

Abstract
Background
A complete understanding of the role of the liver in burn-induced hypermetabolism is lacking. We investigated the acute effect of severe burn trauma on liver mitochondrial respiratory capacity and coupling control as well as the signaling events underlying these alterations.
Methods
Male BALB/c mice (8–12 weeks) received full-thickness scald burns on ~30% of the body surface. Liver tissue was harvested 24 hours post injury. Mitochondrial respiration was determined by high-resolution respirometry. Citrate synthase activity was determined as a proxy of mitochondrial density. Male Sprague-Dawley rats received full-thickness scald burns to ~60% of the body surface. Serum was collected 24 hours post injury. HepG2 cells were cultured with serum-enriched media from either sham or burn treated rats. Protein levels were analyzed via western blot.
Results
Mass-specific (p=0.01) and mitochondrial-specific (p=0.01) respiration coupled to ATP production significantly increased in the liver after burn. The respiratory control ratio for ADP (p=0.04) and the mitochondrial flux control ratio (p=0.03) were elevated in the liver of burned animals. Complex III and Complex IV protein abundance in the liver increased after burn by 17% and 14%, respectively. Exposure of HepG2 cells to serum from burned rats increased the pAMPKα:AMPKα ratio (p<0.001) and levels of SIRT1 (p=0.01), Nrf2 (p<0.001), and PGC1α (p=0.02).
Conclusions
Severe burn trauma augments respiratory capacity and function of liver mitochondria, adaptations that augment ATP production. This response may be mediated by systemic factors that activate signaling proteins responsible for regulating cellular energy metabolism and mitochondrial biogenesis.
Introduction
Severe burns covering more than 30% of the total body surface area (TBSA) incur a hypermetabolic stress response that persists for years post injury (1). This hypermetabolic response is associated with protein catabolism and cachexia, thereby contributing to morbidity and mortality (1). The physiological mechanisms underlying burn-induced hypermetabolism are not fully understood. Whole-body O2 consumption and ATP turnover increase in response to burn injury. The majority (>90%) of whole-body O2 consumption and ATP production occurs within mitochondria (2), suggesting that these subcellular organelles mediate burn-induced hypermetabolism. ATP-dependent processes such as protein synthesis and gluconeogenesis account for approximately 50–60% of the hypermetabolic response to burns (3), with mitochondrial thermogenesis thought to be responsible for the remaining hypermetabolism (4–8).
Interestingly, the contribution of ATP-dependent and ATP-independent pathways to burn-induced hypermetabolism appears to be organ-specific (9). Indeed, we have recently demonstrated that skeletal muscle (5, 7, 8) and adipose tissue (4, 6, 10) mitochondria become thermogenic in response to burns. While muscle and adipose tissue may contribute to burn-induced hypermetabolism via increased thermogenesis, it is likely that central organs, such as the liver, influence burn-induced hypermetabolism through an increase in oxidative phosphorylation. The liver is responsible for approximately 25% of the metabolic rate in burned individuals (11). Moreover, the liver is the principle site of acute-phase protein synthesis and gluconeogenesis, processes that are upregulated after burn (12). Perhaps not surprisingly then, liver O2 consumption significantly increases after burns (11). Interestingly though, a robust mechanistic understanding of burn-induced hypermetabolism at the level of the liver is lacking.
In this study, we investigated the acute effect of severe burn trauma on liver mitochondrial respiratory capacity and coupling control as well as the signaling events underlying altered hepatic bioenergetics after burn. We hypothesized that severe burn injury augments liver mitochondrial function acutely, increasing the capacity for and efficiency of oxidative phosphorylation.
Materials and Methods
Rodent models of full-thickness scald burn injury
All animal research procedures adhered to the National Institutes of Health guidelines for experimental animal use and were approved by the Institutional Animal Care and Use Committee at the University of Texas Medical Branch (Galveston, TX). Animals were allowed to acclimate for 1 week before experimentation and received food and water ad libitum throughout the study. Animals were kept on a 12:12 light-dark cycle. Temperature was maintained at ~22°C in all animal holding and procedure rooms.
