperature at 60°C. PCR products were monitored in real time by measuring the increase in fluorescence caused by the binding of SYBR Green I Dye.
pGP Inhibition Assay
Briefly, A549 cells were plated in 20,000 cells/well in a 24-well plate and allowed to attach overnight. The cells were treated with different concentrations of pshPFKFB3, and incubated for 24 h at 37°C. Wells were treated with Rh123 alone or with PSH-L /verapamil and incubated for 60 min and then washed twice with PBS. Fluorescence microscopic images were taken to observe the uptake of Rh123 in the presence of pshPFKFB3 and verapamil.
Apoptosis Analysis AO/EB Double-Staining and Fluorescent Microscopy
Briefly, A549 and H460 cells were seeded at a density of 10,000 cells/well plate in a 96-well plate and allowed to attach for 24 h. The cells were treated with or without pshPFKFB3, DL and PSH-DL complexes and incubated for 24 h at 37°C. A control was maintained as untreated cells. The cells were washed twice with PBS and incubated with 3 mg/ml acridine orange (AO) and 3 mg/mL ethidium bromide (EB) for 30 min. The cells were washed twice with PBS before observing them with fluorescence microscopy using an Olympus BX40 fluorescence microscope.
Western Blot
Western blotting was carried out on cells treated with different formulations for analyses of various protein markers Cdk2, Survivin, Bcl2, Cleaved Caspase 3, JNK, pJNK, p38 and p-p38 and PFKFB3. Protein lysates were prepared from A549 and H460 cells. Briefly, after 48 h post incubation with PSH-L, DL and PSH-DL; the cells were incubated in RIPA buffer (50 nM Tris-HCl, pH 8.0, with 150 nM Sodium Chloride, 1.0% Igepal CA-630 (NP-40), 0.5% Sodium Deoxychlorate, and 0.1% sodium dodecyl sulfate) with protease inhibitor (500 mM phenylmethylsulfonyl fluoride). Protein concentrations were determined according to BCA protein assay using manufacturer’s protocol (Rockford, IL) and the standard plot was generated by using bovine serum albumin (BSA). Briefly, 50 μg of protein from the control sample and all the different treatment samples were denatured by boiling at 100°C for 5 min in SDS sample buffer and subsequently electrophoresed in 10% SDS-PAGE gel and then transferred into nitrocellulose membranes followed by blocking with 5% BSA in tris-buffered saline with Tween 20 (10 mM tris-HCl (pH 7.6), 150 mM NaCl, and 0.5% Tween), and probed with antibodies against Cdk2, Survivin, Bcl2, Cleaved Caspase 3, JNK, pJNK, p38 and p-p38 at 1:500 and PFKFB3 at 1:200 for the lung cancer cells. Proteins were detected with HRP conjugated secondary antibodies using SuperSignal West pico chemiluminescent solution (PIERCE, Rockford, IL). Densitometric analysis of bands was performed as per our previous methods using Chemi-Doc XRS+ Imaging system (Bio-Rad). Results were expressed as percentage ratios of protein expression to beta-actin (set to 100%) and plotted against the relative intensity compared to the control.
In Vivo Anti-Tumor Activity
The antitumor efficacy of different formulations of PSH-L, DL and PSH-DL were, investigated using A549 tumor xenograft model in nude mice already established in our laboratory. BALB/c athymic nudes were purchased from Charles River Laboratories (Wilmington, Maryland) and were kept in specific pathogen-free conditions in a facility approved by the American Association for Accreditation of Laboratory Animal Care (AAALAC). All experiments were done in accordance with the guidelines of the Institutional Animal Care and Use Committee (IACUC) at Florida A&M University. For this, 4×106 A549 human lung cancer cells were subcutaneously injected into the right flank of the mice. When tumor volume approximately reached 150 mm3, respective formulations were given intravenously at a dose of 5 mg/kg of DTX, once every 3 days for 2 weeks. Subsequently, tumor volume was measured at specified time using Vernier caliper in two dimensions. Tumor volume (V) was measured by the formula: V = (L×W2)/2, where length (L) is the longest diameter and width (W) is the shortest diameter perpendicular to length.
RESULTS
Preparation and Characterization of Dual Functioning Liposomes
Preparation of DTX Liposomes
DTX liposome formulation was prepared by solvent evaporation method by using different ratios of phospholipid mixtures. Several batches of DTX liposomes were prepared according to the DoE studies using the Box Behnken design with the quadratic model. The results are shown in the Supplementary information S1. The particle size of DTX liposomes ranged from 84.0 nm to 133.7 nm and the entrapment efficiency of DTX ranged from 43.6% to 98.9%. Optimized DTX liposome (using Desirability index based selection method) was made based on lowest particle size and highest entrapment efficiency and the composition of optimum batch was 10 mg DTX, 150 mg DOPC, 40 mg cationic lipids (DOTAP and DDAB), 40 mg DOPE and 30 mg DSPE-mPEG2000. This formulation had particle size of 91.2 ± 9.4 nm, zeta potential of +22.3 ± 1.8 mV, entrapment of 87.3 ± 2.6% as shown in supplementary information S1. Docetaxel loading in total lipids used is 3.36 ± 0.09% w/w. The optimized lipid composition was further used for complexation with pshPFKFB3 or with NC-pDNA. Particle size distribution, zeta potential and entrapment efficiency data of different liposomal formulations are shown in Table I.
Table I.
Characteristics of Docetaxel Liposome Formulation and the Corresponding Placebo
| Formulation | Particle size (nm) | Polydispersity | Zeta potential (mV) | Entrapment efficiency (%) |
|---|---|---|---|---|
| Blank liposomes (without DTX and pshPFKFB3) | 87.3 ± 2.3 | 0.204 ± 0.009 | +21.6 ± 1.9 | – |
| DTX liposomes (From DoE) | 91.2 ± 9.4 | 0.269 ± 0.012 | +22.3 ± 1.8 | 87.3 ± 2.6 |
| DL | 105.6 ± 7.4 | 0.265 ± 0.021 | +17.8 ± 2.2 | 89.7 ± 4.1 |
| PSH-DL | 110.3 ± 8.3 | 0.285 ± 0.019 | +18.5 ± 0.9 | 88.9 ± 3.4 |
Preparation of PSH-DP
Briefly, pshPFKFB3 complexed liposomes were prepared using the lipid composition utilized for DTX liposomes. The results are shown in Fig. 1. The complexation efficiency of liposomes was evaluated by centrifugation/UV spectrophotometric analyses at different w/w ratios of cationic lipids to pDNA. The weight ratio that allowed complete complexation of the pDNA was found to be 2:1 and it corresponded to the N/P ratio of 2.2 indicating 2.2 M excess of cationic lipids over phosphates of pDNA. The weight ratio higher than 2.2 did not significantly increase the pDNA complexation. Similarly, DTX lipoplexes complexed with NC-pDNA were prepared at the same weight ratio. The particle size and zeta potential of the complexed liposomes were found to be 110.3 ± 8.3 nm and 18.5 ± 0.9 mV. Complexation of the pDNA did not cause any significant increase in the size of the liposomes indicating that the liposomal bilayer retained its integrity upon complexation with pDNA and maintained the small unilamellar vesicular structure. The marginal increase in the mean particle size can be accounted to the pDNA complexation to the liposomes. Moreover, as expected, reduction in the zeta potential of the liposomes was in line with the pDNA complexation to the cationic liposomes.
