

Abstract
Median survival for glioblastoma (GBM) remains <15 months. Human Cytomegalovirus (CMV) antigens have been identified in GBM but not normal brain, providing an unparalleled opportunity to subvert CMV antigens as tumor-specific immunotherapy targets. A recent trial in recurrent GBM patients demonstrated the potential clinical benefit of adoptive T cell therapy (ATCT) of CMV phosphoprotein 65 (pp65)-specific T cells. However, ex vivo analyses from this study found no change in the capacity of CMV pp65-specific T cells to gain multiple effector functions or polyfunctionality, which has been associated with superior antitumor efficacy. Previous studies have shown that dendritic cells (DC) could further enhance tumor-specific CD8+ T-cell polyfunctionality in vivo when administered as a vaccine. Therefore, we hypothesized that vaccination with CMV pp65 RNA-loaded DC would enhance the frequency of polyfunctional CMV pp65-specific CD8+ T cells after ATCT. Here we report prospective results of a pilot trial in which 22 patients with newly-diagnosed GBM were initially enrolled of which 17 patients were randomized to receive CMV pp65-specific T cells with CMV-DC vaccination (CMV-ATCT-DC) or saline (CMV-ATCT-Saline). Patients who received CMV-ATCT-DC vaccination experienced a significant increase in the overall frequencies of IFNγ+, TNFα+, and CCL3+ polyfunctional, CMV-specific CD8+ T cells. These increases in polyfunctional CMV-specific CD8+ T cells correlated (R = 0.7371, p= 0.0369) with overall survival, although we cannot conclude this was causally related. Our data implicate polyfunctional T-cell responses as a potential biomarker for effective antitumor immunotherapy and support a formal assessment of this combination approach in a larger randomized study.
Keywords: cytomegalovirus pp65, glioblastoma, dendritic cell vaccines, polyfunctional T cells, adoptive T cell therapy
Introduction
Glioblastoma (GBM) is the most common primary malignant brain tumor and has a median survival of <15 months despite an aggressive clinical standard of care, including maximal surgical resection, high-dose radiation, and dose-intensified temozolomide (TMZ) chemotherapy (1). Novel therapies are urgently needed, and immunotherapy has recently emerged as a highly promising therapeutic approach for cancer.
We and others have previously reported the presence of Cytomegalovirus (CMV) antigens in 90% of GBMs but not in normal brain (2–4). The presence of these unique and immunogenic antigens presents an opportunity to leverage CMV-specific immunity against GBM while minimizing the potential for toxicity. In maximizing anti-tumor T-cell responses, it is becoming increasingly clear that polyfunctional T cells, which simultaneously express more than one effector function, are proving critical for effective anti-cancer immunity. Recently, Crough et al. also demonstrated that CMV-specific T cells in patients with GBM have attenuated abilities to generate multiple cytokines and chemokines, which is uncharacteristic of CMV-specific T cells in healthy virus carriers (5). When cultured ex vivo with HLA-matched CMV peptides and IL-2 though, these T cells became polyfunctional and appeared to induce antitumor immunity when transferred back into a single patient with recurrent GBM (5). Moreover, another recent clinical trial investigated adoptive immunotherapy with CMV-specific T cells in patients with recurrent GBM and showed that 11 patients infused with ex vivo expanded CMV-specific T cells had a promising median overall survival (OS) of 13.4 months and a median progression free survival (PFS) of approximately 8.1 months (6). This suggests that adoptive T cell therapy (ATCT) may also be a promising approach for recurrent GBM (6). Importantly, however, ex vivo analyses from this study found no remarkable change in the polyfunctionality of CMV-specific T cells.
Dendritic cells (DCs) are potent antigen presenting cells, play a central role in controlling immunity, and are among the most frequently used cellular adjuvants in experimental immunotherapy trials. Prior work has shown that DCs can positively impact the polyfunctionalilty of T cells (7,8). Moreover, a recent retrospective study by Wimmers et al. suggested a link between polyfunctional T-cell responses induced by DCs and long-term tumor control in end-stage melanoma patients (9). With these studies in mind, we hypothesized that vaccination with CMV phosphoprotein 65 (pp65) RNA-loaded DCs would enhance the frequency of polyfunctional CMV-specific T cells after ATCT and therefore improve outcomes of GBM patients.
Herein, we report the safety and feasibility of using CMV pp65 RNA-pulsed DCs to enhance the polyfunctionality of adoptively transferred CMV pp65-specific T cells in a randomized pilot trial in patients with newly-diagnosed GBM. Immunotherapy targeted the immunodominant CMV antigen pp65. Patients randomized to receive CMV pp65-specific T cells and CMV pp65 RNA-loaded DCs (CMV-ATCT-DC) had a significant increase in the overall frequencies of polyfunctional CMV pp65-specific CD8+ T cells capable of simultaneously expressing IFNγ, TNFα, and CCL3. Furthermore, within this treatment group, the increase in polyfunctional CMV pp65-specific CD8+ T cell frequency did correlate with overall survival confirming the results found by Wimmers et al. in melanoma although we cannot conclude this was causally related.