Mouse model
8–12 weeks old male BALB/c mice (25–30g) were used. Prophylactic analgesia (buprenorphine, 2 mg/kg, s.c.) was administered 30 to 60 minutes prior to general anesthesia (3–5% isoflurane). The dorsum was shaved with electric clippers, and 1 mL of room temperature Lactated Ringer’s (LR) solution was injected under the skin to protect the underlying spinal column. An ~30% TBSA burn was created on the dorsum by placing the mouse in a protective polyvinyl chloride mold and immersing the exposed dorsum in 95 to 100°C water for 10 seconds to produce a full-thickness injury (4, 8). Burned mice were immediately resuscitated with 1 to 2 mL of room temperature LR solution injected intraperitoneally. Sham animals were treated exactly as described above but were not immersed in hot water. Burn- and sham-treated mice were housed individually throughout the experimental period. Mice were sacrificed 24 hours post injury. Blood and liver samples were collected for biochemical analysis.
Rat model
Rats were used as a source of serum for in vitro studies because they have an approximately 10-fold greater blood volume than mice. A well-established method for the induction of a 60% TBSA full-thickness burn was used (13). Briefly, male Sprague-Dawley rats (325–350 g) were anesthetized with general anesthesia (ketamine, 40 mg/kg, i.p. and xylazine, 5 mg/kg, i.p.) and received analgesia (buprenorphine, 0.05 mg/kg, s.c.). Rats were placed within a protective mold that exposed ~30% of the TBSA and submerged in 95 to100°C water to induce a scald burn. The dorsum was immersed for 10 seconds and the abdomen for 2 seconds, resulting in a 30% TBSA injury (60% TBSA burn in total). Room temperature LR solution (40 mL/kg, i.p.) was administered immediately after the burn for resuscitation. Rats received oxygen during recovery from anesthesia. After 24 hours, rats were humanely euthanized and serum was collected. The serum was collected from two sham treated and two burn treated rats and used to treat a HepG2 hepatocyte cells (see: in vitro model of a severe scald burn).
High-resolution tissue respirometry
Immediately after being harvested, liver samples (5–10 mg) were submerged in approximately 2 to 3 mL of an ice-cold preservation solution that contained 10 mM CaK2-EGTA; 7.23 mM K2-EGTA; 20 mM imidazole; 20 mM taurine; 50 mM K-MES; 0.5 mM dithiothreitol; 6.56 mM MgCl2; 5.77 mM ATP; and 15 mM creatine phosphate (pH 7.1). Respirometry measurements were performed approximately 90 to 180 minutes after tissue collection. Liver samples were first minced in chilled preservation buffer using forceps and a scalpel. Liver tissue (~2–3 mg) was then blotted onto filter paper and weighed on a precision microbalance (Mettler-Toledo, Zaventem, Belgium). Tissue was then placed into an Oxygraph-2K respirometer (Oroboros Instruments, Innsbruck, Austria) with 2 mL of pH-adjusted (7.1) respiration solution composed of the following: 0.5 mM EGTA, 3 mM MgCl2, 60 mM K-lactobionate, 20 mM taurine, 10 mM KH2PO4, 20 mM HEPES, 110 mM sucrose, 1 mg/mL essential fatty acid–free bovine serum albumin. During all respirometry experiments, temperature was maintained at 37°C and O2 concentration within the range of 300 to 450 μM. Mitochondrial O2 flux (pmol/s/mg) was recorded at 2- to 4-second intervals (DatLab; Oroboros Instruments, Innsbruck, Austria).
All respiratory states were determined sequentially within the same sample during a respirometry experiment that typically lasted between 45 and 60 minutes. Liver mitochondrial respiratory capacity and function were assayed by the addition of substrates (1.5 mM octanoyl-l-carnitine, 5 mM pyruvate, 2 mM malate, and 10 mM glutamate) to support state 2 respiration (S2) with electron flow primarily through complex I of the electron transport chain (ETC). ADP (5mM) was then titrated into the oxygraph chamber to couple respiration to ATP production (state 3 respiration supported by complex I of the ETC; S3I). Thereafter, 10 mM succinate was titrated into the oxygraph chamber to induce coupled state 3 respiration with electron input from both complex I and II of the ETC (S3I+II). To ensure that the mitochondrial membranes were intact during the respiration assay, we titrated 10 μM cytochrome C into the oxygraph chamber; no/minimal change in respiration following cytochrome C addition indicated intact outer mitochondrial membranes. The ionophore carbonyl cyanide m-chlorophenyl hydrazone was then titrated into the chamber to a final concentration of 5 μM to uncouple oxidative phosphorylation, thereby inducing maximal uncoupled (state 3U) respiration. Finally, mitochondrial respiration was completely inhibited by the addition of 5 μM antimycin A; residual O2 consumption represented non-mitochondrial respiration.