Fig. 1.
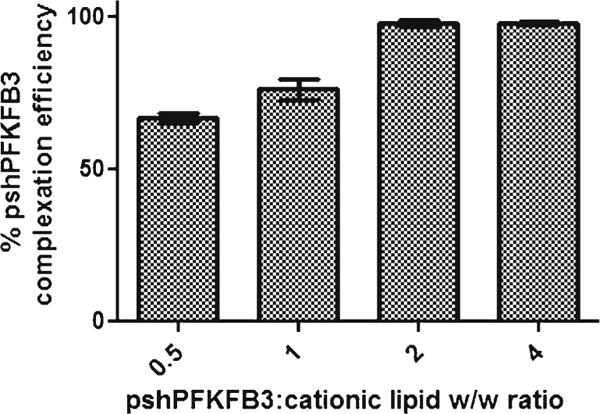
Complexation efficiency of pshPFKFB3 with DL at different w/w ratios of plasmid to cationic lipids. The results show mean ± SEM of three independently performed experiments.
Cytotoxicity of DTX and pshPFKFB3
Optimized liposomes of DTX and pshPFKFB3 were first evaluated for their cytotoxicity against NSCLC cells in vitro. Initially, individual cytotoxicity of the DTX liposomes complexed with NC-pDNA (negative control pDNA) and PSH-L without DTX loading were formulated to determine their individual effects.
Cytotoxicity of DTX Liposome
The cytotoxicity of DL was evaluated against DTX solution against A549 and H460 cells by incubating cells with various concentrations of DTX for 24 and 48 h. In-vitro cytotoxicity results showed that after 48 h of exposure to DTX solution, the IC50 values of A549 and H460 cells were 0.43 μM and 0.37 μM and were decreased to 0.32 μM and 0.20 μM, respectively when treated with DTX liposomes. The time dependent cytotoxicity by DTX observed with the DTX-liposomal formulation was possibly due to cell internalization of nanoparticles over a period of time (Table II).
Table II.
IC50 of Different Lung Cancer Cells for Docetaxel Solution and Docetaxel Liposome over a Period of 24 and 48 h
| Treatment | Cell line | 24 H | 48 H |
|---|---|---|---|
| DTX Solution (μM) | A549 | 0.95 | 0.43 |
| H460 | ˃2 | 0.37 | |
| DTX Liposome (μM) | A549 | 0.23 | 0.32 |
| H460 | ˃2 | 0.20 |
Cytotoxicity of Cationic Lipids on Normal Cells
The cytotoxicity of the blank liposomes was evaluated against HEK293 cells to determine the safety of the formulated delivery systems (23) (Fig. 2). The cells were treated with different concentrations of cationic lipids and incubated for 48 h. The cells showed a viability of approximately 60% even in the highest concentration of 6 mg/mL of cationic lipid and up to 3 mg/mL concentration, cells retained ≥80% viability.
Fig. 2.
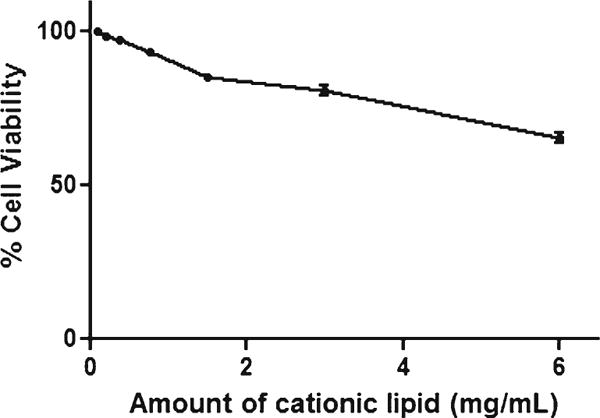
In vitro cell viability of blank liposome for HEK293 cells.
Cytotoxicity of DTX-pshPFKFB3 in Combination
The effect of PSH-L was evaluated with A549 and H460 cells and it was observed that cytotoxicity of pshPFKFB3 was 3.3% and 3.8% for A549 and H460, respectively; but when combined with DL at concentrations of 0.25 μM, 0.50 μM and 1.0 μM, the cytotoxicity increased to 66.6%, 72.5% and 85.4% for A549 NSCLC cells; 30.8%, 52.4% and 70.4% for H460 NSCLS cells respectively (Fig. 3). Both A549 and H460 showed dose dependent increase in cellular toxicity.
Fig. 3.
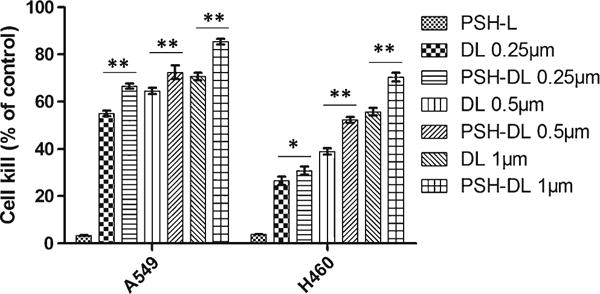
Cytotoxicity of PSH-DL complexes against A549 and H460. The results show mean ± SEM of three independently performed experiments. ns, statistically non-significant, compared with DL. *, P < 0.05, compared with DL. **, P < 0.01, compared with DL.
Effect of pshPFKFB3 on the Anticancer Activity of DTX Liposome
Inhibition of PFKFB3 mRNA Levels by pshPFKFB3
For quantitative estimation of effect of formulations on PFKFB3 mRNA expression, total RNAs from A549 cells were analyzed by RT- PCR after reverse transcription of the RNA into cDNA. The results are shown in Fig. 4. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA was kept as a housekeeping gene. Liposomes prepared with negative control plasmid (NC-pDNA-L) did not show any silencing of the PFKFB3 mRNA. It was observed that the PFKFB3 mRNA levels decreased by approximately 11% at 20 ng and 35% at 40 ng plasmid level as compared to the control which was significantly lower than control. Interestingly, there was no significant decrease (p > 0.05) in the PFKFB3 mRNA level when concentration of plasmid was increased from 40 ng to up to 100 ng per 10,000 cells. Therefore, inhibition of PFKFB3 mRNA expression by developed PSH-L was found to be effective between 40 ng - 100 ng of the pshPFKFB3 complexed with liposomes.
Fig. 4.