Materials and Methods
Study design and participants
We conducted a randomized, parallel, single-blind, single-institution pilot clinical trial at Duke University Medical Center (North Carolina, USA). The study schematic is summarized in Fig. 1. This protocol was reviewed and approved by the U.S. Food and Drug Administration and the Institutional Review Board at Duke University. This study was conducted according to the Declaration of Helsinki, Belmont Report, U.S. Common Rule guidelines, and the International Ethical Guidelines for Biomedical Research Involving Human Subjects (CIOMS). All patients signed a written informed consent before study inclusion.
Figure 1. Trial design.
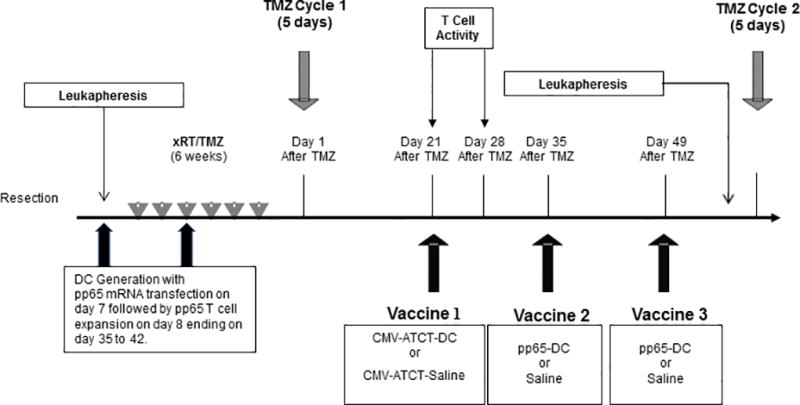
As per the clinical standard of care, patients underwent surgical resection and received xRT with concurrent TMZ (75mg/m2) over a 6-week period. 3–4 weeks after xRT/TMZ, patients received cycle 1 of a TMZ (200 mg/m2/d) daily for 5 days starting on Day 1. Leukapheresis was performed prior to chemoradiation. DC generation begins at leukapheresis followed by pp65 mRNA transfection on Day 7 of culture. Expansion of T cells begins on Day 8 ending on Day 35 to 42. On Day 21, patients received CMV pp65-specific T cells with either CMV pp65 RNA-loaded DCs or saline by random assignment. T cell activity was measured pre Vaccine 1on Day 21 and 7 days after Vaccine 1 at Day 28. A total of three CMV pp65-loaded DC or saline infusions were administered in 14-day intervals. All patients underwent leukapheresis after Day 49, and then received a cycle of TMZ every 28 days for at least 6 cycles. Imaging was performed bimonthly, and upon tumor progression, suitable participants underwent stereotactic biopsy or resection as standard of care. Patients were followed until death. xRT, External beam radiotherapy. TMZ, Temozolomide.
This trial recruited 22 CMV-seropositive patients with confirmed World Health Organization (WHO) grade IV GBM and a Karnofsky Performance Scale (KPS) score ≥ 80. Patients underwent gross total resection and subsequent leukapheresis for CMV pp65-specific T cells and CMV pp65 RNA-loaded DC generation. Patients then received conformal external beam radiotherapy with concurrent temozolomide (xRT/TMZ) (75 mg/m2/d) over a six-week timeframe (Fig. 1). TMZ was discontinued in one patient due to intolerability. 21–28 days post xRT/TMZ, patients received a five-day cycle of TMZ at a dose of 200mg/m2/d as per standard of care. After completion, patients were screened by CT or MRI for evidence of progressive disease, and five patients were excluded prior to immunotherapy initiation on this basis. Patients were regularly monitored for clinical decline or progression, and were removed from study if either occurred before experimental therapy was complete. Two patients began experimental treatment before progression was confirmed radiographically, and were immediately removed from study and excluded from immune monitoring and clinical analyses, but were included in safety analyses. A total of 17 patients were randomly assigned to receive CMV pp65-specific T cells with either CMV pp65 RNA-loaded DC (n=9) or saline (n=8) and fifteen patients received all 3 vaccinations (Supplementary Fig. 1).
In an attempt to treat these tumors by targeting CMV antigens, we conducted an adoptive T cell therapy of ex vivo expanded CMV pp65-specific T cells and then randomized patients to receive either DCs loaded with pp65 RNA (n=8) or saline (n=7) to determine if a DC vaccine could enhance the function of the transferred T cells. Patients received three CMV pp65 RNA-loaded DC or saline infusions in 14-day intervals. Following the third administration, all patients underwent leukapheresis or blood draw, and later received five-day cycles of TMZ every month (150–200mg/m2/d) for 6–25 cycles. In certain cases, TMZ dosing was modified during maintenance cycles as determined by the attending physician. MRI was performed bimonthly to monitor disease, and upon progression, suitable participants underwent stereotactic biopsy or resection as standard of care. Radiographic progression was defined by the Response Assessment in Neuro-Oncology (RANO) criteria. Clinical progression was defined by significant change in overall neurological status, a change in KPS of ≥ 30 points, or the development of a new focal neurologic deficit. The study schematic is summarized in Fig. 1. Baseline patient characteristics are summarized in Table 1. Adverse events with possible relation to experimental treatment are summarized in Table S1.