Mitochondrial coupling and flux control ratios were calculated from mass-specific respiration data to provide indices of mitochondrial quality. First, the respiratory control ratio (RCR) for ADP was calculated by dividing S3I by S2 respiration. The RCR provides an index of how coupled mitochondria are. The flux control ratio (FCR) was determined by normalizing coupled S3I+II respiration to maximum uncoupled S3U respiration. The FCR provides an index of the efficiency of the oxidative phosphorylation system. Finally, total respiration and its components (non-mitochondrial respiration and leak, coupled and reserve respiration) were determined per milligram of tissue as described elsewhere (14) and are presented as a percentage of total respiration.
Citrate synthase activity
Citrate synthase (CS) activity was determined to provide a proxy of mitochondrial volume density (15). Liver homogenates (5 mg/mL) were prepared in a 175 mM KCl buffer containing 2 mM EDTA and 1% Triton. CS activity was assayed in extracts diluted in a 100 mM phosphate buffer (pH 7.1) containing 30 mM acetyl-CoA, 10 mM 5,5′-dithiobis-(2-nitrobenzoic acid), and 10 mM oxaloacetate (16). The resultant change in light absorbance due to the condensation of oxaloacetate with acetyl-CoA was determined spectrophotometrically (BioTek Instruments, Winooski, VT, USA). CS activity is expressed as nmol/s/g (wet weight).
Electron Transport chain protein quantification
Murine livers were homogenized using the Kinematic Polytron PT 10/35GT (Thermo Fisher Scientific, Inc.), per the manufacturer’s instructions, in modified RIPA buffer [50 mM Tris tris(hydroxymethyl)aminomethane] base, pH 7.5; 150 mM NaCl; 1 mM EDTA; 1% NP-40; 0.5% sodium deoxycholate; and 0.1% sodium dodecyl sulfate (SDS)] containing 1% Halt protease inhibitor cocktail and 1% Halt phosphatase inhibitor cocktails (Thermo Fisher Scientific, Inc., Waltham, MA). Western blot analysis was performed as previously described (15). Briefly, protein concentration was quantified using the Bradford method. Proteins (10–30 μg) were fractionated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) (Life Technologies Corporation, Carlsbad, CA) under denaturing conditions and then electro-transferred to a polyvinylidene fluoride membrane. After being blocked with blocking buffer (LI-COR, Inc., Lincoln, NE) the membrane was probed with Total OXPHOS Rodent WB Antibody (AB) Cocktail (Abcam [ab110413], Cambridge, MA) diluted with blocking buffer. Membranes were washed three times with phosphate-buffered saline with 0.1% Tween 20 (PBST) and incubated 1 hour with IRDye 680-conjugated anti-mouse μ or IRDye 800-conjugated anti-rabbit Ab (LI-COR, Inc., Lincoln, NE). Finally, the membranes were washed three times with PBST, and signals were detected using the Odyssey Infrared Imaging System (LI-COR, Inc., Lincoln, NE).
In vitro model of a severe scald burn
All cell culture media and trypsin were purchased from Life Technology Corp. (Carlsbad, CA). The human immortalized hepatocyte cell line HepG2 was purchased from American Type Cell Culture (ATCC, Manassas, VA). Cells were cultured for 24 hours in Dulbecco’s Modified Eagle’s Medium containing 4 mM L-glutamine, 4,500 mg/L glucose, 1 mM sodium pyruvate, and 1,500 mg/L sodium bicarbonate (ATCC [#30-2002], Manassas, VA) supplemented with either 5% serum from a sham treated rat or 5% serum from a rat with a 60% TBSA scald burn. All analysis was repeated using serum from different sham and burn treated rats.
Proteins were isolated and Western blots performed as described previously (17). We used the following primary antibodies: total AMPKα (recognizes phosphorylated and non-phosphorylated forms of AMPKα) [#5831], phosphorylated-AMPKα (Thr172) [#2535] and Sirt1 [#2496] purchased from Cell Signaling Technology (Danvers, MA), PGC-1α (Abcam [ab722-30], Cambridge, MA) and Nrf2 (Santa Cruz [sc-722], Santa Cruz, CA).
Statistical analysis
All data are presented as group means ± the standard error. Unpaired Student’s t-tests were used to detect statistically significant differences between sham and burn groups. Significance was accepted at p≤0.05. Statistical analysis was performed using GraphPad Prism Version 7 (GraphPad, La Jolla, CA).