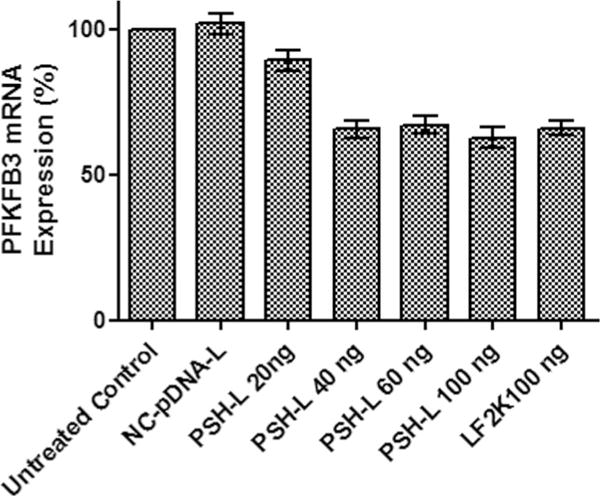
Impact of PSH-L at different doses of pshPFKFB3 on PFKFB3 mRNA expression in A549 cells. Bar graph depicts real-time RT-PCR expression of PFKFB3 as compared to untreated control cells. The results show mean of three independent experiments with SEM. NC-pDNA-L liposomes with 100 ng of NC-pDNA, LF2K100 = Lipofectamine-2000 complexed with pshPFKFB3 100 ng, PSH-L20, 40, 60 and 100 = PSH-L with 20 ng, 40 ng, 60 ng and 100 ng of pshPFKFB3.
Fluorescence Microscopy for Expression of pshPFKFB3
Cell internalization and expression of GFP by PSH-L was visualized by fluorescence microscopy. The results of A549 lung cancer cells with different test formulations and control cells are presented in Fig. 5. Fluorescence from the control cells eliminated the possibility of background fluorescence from the cells. Naked pshPFKFB3 was not able to transfect the cells and hence did not show any GFP expression which goes in line with the RT-PCR studies. This fluorescence may result from some cell transfection via nonspecifically captured plasmid-bearing liposomes. Fluorescence microscopy images also revealed that the treatment of A549 cells with PSH-L resulted in an increase in cellular fluorescence. Hence, the liposomes were being internalized in the cells and could express GFP. Expression of GFP from the vector also suggests the expression of the shPFKFB3 as well. The cells treated with the complexes demonstrated a substantially higher fluorescence, i.e., a higher transfection outcome at a concentration of 40 ng per 10,000 cells. At similar conditions, 100 ng pshPFKFB3 complexes prepared with Lipofectamine-2000 (LF2K100), a commercial transfection reagent, provided higher extent of fluorescence level compared with the PSH-L40.
Fig. 5.
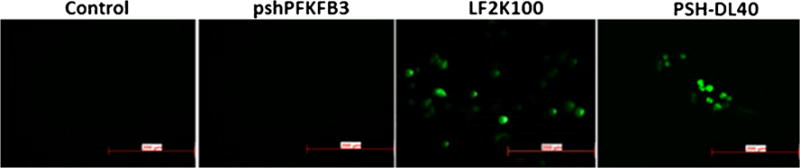
In vitro expression of GFP after pshPFKFB3 plasmid transfection. pPFKFB3 plasmid contains GFP expressing sequence that can express GFP after transfection. Control indicates cells without any treatments. pshPFKFB3 indicates cells treated with naked pshPFKFB3. LF2K100 = Lipofectamine-2000 complexed with 100 ng pshPFKFB3, PSH-L 40 = PSH-L with 40 ng of pshPFKFB3. Scale bar is of 200 μm size.
Effect of pshPFKFB3 on pGP Activity
Rhodamine123 exclusion assay was performed for demonstration of pGP activity after PSH-L treatment of A549 cells. The microscopic images are shown in Fig. 6. Verapamil with rhodamine was used as a positive control, Rh123 as a negative control and lipofectamine-2000 as reference control. The accumulation of Rh123 was noted to differ for different formulations or controls indicating differential pGP activity in the cells after different treatments. Verapamil being a pGP inhibitor led to highest accumulation of Rh123 in the cells as seen by the highest green fluorescence in the cells. Also, pshPFKFB3 liposomes led to reduced exclusion of Rh123. It was visually discernible that there was reduction in Rh123 efflux after treatment with pshPFKFB3 liposomes from 20 ng/well to 40 ng/well. However, increasing levels of pshPFKFB3 i.e. 60 ng and 100 ng did not cause any visually apparent increase in the fluorescence compared to 40 ng level and the Rh123 accumulations were almost similar to 40 ng treatment.
Fig. 6.
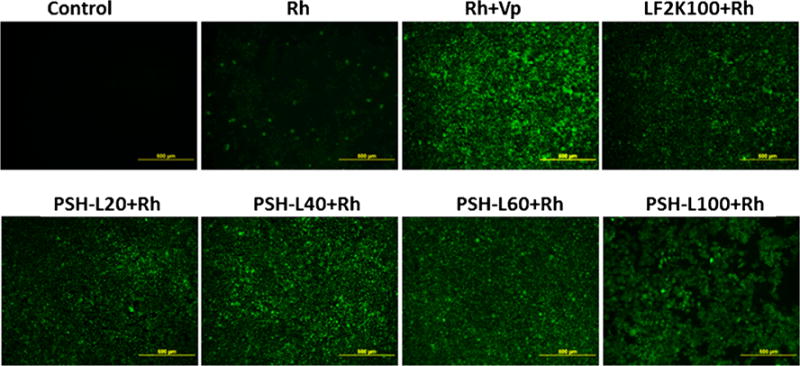
pGP inhibition assay using Rh123. Fluorescence microscopy images of A549 lung cancer cells incubated with Rh123 after treatment with different formulations. Rh = Rh123, Vp = verapamil, LF2K100 = Lipofectamine-2000 complexed with pshPFKFB3 100 ng, PSH-L20, 40, 60 and 100 = PSH-L with 20 ng, 40 ng, 60 ng and 100 ng of pshPFKFB3.
Apoptosis Analysis by AO/EB Double-Staining and Fluorescent Microscopy
After treatment with PSH-L, DL and PSH-DL complexes for 48 h, apoptosis induction was demonstrated using acridine orange and ethidium bromide on A549 and H460 cells. As shown (Fig. 7) in the control group (no treatment), a scarce number of apoptotic cells were visible. Same observation was there with the PSH-L as well; while in DL group and PSH-DL group, the number of apoptotic cells increased. PSH-DL showed highest number of apoptotic cells to the magnitude of 2–3 folds higher when co-loaded liposomes were used. A similar trend was observed in the H460 cells as well which had the highest number of apoptotic cells in the PSH-DL group.
Fig. 7.
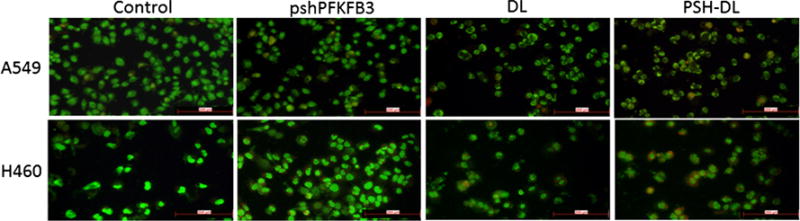
Apoptosis by AO/EB dual staining. Fluorescent microscopic images of A549 and H460 NSCLC cells after different treatments.