Table 1.
Baseline characteristics of patients included in the immune monitoring and efficacy analyses
| Patient ID | Age (y) | Race | Sex | MGMT promoter status |
Random assignment |
KPS | CMV serostatus |
|---|---|---|---|---|---|---|---|
| ER1 | 35 | C | M | Unmethylated | CMV-ATCT-Saline | 90 | + |
| ER2 | 71 | C | F | Unmethylated | CMV-ATCT-DC | 80 | + |
| ER3 | 68 | C | M | Unmethylated | CMV-ATCT-Saline | 90 | + |
| ER4 | 50 | C | M | Methylated | CMV-ATCT-DC | 90 | + |
| ER5 | 54 | C | M | Methylated | CMV-ATCT-DC | 90 | + |
| ER6 | 73 | C | F | Unmethylated | CMV-ATCT-Saline | 90 | + |
| ER7 | 46 | C | F | Not Done | CMV-ATCT-Saline | 90 | + |
| ER8 | 73 | C | M | Unmethylated | CMV-ATCT-DC | 90 | + |
| ER9 | 57 | C | M | Unmethylated | CMV-ATCT-DC | 90 | + |
| ER10 | 47 | C | M | Unmethylated | CMV-ATCT-DC | 90 | + |
| ER11 | 43 | C | M | Unmethylated | CMV-ATCT-Saline | 90 | + |
| ER12 | 61 | AA | M | Unmethylated | CMV-ATCT-DC | 80 | + |
| ER13 | 51 | C | M | Unmethylated | CMV-ATCT-Saline | 80 | + |
| ER14 | 55 | C | F | Unmethylated | CMV-ATCT-DC | 80 | + |
| ER15 | 59 | C | M | Unmethylated | CMV-ATCT-Saline | 90 | + |
Abbreviations: MGMT, O-6-methylguanine DNA methyltransferase. KPS, Karnofsky Performance Score. CMV, Cytomegalovirus. ATCT, Adoptive T cell therapy. C, Caucasian. AA, African American. ER, ERaDICATe. Age was recorded at time of patient consent.
In vitro generation of CMV pp65-specific T cells and CMV pp65-loaded DCs
To generate CMV pp65-specific T cells and CMV pp65 RNA-loaded DCs, PBMCs were obtained by leukapheresis. Monocyte precursors for dendritic cell generation were isolated by plastic adherence in AIM V media containing 2% human AB serum (HABS) for 1 hour at 37°C, 5% CO2. After 1 hour, non-adherent cells were collected and cryopreserved to be used for CMV pp65-specific T cell generation. The adherent cells were incubated for seven days at 37°C, 5% CO2 in AIM V media containing 800 units of GM-CSF and 500 units of IL-4 per mL. After 7 days, immature DCs were harvested and transfected with CMV pp65-mRNA by electroporation. The transfected DCs were cultured with a maturation cocktail of TNFα (10 ng/mL), IL-1β (10 ng/mL), and IL-6 (1000 units/mL) in AIM V containing GM-CSF and IL-4 for 18–24 hours at 37°C, 5% CO2. DCs were harvested and cryopreserved in 80% HABS, 10% DMSO and 10% dextrose. Patients randomized to the CMV-ATCT-DC cohort received three separate infusions of 2×107 DCs. according to the treatment schedule shown in Fig. 1.
For CMV pp65-specific T cell generation, the previously cryopreserved non-adherent (NA) cells were thawed, and cells were mixed at a ratio of 1:10 pp65-loaded DCs:NAs in AIM V media with 2% HABS and IL-2 was added at a concentration of 100 IU/mL on Day 3. Cells were adjusted to 1–5×106/mL during expansion with fresh media and IL-2. Cells were cryopreserved after a dose of 3×107 was achieved. Prior to infusion, CMV pp65-specific T cells were thawed and resuspended in saline containing 1% human serum albumin at a concentration of 2.5 ×107 cells/mL. Patients received an intended dose of 3 ×107 CMV pp65-specific T cells/kg. CMV pp65-specific T-cell responses were measured by direct ex vivo IFNγ ELISpot assay as previously described (10). Results were expressed as the mean spot-forming cells (SFC)/106 PBMC after subtraction of counts from cells cultured with no peptide. Polychromatic flow cytometry was used to identify cell populations present in pre-and post-expanded CMV pp65-specific T cells prior to CMV pp65-specific T cell infusion. Approximately 300,000 total events were collected per sample. Lymphocyte-gated events ranged between 75,000–250,000 events in data shown.