Results
Mass-specific mitochondrial respiration is increased in liver following burn injury
Mass-specific liver mitochondrial respiration data (respiration per mg of tissue) for the burn and sham groups are shown in Figure 1A. Basal (prior to substrate titration) and leak respiration (S2) were not different between groups. Coupled ADP-dependent respiration supported by electron flow through complex I (S3I) was higher in the burn group than the sham group (73.5±6.8 vs. 53.1±3.9 pmol O2/s/mg; p=0.02). Similarly, coupled ADP-dependent respiration supported by electron flow through complex I and complex II (S3I+II) was higher in the burn group than the sham group (114.7±11.2 vs. 76.3±8.3 pmol O2/s/mg; p=0.01). Cytochrome c had a minimal effect on respiration in both the sham (4±1% increase) and burn groups (7±1% increase), suggesting that the outer mitochondrial membranes were largely intact during respirometric assays. Maximal respiration (S3U) was greater in the burn group than the sham group (135.1±13.0 vs. 105.7±7.8 pmol O2/s/mg; p=0.01).
Figure 1.
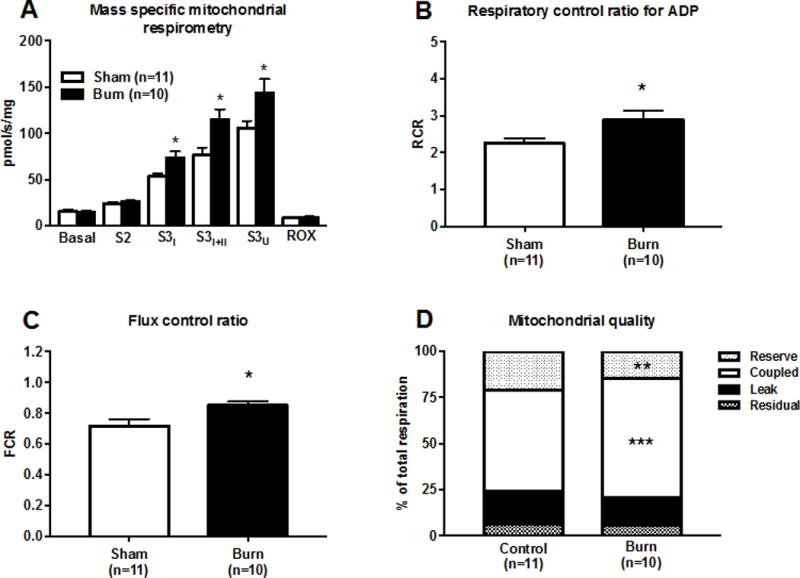
A 30% TBSA scald burn increases the respiratory capacity of mouse liver mitochondria. Mass-specific mitochondrial respiration in panel A. State 2 (S2) respiration was recorded following titration of substrate. State 3 respiration supported by complex I was recorded following titration of ADP (S3I), and state 3 respiration supported by complex I and II (S3I+II) was recorded thereafter following titration of succinate. Respiration was uncoupled by the titration of an ionophore and maximal uncoupled respiration (S3U) recorded. Antimycin A was titrated to inhibit mitochondrial respiration so that residual oxygen consumption (ROX) could be recorded. The respiratory control ratio (RCR) for ADP (S3I/S2) is shown in panel B. The flux control ratio (RCR) for ADP (S3I+II/S3U) is shown in panel C. Rates of residual, leak, coupled, and reserve respiration normalized to total respiratory capacity (S3U) are shown in panel D. For all data presented, n=11 (sham) and n=10 (burn), respectively. All values are group means ± SE, *p<0.05, **p<0.01, and ***p<0.001.