Western Blot Analyses
Western blotting was performed to see the effects of different formulations on expression of various cell apoptosis, survival and stress markers. Densitometric analysis of bands showed that PFKFB3 was significantly downregulated as compared to control in the PSH-L and similar results were observed with PSH-DL treated cells (Fig. 8). Further, the densitometric analysis of western blot bands revealed that PFKFB3 relative band density was reduced to 0.41 for the PSH-DL in comparison to untreated control cells for both, A549 and H460 cells. A significant down regulation of Cdk2 was observed in both the NSCLC. The combination caused a decrease in the relative intensity to 0.43 and 0.66 in A549 and H460 cells respectively, as compared to the control. Cell survival marker, survivin, expression also decreased significantly (p < 0.05) to 0.31 and 0.24 in A549 and H460 respectively in the PSH-DL complexes as compared to the control. The expression of Bcl2, an apoptotic marker, was significantly down regulated (p < 0.05) to 0.04 and 0.06 in the PSH-DL complexes for A459 and H460 cells respectively compared to control cells (Fig. 8). A significant up-regulation of cleaved caspase-3 was also found in the PSH-DL complexes. The relative intensity increased to 2.39 (p < 0.05) and 2.54 (p < 0.01) for A549 and H460, respectively. Up-regulation of p-JNK and p-p38 was also observed indicating that activation of stress-related pathways may have triggered apoptosis.
Fig. 8.
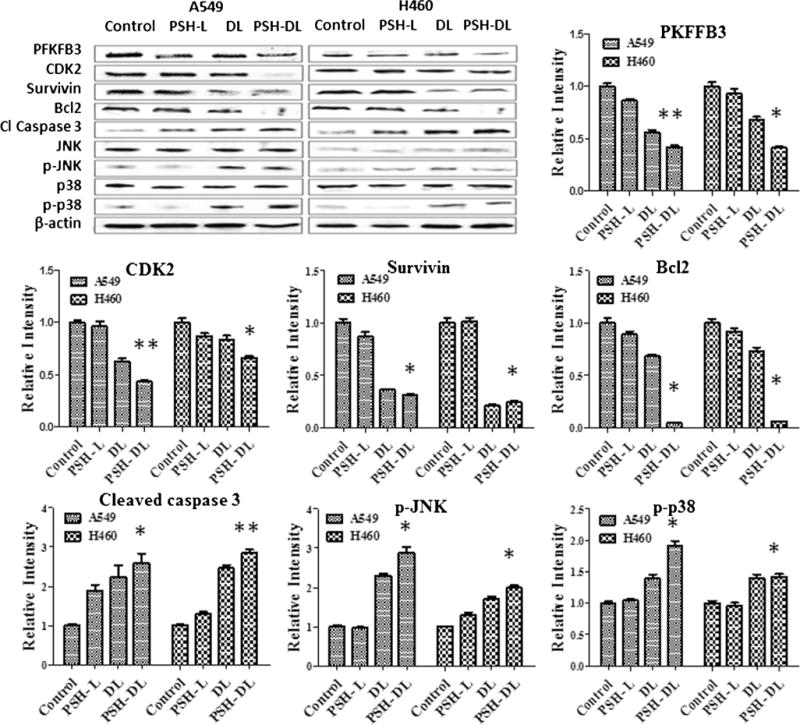
A. Western blot densitometric analysis of PFKFB3, various cell survival markers, apoptotic markers and stress related markers. The results show mean of three independent experiments. * indicates p < 0.05 compared with untreated controls; ** indicates p < 0.01 compared to untreated controls.
In Vivo Anti-Tumor Activity
Tumor volume significantly decreased after treatment with DL or PSH-DL complexes compared to control (Fig. 9). PSH-L did not show a significant decrease (p > 0.05) in tumor volume. Tumor volume for the combination treatment averaged 848 mm3compared with 1597 mm3 for DL treatment or 3207 mm3for PSH-L treatment on day 21 post tumor cell implantation. It is evident that combination treatment was most effective in inhibiting tumor growth compared to DTX or pshPFKFB3 treatments.
Fig. 9.
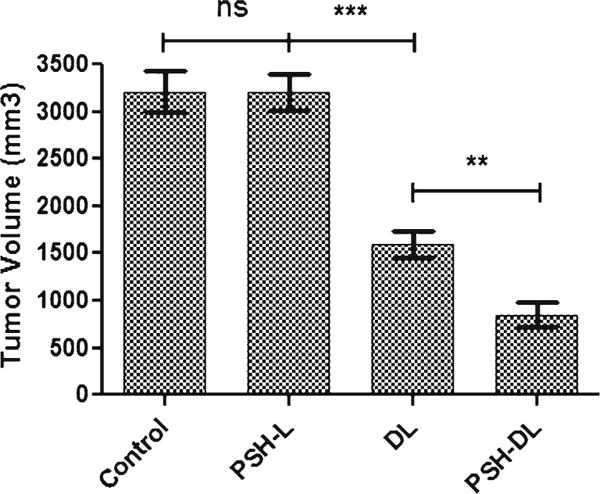
Effects of different liposomes on human A459 lung tumor xenograft model tumor volume. Measurements were made on day 21 after tumor cell implantation. Tumor volumes are captured in mm3. The results show mean of three independent experiments with the SEM. ns, statistically nonsignificant, compared with untreated control; ** indicates p < 0.01, compared to DL; and *** indicates p < 0.001, compared with untreated controls.
DISCUSSION
The present study of development of co-delivery system for plasmid for expression of shRNA against PFKFB3 mRNA along with docetaxel was hypothesized based on cancer tumor’s two basic characteristics that are flaunted by their different physical attributes a) outer highly proliferating mass of cells and b) deep-lying comparatively slow proliferating hypoxic mass of cells. This makes the use of single agent therapeutics to be less effective in eradicating the cancer cells due to their higher effectiveness against these highly proliferating cells as compared to the hypoxic low proliferating cancer cells. Hence, targeting the pathways that are specific to hypoxia along with use of chemotherapeutic agents is a better way to treat cancer. Glycolysis being the predominant energy production mechanism in cancer cells, it can be targeted using several of glycolytic inhibitors which target specific enzymes of glycolytic pathways (24). PFKFB3 being a rate limiting enzyme in glycolytic pathway, it has been recently explored as a therapeutic target for cancer treatment using small molecule inhibitors of PFKFB3 such as 3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one (3PO) and 1-(4-pyridinyl)-3-(2-quinolinyl)-2-propen-1-one (PFK15) (25,26).