Immune monitoring and T-cell profiling
We collected blood samples from patients before and seven days after CMV pp65-specific T cell infusion and the first CMV pp65 RNA-loaded DC or saline administration. PBMC were separated within six hours of collection by density centrifugation on Histopaque (Sigma 1077), frozen to −80°C at a rate of 1°C /min, and stored in liquid nitrogen. On day of testing, PMBC were thawed at 37°C, washed, resuspended in R-10 medium containing serum, and cell number and viability were measured by Guava Counter (Millipore, Billerica, MA). All samples recorded a viability >75% after thawing. CMV pp65-specificity of T cells was confirmed by CMV pp65-specific IFNγ ELISpot assay. Polyfunctionality of CMV pp65-specific CD8+ T cells was evaluated using polychromatic intracellular flow cytometry as previously described (10). The frequency of antigen-specific CD8+ T cells producing one, two, and/or three cytokines was calculated using FlowJo software and analyzed. The data analysis software Simplified Presentation of Incredibly Complex Evaluations (SPICE) was used to analyze and produce representations of high content and multivariate data sets.
Statistical Analyses
OS was computed from the date of surgical resection to date of death. All patients were followed until death. Survival distributions are described using Kaplan-Meier methods, and associations of survival outcomes with polyfunctional T-cell frequencies were assessed using the Pearson correlation coefficient. The log-rank test was used to compare survival distributions between the two treatment groups. Wilcoxon signed-rank tests were used to assess changes in cell frequencies before and after immunotherapy. A Kruskal-Wallis non-parametric ANOVA followed by Dunn’s pairwise comparison was used to assess differences in IFNγ MFI among functional subsets of CMV pp65-specific CD8+ T cells.
As an exploratory effort to assess the impact of CMV pp65 RNA-pulsed DCs in enhancing T cells, this pilot study was not designed to detect clinically important differences between randomized groups with reasonable power. Without adequate power, a hypothesis test lacking statistical significance does not eliminate or minimize the possibility that a clinically important difference may truly exist.
Results
Patient Characteristics and Safety
Twenty-two CMV-seropositive patients with newly-diagnosed GBM were consented into the clinical trial Evaluation of Recovery From Drug-Induced Lymphopenia Using Cytomegalovirus-specific T-cell Adoptive Transfer (ERaDICATe) after undergoing surgery. All of these patients met protocol eligibility with residual radiographic contrast enhancement on post-resection computerized axial tomography (CT) or magnetic resonance imaging (MRI) of <1 cm in maximal diameter in any axial plane. Leukapheresis was performed prior to standard-of-care chemoradiation (see Materials and Methods) as shown in the accompanying trial schema (Fig. 1) and patient flow diagram (Fig. S1). Five of the 22 consented patients exhibited clinical decline or progressive disease during or after completion of chemoradiation and were removed from study evaluations before randomization. The remaining 17 patients were randomly assigned to receive either CMV-ATCT-DC (n=9) or CMV-ATCT-Saline (n=8). Two of these 17 patients (1 in each arm) initiated treatment even though pre-vaccination MRI findings were inconclusive and did not definitively rule out disease progression. In these two patients, follow-up MRI was conducted one month later. At that time, progression was confirmed and these two patients were withdrawn from study participation. During this month, the patient in the CMV-ATCT-Saline arm received one treatment, and the patient in the CMV-ATCT-DC arm received two vaccinations. These patients were not included in efficacy and immune monitoring analyses. Hence, efficacy and immune monitoring analyses were limited to the 15 patients who completed treatment while safety analyses included all 17 patients.
Baseline demographics and patient characteristics were comparable between treatment arms (Table 1). The first patient was consented on 8/1/2008 and the last patient expired on 4/7/2015. The trial was ended after the first cohort of at least twelve patients successfully completed the trial.
In general, immunotherapy was safe, well tolerated, and produced only minor adverse events (AEs) consistent with those expected in this patient population following the clinical standard of care. Toxicity grading was assigned according to the NCI Common Terminology Criteria for Adverse Events (Version 3.0). No severe adverse events (SAEs) were produced by treatment. Patients were monitored by the attending physician and medical staff and were managed with routine clinical practice as required. AEs are summarized in Table S1.
In vitro expansion of patient PBMCs enriches for CMV pp65-specific CD8+ T cells
Adoptive T-cell immunotherapy and DC vaccination were designed to target the immunodominant CMV antigen pp65. We established and qualified a protocol (see Materials and Methods) to selectively expand CMV pp65-specific T cells and DCs-loaded with pp65 RNA from patient-derived peripheral blood mononuclear cells (PBMCs) to clinical scale. Both cellular products were generated prior to planned randomization for all patients initially recruited for study, and were manufactured with sufficient quantity and quality to meet release criteria in 100% of patients (n=22).