In the present study, we have developed a novel liposomal delivery system for the co-delivery of pshPFKFB3 (plasmid for shRNA against PFKFB3) and DTX. Selection of plasmid for shRNA over siRNA itself was based on the stability of the plasmid in vivo milieu and more importantly, the expression of plasmid would provide constant supply of shRNA over time which would exert its silencing activity on the PFKFB3 mRNA. Such shRNA based targeting offers additional advantages over RNAi by siRNA due to low likelihood of off-target effects and low immunological reactivity and toxicity (14,27). Moreover, the co-delivery of pshPFKFB3 with DTX could act in synergy through i) energy deprivation of cells leading to either cytostasis/cytotoxicity, ii) inhibition of energy dependent pathways and processes iii) sensitization to/synergism with chemotherapeutic agents through reduction of hypoxia-linked resistance or ATP dependent MDR (24). Hence, our formulation strategy would make it possible to act on both hypoxic and non-hypoxic tumor cells.
We have used unsaturated lipids such as DOPC, DOPE, and DOTAP all of which have unsaturated oleoyl chains and provide bilayer deformities to accommodate the drug in the lipid bilayer. Moreover, DOPE was used for its endosomal escape capabilities through transition of bilayer phase to inverted hexagonal phase under acidic condition of endosomes leading to cytosolic release of nucleic acid cargo (28,29). Furthermore, development of a co-delivery system through co-loading of gene and drug in same liposomal carrier gives advantage over other polymeric system in their more biocompatibility and possibility for co-loading hydrophilic/hydrophobic drugs in liposomal core/bilayer without extensive modifications of the system.
Liposomes were initially optimized for the drug loading and particle size using DoE approach using a Box-Benken design (See supplementary information-1). Using the same lipid composition, pshPFKFB3 complexed liposomes – PSH-L and PSH-DL were prepared. We separately evaluated the effect of PSH-L and DL. For co-delivery, optimized liposomal formulation of DTX was further used for complexation with pshPFKFB3. Developed DTX-pshPFKFB3 liposomes were 110.3 nm with a positive surface charge of 18.5 mV and had an entrapment efficiency of ~89% which is equivalent to drug to lipid ratio of ~3.7 mol%. PEGylation which would impart long circulation character to liposomes and nano range particle size infers that the developed liposomes would be able to provide tumor tissue penetration through enhanced permeation and retention (EPR) effect in the lung tumors (30). Reduced zeta potential of PSH-DL compared to DTX liposomes is due to the complexation of cationic liposomes with pshPFKFB3 which makes ionic complex through its phosphates with cationic head groups of DOTAP and DDAB (31). Moreover, the reduction in the zeta potential is also in part due to the surface masking as a result of PEGylation. The complexation N/P ratio of 2.2:1 suggests that there is two times molar excess of cationic nitrogens of DOTAP and DDAB over phosphates of the pDNA which goes in concordance with the literature data. This supports the external model of complexation of pDNA with liposomes (i.e. complexation of pDNA on the external liposomal surface) with cationic lipids on the inner side of the bilayer not used for complexation (32).
We assessed the extent of cancer cell killing by the DTX solution, blank cationic liposomes and DTX liposomes complexed with NC-pDNA and pshPFKFB3 liposomes on A549 and H460 NSCLC cell lines. Blank liposomes showed negligible toxicity on all the cancer cell lines used in the cytotoxicity studies (<5%). DTX liposomes complexed with NC-pDNA were used for cytotoxicity studies as DTX liposomes alone would be taken up more efficiently than the pDNA complexed liposomes. Hence, DTX liposomes complexed with NC-pDNA (DL) were used for cytotoxicity studies and other studies to represent the actual liposomal formulation which considers the effect of complexation of pDNA on cell uptake and toxicity. Moreover, the NC-pDNA-L did not show any cytotoxicity to cells indicating there is no carrier dependent cytotoxicity nor there is any cytotoxicity due to off-target effects by the plasmid vector. Hence, the toxicity rendered by the liposomes is due to DTX only. For both DTX formulations i.e. DTX solution and DL, we observed a decrease in IC50 with time i.e. 24 h vs. 48 h showing the time dependent cytotoxicity by DTX which goes in line with the increasing cellular mitotic spindle formation defects induced by DTX on longer duration of exposure (33). However, there was a statistically significant improvement in the cytotoxicity of the DL against the DTX solution which is due to the improved uptake of liposomes in the cells compared to free drug. Moreover, this study established that the developed liposomal delivery is able to deliver DTX to lung cancer cells more effectively over solution.
Our developed liposomes are cationic systems with membrane destabilizing lipids, hence we evaluated toxicity of the liposomes on normal epithelial cell line (HEK cell line) which would give an idea about the toxicity of liposomes to other normal cells of the body (34). HEK cells were treated with blank liposomes and cytotoxicity estimations were made. Results revealed that even up to a concentration of 3 mg/mL of cationic lipids, liposomes showed more than 80% cell viability suggesting that at therapeutically effective concentration of DTX and pshPFKFB, there would be negligible toxicity posed by cationic lipids.
Next, we evaluated the effect of pshPFKFB3 on the cellular PFKFB3 expression in A549 cells. The level of expression of shRNA was confirmed through RT-PCR studies in which the down-regulation of pshPFKFB3 was monitored through RT-PCR for PFKFB3 mRNA. No PFKFB3 mRNA silencing by NC-pDNA liposomes suggested there is no nonspecific silencing by the NC plasmid and by the vector used in the study. This concludes that the silencing of the PFKFB3 mRNA is due to the pshPFKFB3 only. With the PSH-L, the levels of PFKFB3 mRNA gradually decreased with increasing concentration of pshPFKFB3 up to 40 ng level. However, further increase up to 100 ng in the pshPFKFB3 level did not cause any apparent down-regulation in PFKFB mRNA. This suggests that the level of expressed PFKFB3 mRNA reaches a plateau after certain levels of expression of shRNA. It can be deduced from these results that increased expression of shRNA will not be effective to further down-regulate the PFKFB3 mRNA due to saturation of the RNAi machinery required for effectiveness of shRNA which will ultimately lead to saturation of the efficacy of pshPFKFB3 (26,35). Further, we evaluated the expression of plasmid through fluorescence microscopy studies using GFP expression and we also evaluated if the naked plasmid itself is able to do any transfection. Results confirmed that naked plasmid has been reported in literature, is not able to transfect the cells due to the high molecular weight and negative charge. However, both the liposomal carriers i.e. lipofectamine-2000 and developed PSH-L could transfect cells effectively as it can be seen from the fluorescence inside the cells due to GFP expression. Results also showed that LF2K complexed with pshPFKFB3 at 100 ng of plasmid showed higher expression of GFP compared to GFP expression by PSH-DL at 40 ng plasmid level. This indicates that the expression of mRNA for GFP and hence, mRNA for shRNA from plasmid might be higher at higher pshPFKFB3 levels (60 and 100 ng). However, as speculated in the mRNA expression studies, saturation of the shRNA silencing machinery would be the reason for the plateaued silencing after 40 ng plasmid level. However, the cellular GFP expression studies undoubtedly conclude that developed liposomal vector is effective in expressing the mRNA either for GFP expression or for further metabolism to convert to shRNA for silencing PFKFB3. Based on this study, the treatment level of pshPFKFB3 was kept constant at 40 ng/well for 96 well plate and appropriate scaling of pshPFKFB3 level was done to keep this level constant for further experimentation in the 24 well plates, 12 well plates and 25 cm2 T-flasks for use with different levels of DTX.
Confirming the amount of pshPFKFB, we next evaluated the cytotoxicity of PSH-L as well as PSH-DL compared to DL in both A549 and H460 NSCLC cell lines to validate the results that were observed in aforementioned studies. For this, cytotoxicity studies were carried out on A549 cells with PSH-L, DL and PSH-DL keeping 40 ng/well (for 96 well plate or for 10,000 cells) of pshPFKFB3 constant. It was observed that PSH-L had negligible effect on the cytotoxicity of A549 and H460 cells suggesting that glycolytic seizure rendered at the selected dose of pshPFKFB3 is not causing cell killing. This might be due to the cytostatic and not cytotoxic effect of pshPFKFB3 on the cancer cells. Similar results have been observed for certain glycolysis inhibitors where cell proliferation was arrested instead of cell killing (11,26). However, our results show that in A549 and H460 cells, statistically insignificant improvement of mean cancer cell death was there (p > 0.05). Moreover, cytotoxicity of PSH-DL was significantly (p < 0.05) higher compared to PSH-L and DL. This deduces that even at the concentration of pshPFKFB3 which did not show effective cell killing, it enhanced the cytotoxicity of DTX liposomes.
It is known that the epidermal cells enter division only in the presence of glucose (36). This contention can be extended to proliferation of all epithelial cells including epidermal cells i.e. lung epithelial cells. Moreover, NSCLC cells bearing a cancerous genotype exhibit higher glucose requirements for proliferation and growth. Bullough analyzed and established that one of the rate limiting step that controls energy production as well as mitosis is PFKFB mediated (36,37). Hence, the pshPFKFB3 is effective in blocking the energy supply in the cells that provides ATP supplies to the energy dependent processes of the cell. As pshPFKFB3 transfection alone is not cytotoxic to cancer cells, improvement in the cytotoxicity of DTX liposomes in presence of pshPFKFB3 might be due to a mechanism other than the energy deprivation of cells due to glycolysis inhibition. This can be justified by the effective blocking of glycolysis that provides for the energy requirement for mechanisms other than just inhibition of proliferation due to energy deprivation of cancer cells (38). We hypothesized this mechanism to be energy dependent pGP efflux of drug which is active in the NSCLC cells and gets ceased with the pshPFKFB3 transfection. Moreover, other biochemical processes involved in cellular response to damage caused by anticancer drugs are energy dependent inhibition of processes which due to diminution of PFKFB3 mRNA by pshPFKFB3 has enhanced the cytotoxicity of the DTX on cancer cells.
To confirm the aforementioned hypothesis and for the direct measurement of activity of PFKFB3 shRNA expressed from pshPFKFB3 on pGP, we carried out the pGP inhibition assay with Rhodamine123 (Rh123) which is a pGP substrate (39). Our studies confirmed that the inhibition of glycolysis by pshPFKFB3 liposomes reduced the activity of the pGP which ultimately leads to higher accumulation of Rh123 in cells. Due to the active pGP efflux in A549 cells, accumulation of Rh123 and hence fluorescence was of low intensity when cells were treated only with Rh123. Verapamil which is used as the positive control, showed highest accumulation of the Rh123 due to its blocking activity on pGP efflux. All the pshPFKFB3 liposomal formulations showed increasing fluorescence as compared to control indicating inhibition of glycolysis by pshPFKFB3 was able to reduce the pGP based efflux of Rh123. Hence, it confirms that increased cytotoxicity seen with the lipoplexes is due to the reduced DTX efflux by pGP due to simultaneously inhibited PFKFB3 mRNA by the pshPFKFB3.
In order to identify and confirm various markers involved in the cytotoxicity by the co-delivery, we carried out the western blotting for apoptotic markers in A549 and H460 cells (40). Protein markers of cell survival and apoptosis were evaluated. Activation of the caspase-3 pathway is a hallmark of apoptosis (41). Our liposome formulation was successful in up-regulating the cleaved caspase 3 in the lung cancer cells. Moreover, other apoptotic markers such as p38 and JNK were also up-regulated including their activated (phosphorylated) forms. JNK-p38 pathway is also a well-established indicator of apoptosis in cancer cells (42). Many chemotherapeutic agents including DTX require p38 activity for the induction of apoptosis (43). Once activated, p38 proteins translocate from the cytosol to the nucleus causing cellular responses through the phosphorylation of its downstream transcription factors. Our liposomal formulation have shown to reduce the expression of Cdk2 which is critical during the G1 to S phase transition (44). Moreover, reduction in expression of Bcl-2 and survivin are also involved in apoptosis induction in lung cancer cells. Ultimately, the formulation was evaluated in the in vivo model of NSCLC. The in vivo therapeutic activity was evaluated in the A549 tumor xenograft model. The results further confirmed that the plasmid alone was not able to reduce the tumor size in the animal model. However, the co-loaded liposomes PSH-DL were showing significantly higher tumor volume reduction. The results of in vivo study are in line with and validate the results of in vitro studies. PSH-L alone is ineffective in the tumor volume reduction while PSH-DL demonstrated statistically significant reduction in the tumor volume with co-delivery compared to DL. This suggest that the liposomal formulation is effective in penetrating the tumor and delivering the pshPFKFB3 and DTX to the cancer cells that works in a synergistic fashion with the DTX.
CONCLUSION
Overall, we demonstrated for the first time that the combination of pshPFKFB3 with docetaxel can be effectively used for lung cancer treatment. The liposomes carrying the pshPFKFB3 and DTX demonstrated anticancer activity by increasing apoptosis and cellular stress. In summary, the present findings establish that the co-delivery of docetaxel and pshPFKFB3 can prove to be of great therapeutic value for lung cancer treatment due to the ability of RNAi based glycolytic inhibitor to enhance the therapeutic efficacy of docetaxel. Moreover, our study provides a proof-of-concept for simultaneous delivery of synergistic combination of gene and drug to cancer cells through co-loading of gene and drug in the same liposomal system. Furthermore, such treatment needs to be evaluated in more bio-relevant in vivo models such as PDX models and also utility of this formulation can be evaluated for the treatment of NSCLC not responding to or resistant to traditional therapies. In summary, chemosensitization of NSCLC cells through glycolysis inhibition could possibly a better mechanism of an effective lung cancer treatment.
Supplementary Material
Acknowledgments
This research was supported with funding from: the National Institutes of Health’s Minority Biomedical Research Support (MBRS)-SC1 program (Grant #SC1 GM092779–01); and the Department of Defense (DOD) Breast Cancer Program(Grant #W81XWH-11–1–0211).