In vitro culturing conditions expanded the mean frequency of CD3+ T cells from 77.42% to 97.27% of total cells (Fig. 2A, p = 0.0002). The mean frequency of CD4+ T cells was reduced from 53.11% to 19.79% (Fig. 2A, p = 0.0005), while CD8+ T cells comprised 72.16% of the post-expansion product compared to just 39.04% before expansion (Fig. 2A, p = 0.0010). The final infusion products were characterized prior to infusion; the mean frequencies of CD8+ T cells, CD4+ T cells, regulatory T cells (Tregs), B cells, and NK cells were 70.36%, 19.09%, 6.23%, 0.17%, and 0.85%, respectively. Importantly, the expanded lymphocyte fraction was functionally responsive to CMV pp65 antigen stimulation as measured by IFNγ Enzyme-Linked ImmunoSpot (ELISpot) assay (Fig. 2B, p = 0.0002). All patients, with the exception of two, received a single infusion of 3×107 cells/kg as in vitro generated CMV pp65-specific T cells. These two patients received 71% and 72% of the intended doses as shown in Supplementary Table S2. Separately, CMV pp65 RNA-loaded DCs generated from autologous DCs transfected with pp65-lysosomal associated membrane protein mRNA were matured and infused intradermally at a dose of 2×107 cells in patients randomized to this arm (CMV-ATCT-DC).
Figure 2. Characterization and responsiveness of in vitro expanded CMV pp65-specific T cells.

Patient PBMCs were co-cultured with autologous DCs loaded with CMV pp65-encoding RNA and expanded in vitro in the presence of IL-2. (A) Peripheral blood lymphocytes (PBLs) were phenotyped before and after in vitro expansion. (B) Magnitude of CMV pp65-specific T cell responses were measured before and after expansion by CMV pp65 IFNγ ELISpot assay after stimulation. SFC; spot forming cell. Due to insufficient numbers of cells from 2 of 15, 13 patients’ cells were analyzed for phenotype and function. Statistical significance was determined by Wilcoxon signed-rank test. PBMCs, peripheral blood mononuclear cells. PBLs, peripheral blood lymphocytes.
CMV pp65 RNA-loaded DC enhances T-cell polyfunctionality of CMV pp65 T cells
In order to assess whether vaccination with CMV pp65 RNA-loaded DCs impacted T-cell responses in peripheral blood, we performed ex vivo analyses on circulating CMV pp65-specific CD8+ T cells before and seven days after patients were administered CMV-ATCT-Saline or CMV-ATCT-DC. We found that patients who received saline following CMV pp65-specific T cells had no significant change in the frequency of cells positive for one marker (Fig. S2 A, C, E, p = 1.0000, NS), whereas patients in the CMV-ATCT-DC cohort exhibited significantly enhanced frequencies of CMV pp65-specific T cells single positive for IFNγ, TNFα, or CCL3 (Fig. S2 B, D, F, p = 0.0078).
Next, we determined if CMV-ATCT-DC elicited a change in the frequency of circulating polyfunctional T cells, which are T cells that are capable of simultaneously generating IFNγ, TNFα, and CCL3 at the single-cell level. Patients in the cohort that received saline with ex vivo expanded CMV pp65-specific T cells (CMV-ATCT-Saline) had no increase in IFNγ+ TNFα+ CCL3+ triple-positive CMV pp65-specific T cells (Fig. 3A, p = 1.000 NS) while patients randomized to receive CMV pp65-specific T cells and CMV pp65 RNA-loaded DCs (CMV-ATCT-DC) had a significant increase in the mean frequency of IFNγ+ TNFα+ CCL3+ CMV pp65-specific CD8+ T cells (Fig. 3B, p = 0.0078). To confirm detection of polyfunctional CMV-specific T cells, we compared the median fluorescence intensity (MFI) of IFNγ expression in T cells defined by 3, 2, and 1 function(s) in a validated assay as previously described (10). As expected, CMV pp65-specific T cells defined by 3 functions exhibited higher MFI of IFNγ than T cells with fewer functions (Fig. 3C–D, p < 0.0001), demonstrating a hierarchy of IFNγ expression between functionally discrete subsets of T cells. This clearly authenticates the presence of bona fide polyfunctionality and substantiates previous reports that T cells with these multiple functions are the most potent effectors. These data collectively support a role for CMV pp65 RNA-loaded DC vaccines in the in vivo expansion and differentiation of polyfunctional CMV pp65-specific T cells.
Figure 3. Assessment of polyfunctionality in circulating CMV pp65-specific CD8+ T cells before and after immunotherapy.

Ex vivo analysis of CMV pp65-specific CD8+ T cells in peripheral blood circulation was performed to measure the frequency of cells simultaneously expressing IFNγ, TNFα, and CCL3 before and after immunotherapy with (A) CMV-ATCT-Saline (p = 1.0000, NS) (n) = 7 or (B) CMV-ATCT-DC (p = 0.0078) (n) = 8. Statistical significance was determined by Wilcoxon signed-rank test. (C–D) To confirm the presence of bonafide polyfunctionality, the MFI of IFNγ expression was compared between CMV pp65-specific CD8+ T cells with 3 functions versus T cells with lesser functions. Statistical significance was determined by Kruskal-Wallis non-parametric ANOVA followed by Dunn’s pairwise comparison.
Finally, we performed polyfunctional flow cytometry aggregate data analyses to catalogue the proportion of CMV pp65-specific CD8+ T cells defined by 3, 2, or 1 function(s) in order to determine how the distribution of these individual populations may have changed relative to one another in response to immunotherapy. Patients in the CMV-ATCT-Saline cohort had no major change in any defined T-cell subsets, whereas patients in the CMV-ATCT-DC cohort experienced enhancements in the mean frequencies of CMV pp65-specific T cells displaying three, two, or one function(s) (Fig. 4A–B).
Figure 4. CMV pp65-specific CD8+ T cells catalogued by quality before and after immunotherapy.

The CMV pp65-specific CD8+ T cell response is composed of distinct responders that vary by functionality. Aggregate data analyses were performed to determine the relative mean distribution of CMV pp65-specific CD8+ T cells expressing one or more functions defined by IFNγ, TNFα, and CCL3 before (gray bar) and after (black bar) immunotherapy in all patients who received (A) CMV-ATCT-Saline, n = 7 or (B) CMV-ATCT-DC, n = 8. Bars represent mean frequencies of CMV pp65-specific CD8+ T cells expressing the particular combination of functions shown. (C–D) The qualitative distribution of CMV pp65-specific CD8+ T cells before and after immunotherapy is shown for a single patient representative of either arm. ER11 and ER14 refer to the ERaDICATe trial patient numbers.
We also assessed T-cell quality before and after immunotherapy individually in all patients (Fig. S3) and highlight two patients representative of the CMV-ATCT-Saline (Fig. 4C) or CMV-ATCT-DC (Fig. 4D) cohort. As shown, ERaDICate (ER) patient 11 (CMV-ATCT-Saline) had no major change in T-cell functionality after immunotherapy, whereas patient ER14 (CMV-ATCT-DC) exhibited a marked reduction in the proportion of T cells monofunctional for IFNγ or TNFα, with a concomitant boost in T cells displaying all three functions. These observations are consistent with the cohorts at large; 4/8 patients (ER2, ER9, ER12, and ER14) receiving CMV-ATCT-DC had a dramatic reduction in the proportion of monofunctional IFNγ secreting cells that was accompanied by an increased proportion of T cells with all three functions (Fig. S3). Only one patient randomized to the saline arm experienced a similar shift (ER15), whereas 6/7 patients had little to no shift in the proportion of monofunctional IFNγ secreting cells following therapy. Importantly, these data collectively show that polyfunctional T cells occupy a significantly increased fraction within the CMV pp65-specific CD8+ T-cell compartment following immunotherapy with CMV-ATCT-DC, indicating that in vivo antigenic stimulation using DCs may be an opportune strategy to favorably shift the functional distribution of tumor-specific T cells to more powerful effector T cells. Notably, patients who received CMV-ATCT-DC also had a significantly higher increase in polyfunctional T cells at day 7 compared to day 1 reflected by a mean-fold change of 2.48 (Fig. 5, p= 0.0401). Whereas patients who received CMV-ATCT-Saline only had a modest mean-fold change in polyfunctional T cells of 1.25.
Figure 5. Assessment of polyfunctionality in circulating CMV pp65-specific CD8+ T cells before and after immunotherapy.

Ex vivo analysis of CMV pp65-specific CD8+ T cells in peripheral blood circulation was performed to measure the frequency of cells simultaneously expressing IFNγ, TNFα, and CCL3 molecules before and after immunotherapy with CMV-ATCT-Saline (white circles) or CMV-ATCT-DC (black circles). Statistical significance was determined by Wilcoxon signed-rank test. The fold change of CMV pp65-specific CD8+ T cells with 3 functions was determined for all 15 patients (p = 0.0401). Statistical significance was determined by the Mann-Whitney test.
Polyfunctional T cells correlate with outcome in vaccinated patients
As polyfunctional T-cell responses are considered a biomarker of protective immunity in acute and chronic viral infections (11–13) we sought to establish their significance in this cancer study. Unlike the negative correlation between the fold change of polyfunctional T cells and improved survival in patients who received CMV-ATCT-Saline (Fig. 6A, R = −0.4835, p= 0.2716), we observed a positive correlation between the fold change of polyfunctional T cells and improved survival in patients who received CMV-ATCT-DC (Fig. 6B, R = 0.7371, p= 0.0369). However, this study was not powered to detect differences between cohorts with regard to the clinical outcomes of PFS and OS so no conclusions can be drawn with regard to the causal association between polyfunctional T cell responses and outcome. To our knowledge, this is the first report to show that DC vaccination positively impacts T-cell responses in peripheral blood after CMV-specific T cell therapy and suggests further studies should examine the clinical significance of enhancing T-cell polyfunctionality in immunotherapy targeting newly-diagnosed GBM.
Figure 6. Correlation of fold change of polyfunctional CMV pp65-specific CD8+ T cells with OS.

Linear regression analysis was performed with the fold change of CMV pp65-specific CD8+ T cells with simultaneous expression of IFNγ, TNFα, and CCL3 and OS for all patients randomized to (A) CMV-ATCT-Saline (R = −0.4835, p = 0.2716 NS) or (B) CMV-ATCT-DC (R = 0.7371, p = 0.0369). (n) = 7 for A, (n) = 8 for B. Associations of survival outcomes with polyfunctional T-cell frequencies were assessed using the Pearson correlation coefficient.