ABBREVIATIONS
- 3PO
3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one
- AO/EB staining
Acridine orange and ethidium bromide staining
- DDAB
Dimethyldioctadecyl-ammonium bromide
- DL
Docetaxel liposomes
- DOTAB
N- (2, 3-Dioleoyloxy-1-propyl) trimethylammonium bromide
- DPPC
1,2-dipalmitoyl-sn-glycero-3-phosphocholine
- DSPE-PEG-2000
1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-methoxy (polyethyleneglycol) 2000
- DTX
Docetaxel
- EPR
Enhanced permeation and retention
- GFP
Green fluorescent protein
- LF2K100
Lipofectamine-2000 complexed with pshPFKFB3 100 ng
- MLV
Multi-lamellar vesicles
- N/P
Nitrogen to phosphate ratio
- NC
Negative control plasmid
- NC-pDNA-DL
Docetaxel loaded iposomes complexed with negative control plasmid
- NC-pDNA-L
Liposomes complexed with negative control plasmid
- NSCLC
Non-small cell lung cancer
- pDNA
Plasmid Deoxyribonucleic acid
- PFK15
1-(4-pyridinyl)-3-(2-quinolinyl)-2-propen-1-one
- PFKFB3
6-Phosphofructo-2-Kinase/Fructose-2, 6-Biphosphatase 3
- pGP
P-glycoprotein
- PSH-DL
Docetaxel loaded liposomes complexed with pshPFKFB3
- PSH-L
Blank liposomes complexed with pshPFKFB3
- pshPFKFB3
6-Phosphofructo-2-Kinase/Fructose-2, 6-Biphosphatase 3 shRNA plasmid
- Rh123
Rhodamine 123
- RNA
Ribonucleic acid
- RNAi
RNA interference
- shRNA
Short hairpin RNA
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s11095-017-2244-x) contains supplementary material, which is available to authorized users.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 2.Laskin JJ, Sandler AB. First-line treatment for advanced non-small-cell lung cancer. Oncology (Williston Park, NY) 2005;19(13):1671–6. discussion 1678-1680. [PubMed] [Google Scholar]
- 3.Warburg O. On the origin of cancer cells. Science (New York, NY) 1956;123(3191):309–14. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 4.Marín-Hernández A, López-Ramírez SY, Del Mazo-Monsalvo I, Gallardo-Pérez JC, Rodríguez-Enríquez S, Moreno-Sánchez R, et al. Modeling cancer glycolysis under hypoglycemia, and the role played by the differential expression of glycolytic isoforms. FEBS J. 2014;281(15):3325–45. doi: 10.1111/febs.12864. [DOI] [PubMed] [Google Scholar]
- 5.Semenza GL. Involvement of hypoxia-inducible factor 1 in human cancer. Intern Med. 2002;41(2):79–83. doi: 10.2169/internalmedicine.41.79. [DOI] [PubMed] [Google Scholar]
- 6.Zheng JIE. Energy metabolism of cancer: glycolysis versus oxidative phosphorylation (review) Oncol Lett. 2012;4(6):1151–7. doi: 10.3892/ol.2012.928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bares R, Klever P, Hauptmann S, Hellwig D, Fass J, Cremerius U, et al. F-18 fluorodeoxyglucose PET in vivo evaluation of pancreatic glucose metabolism for detection of pancreatic cancer. Radiology. 1994;192(1):79–86. doi: 10.1148/radiology.192.1.8208970. [DOI] [PubMed] [Google Scholar]
- 8.Atsumi T, Chesney J, Metz C, Leng L, Donnelly S, Makita Z, et al. High expression of inducible 6-Phosphofructo-2-kinase/fructose-2, 6-Bisphosphatase (iPFK-2; PFKFB3) in human cancers. Cancer Res. 2002;62(20):5881–7. [PubMed] [Google Scholar]
- 9.Pelicano H, Martin DS, Xu RH, Huang P. Glycolysis inhibition for anticancer treatment. Oncogene. 2006;25(34):4633–46. doi: 10.1038/sj.onc.1209597. [DOI] [PubMed] [Google Scholar]
- 10.Chesney J. 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase and tumor cell glycolysis. Curr Opin Clin Nutr Metab Care. 2006;9(5):535–9. doi: 10.1097/01.mco.0000241661.15514.fb. [DOI] [PubMed] [Google Scholar]
- 11.Vuyyuri SB, Rinkinen J, Worden E, Shim H, Lee S, Davis KR. Ascorbic acid and a cytostatic inhibitor of glycolysis synergistically induce apoptosis in non-small cell lung cancer cells. PLoS One. 2013;8(6):e67081. doi: 10.1371/journal.pone.0067081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.O’Neal J, Tapolsky G, Clem B, Telang S, Chesney J. Identification of a PFKFB3 inhibitor suitable for phase I trial testing that synergizes with the B-Raf inhibitor vemurafenib. Cancer Res. 2014;74(19):962. [Google Scholar]
- 13.Boyd S, Brookfield JL, Critchlow SE, Cumming IA, Curtis NJ, Debreczeni J, et al. Structure-based design of potent and selective inhibitors of the metabolic kinase PFKFB3. J Med Chem. 2015;58(8):3611–25. doi: 10.1021/acs.jmedchem.5b00352. [DOI] [PubMed] [Google Scholar]
- 14.Rao DD, Vorhies JS, Senzer N, Nemunaitis J. siRNA vs. shRNA: similarities and differences. Adv Drug Deliv Rev. 2009;61(9):746–59. doi: 10.1016/j.addr.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 15.Patel K, Chowdhury N, Doddapaneni R, Boakye CH, Godugu C, Singh M. Piperlongumine for enhancing oral bioavailability and cytotoxicity of docetaxel in triple-negative breast cancer. J Pharm Sci. 2015;104(12):4417–26. doi: 10.1002/jps.24637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Patel K, Doddapaneni R, Chowdhury N, Boakye CH, Behl G, Singh M. Tumor stromal disrupting agent enhances the anticancer efficacy of docetaxel loaded PEGylated liposomes in lung cancer. Nanomedicine. 2016;11(11):1377–92. doi: 10.2217/nnm.16.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Patel K, Doddapaneni R, Sekar V, Chowdhury N, Singh M. Combination approach of YSA peptide anchored docetaxel stealth liposomes with oral antifibrotic agent for the treatment of lung cancer. Mol Pharm. 2016;13(6):2049–58. doi: 10.1021/acs.molpharmaceut.6b00187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kampan NC, Madondo MT, McNally OM, Quinn M, Plebanski M. Paclitaxel and its evolving role in the management of ovarian cancer. Biomed Res Int. 2015;2015:413076. doi: 10.1155/2015/413076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miele E, Spinelli GP, Miele E, Tomao F, Tomao S. Albumin-bound formulation of paclitaxel (Abraxane (R) ABI-007) in the treatment of breast cancer. Int J Nanomedicine. 2009;4(1):99–105. doi: 10.2147/ijn.s3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Immordino ML, Dosio F, Cattel L. Stealth liposomes: review of the basic science, rationale, and clinical applications, existing and potential. Int J Nanomedicine. 2006;1(3):297–315. [PMC free article] [PubMed] [Google Scholar]