Discussion
Here, we report our ability to produce and safely administer CMV pp65-specific T cells with DC vaccination (CMV-ATCT-DC) or saline (CMV-ATCT-Saline) to patients with newly-diagnosed GBM. The data reported here suggest important principles that warrant a formal assessment within a larger cohort of patients in a follow-up randomized phase II study.
In designing this study, we reasoned that an effective adoptive T cell therapy strategy using CMV pp65-specific T cells would require a qualitative shift of T cells defined by 0–1 function to >1 function in vivo, and we believed this could be achieved by an accompanying DC vaccine based on the success of this combinatorial approach in studies targeting metastatic melanoma and B-cell malignancies (9,14) Here, polyfunctional cells were defined by their ability to simultaneously secrete IFNγ, TNFα, and CCL3. We chose to include CCL3 in our polyfunctional T-cell panel since we recently found CCL3 to be a critical mediator of antitumor immunotherapy in the context of tumor-antigen-specific DC vaccination (15). This study demonstrates that patients with newly diagnosed GBM who are administered CMV-ATCT-DC had significant increases in the overall frequencies of polyfunctional CMV pp65-specific CD8+ T cells, and a greater proportion of these cells were identified to harbor multiple effector functions, compared to those receiving CMV-ATCT-Saline.
Importantly, we observed that augmented polyfunctional T cell presence was detected in patients treated with CMV pp65 RNA-pulsed DCs. These observations suggest that CMV pp65 specific T-cell polyfunctionality may potentially serve as a marker of effective therapeutic response and strengthen support for the role of polyfunctional T cells in the immune system’s effort against GBM and other cancers, in addition to the already established role of polyfunctional T cells in protective immunity against acute and chronic viral infections (11,12). Our findings are consistent with previous data, which described CMV-specific T cells isolated from GBM patients as being deficient in polyfunctionality, but following antigen exposure ex vivo, had restoration of their ability to generate multiple cytokines and were capable of mounting an effective antitumor response in vivo (5). Also analogous to previous reports of GBM patients receiving CMV pp65-specific T cells, patients in our study who were administered CMV-ATCT-Saline did not experience significant shifts in T-cell functionality (6). Incidentally, we were able to obtain samples either at or close to progression on two patients after 9 cycles of TMZ and found that the polyfunctional CD8+ pp65 antigen-specific cytokine response was greatly diminished after TMZ was restarted in one patient and increased in the other. To our knowledge, we are the first to report that DC vaccination enhances polyfunctionality of transferred CMV pp65-specific CD8+ T cells in vivo in patients with newly-diagnosed GBM.
In summary, the findings highlighted here have demonstrated the safety and feasibility of adding CMV pp65 RNA-loaded-DC to CMV pp65-specific T cells in conjunction with the clinical standard of care for patients with newly-diagnosed GBM. Our data also reaffirms earlier experiences targeting CMV pp65 in newly-diagnosed GBM patients by identifying CMV pp65 as a robust anti-GBM antigen. It also provides evidence that CMV pp65-specific T cells and CMV pp65 RNA-loaded DC can be successfully generated from GBM patients, who in general possess cell-mediated immune deficiencies. Since the FDA required the testing of a single antigen at a time for this immunotherapy trial, next steps would be to employ several more antigens in a multiantigenic vaccine that could elicit several different T cell responding populations. Finally, these results indicate that adjuvant DC vaccination enhances transferred T-cell immune responses in vivo, and suggest that ex vivo analyses of T-cell polyfunctionality may equip clinicians with a unique opportunity to help predict patient responses following immunotherapy, which is requisite in this era of individualized antitumor therapy.
Supplementary Material
Acknowledgments
The authors thank the medical staff, including S. Norman, B. Perry, and D. Lally-Goss, who supported this clinical study and provided unparalleled care and comfort to our patients. We also thank S. Janetzki (Zellnet Consulting, Inc, Fort Lee, NJ, and the Cancer Immunotherapy Consortium) for evaluating ELISpot plates and M. Roederer (NIH, Bethesda, MD) for providing access to SPICE software. We would also like to acknowledge each of the patients we’ve encountered who offer an endless source of inspiration. This trial is registered with www.clinicaltrials.gov (NCT00693095) as Evaluation of Recovery From Drug-Induced Lymphopenia Using Cytomegalovirus-specific T-cell Adoptive Transfer (ERaDICATe)
Financial support: This work was supported by grants from the National Institutes of Health (NIH) National Institute of Neurological Disorders and Stroke (R01-NS06703, D.A. Mitchell), National Cancer Institute (R01-CA134844 D.A. Mitchell), and the Department of Defense (W81XWH-10-1-0089, D.A. Mitchell). Additional support was provided by the National Brain Tumor Society (D.A. Mitchell and J.H. Sampson), the American Brain Tumor Association (D.A. Mitchell and J.H. Sampson), Accelerate Brain Cancer Cure Foundation Young Investigator’s Award (D.A. Mitchell), The Kinetics Foundation, (J.H. Sampson) Ben and Catherine Ivy Foundation (J.H. Sampson), and in part by Duke University’s Clinical & Translational Science Awards grant 1UL2 RR024128-01 from the National Institutes of Health National Center for Research Resources.
Footnotes
Disclosure of Potential Conflicts of Interest: J.H.S. and D.A.M. hold ownership interest in patents related to technologies described in this work. S.K.N. has ownership interest (including patents) in Argos Therapeutics. The remaining authors declare no competing financial interests.
References
- 1.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 2.Cobbs CS, Harkins L, Samanta M, Gillespie GY, Bharara S, King PH, et al. Human cytomegalovirus infection and expression in human malignant glioma. Cancer Res. 2002;62:3347–50. [PubMed] [Google Scholar]
- 3.Mitchell DA, Xie W, Schmittling R, Learn C, Friedman A, McLendon RE, et al. Sensitive detection of human cytomegalovirus in tumors and peripheral blood of patients diagnosed with glioblastoma. Neuro Oncol. 2008;10:10–8. doi: 10.1215/15228517-2007-035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dziurzynski K, Chang SM, Heimberger AB, Kalejta RF, McGregor Dallas SR, Smit M, et al. Consensus on the role of human cytomegalovirus in glioblastoma. Neuro Oncol. 2012;14:246–55. doi: 10.1093/neuonc/nor227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crough T, Beagley L, Smith C, Jones L, Walker DG, Khanna R. Ex vivo functional analysis, expansion and adoptive transfer of cytomegalovirus-specific T-cells in patients with glioblastoma multiforme. Immunol Cell Biol. 2012;90:872–80. doi: 10.1038/icb.2012.19. [DOI] [PubMed] [Google Scholar]
- 6.Schuessler A, Smith C, Beagley L, Boyle GM, Rehan S, Matthews K, et al. Autologous T-cell therapy for cytomegalovirus as a consolidative treatment for recurrent glioblastoma. Cancer Res. 2014;74:3466–76. doi: 10.1158/0008-5472.CAN-14-0296. [DOI] [PubMed] [Google Scholar]
- 7.Colleton BA, Huang XL, Melhem NM, Fan Z, Borowski L, Rappocciolo G, et al. Primary human immunodeficiency virus type 1-specific CD8+ T-cell responses induced by myeloid dendritic cells. J Virol. 2009;83:6288–99. doi: 10.1128/JVI.02611-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van Gulck E, Vlieghe E, Vekemans M, Van Tendeloo VF, Van De Velde A, Smits E, et al. mRNA-based dendritic cell vaccination induces potent antiviral T-cell responses in HIV-1-infected patients. AIDS. 2012;26:F1–12. doi: 10.1097/QAD.0b013e32834f33e8. [DOI] [PubMed] [Google Scholar]
- 9.Wimmers F, Aarntzen EH, Duiveman-deBoer T, Figdor CG, Jacobs JF, Tel J, et al. Long-lasting multifunctional CD8+ T cell responses in end-stage melanoma patients can be induced by dendritic cell vaccination. Oncoimmunology. 2016;5:e1067745. doi: 10.1080/2162402X.2015.1067745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bernstein DI, Reap EA, Katen K, Watson A, Smith K, Norberg P, et al. Randomized, double-blind, Phase 1 trial of an alphavirus replicon vaccine for cytomegalovirus in CMV seronegative adult volunteers. Vaccine. 2009;28:484–93. doi: 10.1016/j.vaccine.2009.09.135. [DOI] [PubMed] [Google Scholar]
- 11.Precopio ML, Betts MR, Parrino J, Price DA, Gostick E, Ambrozak DR, et al. Immunization with vaccinia virus induces polyfunctional and phenotypically distinctive CD8(+) T cell responses. J Exp Med. 2007;204:1405–16. doi: 10.1084/jem.20062363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seder RA, Darrah PA, Roederer M. T-cell quality in memory and protection: implications for vaccine design. Nat Rev Immunol. 2008;8:247–58. doi: 10.1038/nri2274. [DOI] [PubMed] [Google Scholar]
- 13.Nebbia G, Mattes FM, Smith C, Hainsworth E, Kopycinski J, Burroughs A, et al. Polyfunctional cytomegalovirus-specific CD4+ and pp65 CD8+ T cells protect against high-level replication after liver transplantation. Am J Transplant. 2008;8:2590–9. doi: 10.1111/j.1600-6143.2008.02425.x. [DOI] [PubMed] [Google Scholar]
- 14.Ding ZC, Huang L, Blazar BR, Yagita H, Mellor AL, Munn DH, et al. Polyfunctional CD4(+) T cells are essential for eradicating advanced B-cell lymphoma after chemotherapy. Blood. 2012;120:2229–39. doi: 10.1182/blood-2011-12-398321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mitchell DA, Batich KA, Gunn MD, Huang MN, Sanchez-Perez L, Nair SK, et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature. 2015;519:366–9. doi: 10.1038/nature14320. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.