- 21.Balazs DA, Godbey W. Liposomes for use in gene delivery. J Drug Deliv. 2011;2011:326497. doi: 10.1155/2011/326497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boakye CH, Patel K, Doddapaneni R, Bagde A, Marepally S, Singh M. Novel amphiphilic lipid augments the co-delivery of er-lotinib and IL36 siRNA into the skin for psoriasis treatment. J Control Release. 2017;246:120–32. doi: 10.1016/j.jconrel.2016.05.017. [DOI] [PubMed] [Google Scholar]
- 23.Boakye CH, Patel K, Doddapaneni R, Bagde A, Behl G, Chowdhury N, et al. Ultra-flexible nanocarriers for enhanced topical delivery of a highly lipophilic antioxidative molecule for skin cancer chemoprevention. Colloids Surf B: Biointerfaces. 2016;143:156–67. doi: 10.1016/j.colsurfb.2016.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scatena R, Bottoni P, Pontoglio A, Mastrototaro L, Giardina B. Glycolytic enzyme inhibitors in cancer treatment. Expert Opin Investig Drugs. 2008;17(10):1533–45. doi: 10.1517/13543784.17.10.1533. [DOI] [PubMed] [Google Scholar]
- 25.Clem BF, O’Neal J, Tapolsky G, Clem AL, Imbert-Fernandez Y, Kerr DA, et al. Targeting 6-Phosphofructo-2-kinase (PFKFB3) as a therapeutic strategy against cancer. Mol Cancer Ther. 2013;12(8):1461–70. doi: 10.1158/1535-7163.MCT-13-0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clem B, Telang S, Clem A, Yalcin A, Meier J, Simmons A, et al. Small-molecule inhibition of 6-phosphofructo-2-kinase activity suppresses glycolytic flux and tumor growth. Mol Cancer Ther. 2008;7(1):110–20. doi: 10.1158/1535-7163.MCT-07-0482. [DOI] [PubMed] [Google Scholar]
- 27.DiGiusto DL, Krishnan A, Li L, Li H, Li S, Rao A, et al. RNA-based gene therapy for HIV with lentiviral vector-modified CD34(+) cells in patients undergoing transplantation for AIDS-related lymphoma. Science Translational Medicine. 2010;2(36):36ra43. doi: 10.1126/scitranslmed.3000931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoekstra D, Martin OC. Transbilayer redistribution of phosphati-dylethanolamine during fusion of phospholipid vesicles. Dependence on fusion rate, lipid phase separation, and formation of nonbilayer structures. Biochemistry. 1982;21(24):6097–103. doi: 10.1021/bi00267a011. [DOI] [PubMed] [Google Scholar]
- 29.Zuhorn IS, Bakowsky U, Polushkin E, Visser WH, Stuart MC, Engberts JB, et al. Nonbilayer phase of lipoplex-membrane mixture determines endosomal escape of genetic cargo and transfection efficiency. Mol Ther. 2005;11(5):801–10. doi: 10.1016/j.ymthe.2004.12.018. [DOI] [PubMed] [Google Scholar]
- 30.Kolate A, Baradia D, Patil S, Vhora I, Kore G, Misra A. PEG—a versatile conjugating ligand for drugs and drug delivery systems. J Control Release. 2014;192:67–81. doi: 10.1016/j.jconrel.2014.06.046. [DOI] [PubMed] [Google Scholar]
- 31.Son KK, Patel DH, Tkach D, Park A. Cationic liposome and plasmid DNA complexes formed in serum-free medium under optimum transfection condition are negatively charged. Biochim Biophys Acta. 2000;1466(1–2):11–5. doi: 10.1016/s0005-2736(00)00176-0. [DOI] [PubMed] [Google Scholar]
- 32.Ferrari ME, Rusalov D, Enas J, Wheeler CJ. Trends in lipoplex physical properties dependent on cationic lipid structure, vehicle and complexation procedure do not correlate with biological activity. Nucleic Acids Res. 2001;29(7):1539–48. doi: 10.1093/nar/29.7.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Orth JD, Loewer A, Lahav G, Mitchison TJ. Prolonged mitotic arrest triggers partial activation of apoptosis, resulting in DNA damage and p53 induction. Mol Biol Cell. 2012;23(4):567–76. doi: 10.1091/mbc.E11-09-0781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yingyongnarongkul BE, Radchatawedchakoon W, Krajarng A, Watanapokasin R, Suksamrarn A. High transfection efficiency and low toxicity cationic lipids with aminoglycerol-diamine conjugate. Bioorg Med Chem. 2009;17(1):176–88. doi: 10.1016/j.bmc.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 35.Kurreck J. RNA interference: from basic research to therapeutic applications. Angew Chem Int Ed Engl. 2009;48(8):1378–98. doi: 10.1002/anie.200802092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gelfant S. The energy requirements for mitosis. Ann N Y Acad Sci. 1960;90:536–49. doi: 10.1111/j.1749-6632.1960.tb23271.x. [DOI] [PubMed] [Google Scholar]
- 37.Boscá L, Corredor C. Is phosphofructokinase the rate-limiting step of glycolysis? Trends Biochem Sci. 1984;9(9):372–3. [Google Scholar]
- 38.Ganapathy-Kanniappan S, Geschwind JF. Tumor glycolysis as a target for cancer therapy: progress and prospects. Mol Cancer. 2013;12:152. doi: 10.1186/1476-4598-12-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Twentyman PR, Rhodes T, Rayner S. A comparison of rhodamine 123 accumulation and efflux in cells with P-glycoprotein-mediated and MRP-associated multidrug resistance phenotypes. Eur J Cancer. 1994;30A(9):1360–9. doi: 10.1016/0959-8049(94)90187-2. [DOI] [PubMed] [Google Scholar]
- 40.Doddapaneni R, Patel K, Chowdhury N, Singh M. Noscapine chemosensitization enhances docetaxel anticancer activity and nanocarrier uptake in triple negative breast cancer. Exp Cell Res. 2016;346(1):65–73. doi: 10.1016/j.yexcr.2016.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McIlwain DR, Berger T, Mak TW. Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol. 2013 Apr 1;5(4):a008656. doi: 10.1101/cshperspect.a008656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sui X, Kong N, Ye L, Han W, Zhou J, Zhang Q, et al. p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett. 2014;344(2):174–9. doi: 10.1016/j.canlet.2013.11.019. [DOI] [PubMed] [Google Scholar]
- 43.Cuadrado A, Nebreda AR. Mechanisms and functions of p38 MAPK signalling. Biochem J. 2010;429:403–17. doi: 10.1042/BJ20100323. [DOI] [PubMed] [Google Scholar]
- 44.Yalcin A, Clem BF, Imbert-Fernandez Y, Ozcan SC, Peker S, O’Neal J, et al. 6-Phosphofructo-2-kinase (PFKFB3) promotes cell cycle progression and suppresses apoptosis via Cdk1-mediated phosphorylation of p27. Cell Death Dis. 2014;5:e1337. doi: 10.1038/cddis.2014.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.