Mitochondrial coupling control is altered in the liver following burn injury
The RCR for ADP, an indicator of mitochondrial coupling control, is shown in Figure 1B. The RCR was higher following burn injury (2.89±0.26 vs. 2.25±0.13; p=0.04), suggesting better coupling of mitochondrial respiration to ATP production in response to burn trauma. As shown in Figure 1C, the FCR was greater in the burn group than the sham group (0.85±0.03 vs. 0.72±0.04; p=0.03), indicating a better matching of mitochondrial electron transfer capacity and ATP production. Absolute rates of leak and coupled respiration were determined by subtracting basal respiration from S2 and S3I+II, respectively. Reserve respiration was calculated from subtracting S3I+II from S3U. Leak, coupled, reserve, and residual respiration were summed to calculate total respiration (data not shown). These four respiratory rates normalized to total respiration are presented in Figure 1D to provide a comprehensive overview of the qualitative effect of acute burn trauma on liver mitochondrial function. Leak respiration accounted for a comparable proportion of total respiration in the liver in both the sham group (13.5±1.2%) and burn group (12.0±0.5%). Respiration coupled to ATP production represented a greater proportion of total liver mitochondrial respiration in the burn group than in the sham group (67.2±2.5% vs. 52.9±3.9%; p=0.001), further suggesting greater capacity for oxidative phosphorylation per mitochondrion. Reserve respiration (spare respiratory capacity) represented a smaller proportion of total liver mitochondrial respiration in the burn group than the sham group (13.9±2.1% vs. 28.9±5.6%; p=0.007). Residual respiration, i.e., non-mitochondrial respiration, accounted for a similar proportion of total liver mitochondrial respiration in the sham group (7.8±0.9%) and burn group (6.9±1.0%).
Mitochondria-specific respiration is increased in liver following burn injury
The mass-specific alterations in mitochondrial respiration shown in Figure 1A are dependent on both mitochondrial volume density and the quality of individual mitochondrion. To further delineate the cause of augmented mitochondrial respiratory capacity in the liver following burn trauma, we measured CS activity as a proxy of mitochondrial protein abundance and normalized mass-specific mitochondrial respiration data (presented in Figure 1A) to CS activity. CS activity was not different between the sham group (15.9±2.5 nmol/s/g) and burn group (13.9±1.7 nmol/s/g; Figure 2A). The mean CS activity value for the sham and burn groups was used to normalize mass specific respiration data reported in Figure 1. Mass specific respiration normalized to CS activity is presented in Figure 2. S3I respiration normalized to CS activity was higher in the burn group than the sham group (5.3±0.5 vs. 3.3±0.3 pmol O2/s/mg/CS activity; p=0.001; Figure 2B). The same was true of S3I+II respiration normalized to CS activity (8.3±0.8 vs, 4.8±0.6 pmol O2/s/mg/CS activity; p=0.002; Figure 2C) and S3U respiration normalized to CS activity (9.7±0.9 vs. 6.7±0.5 pmol O2/s/mg/CS activity; p=0.01; Figure 2D).
Figure 2.
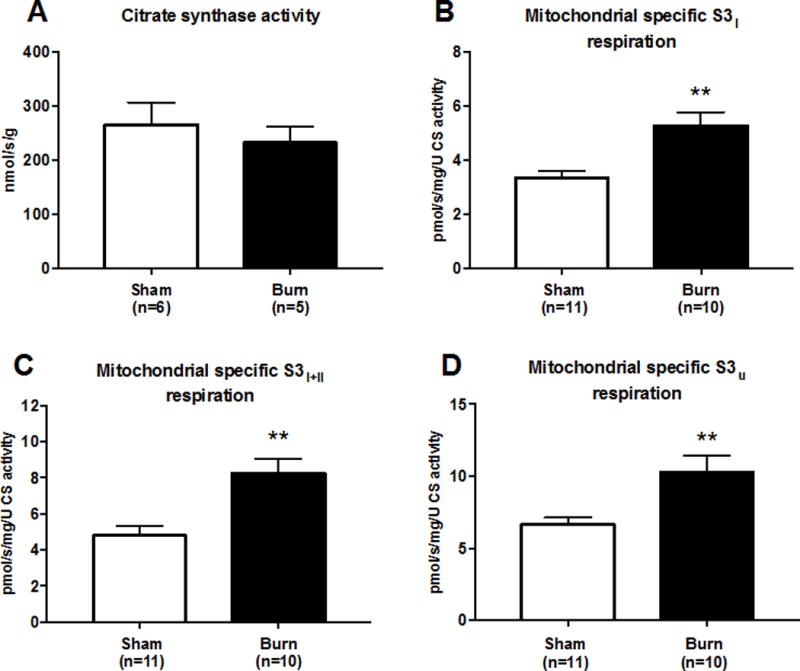
A 30% TBSA scald burn increases the respiratory function of mouse liver mitochondria. Citrate synthase (CS) activity is shown in panel A. S3I respiration (Figure 1A) normalized to CS activity is shown in panel B. S3I+II respiration (Figure 1A) normalized to CS activity is shown in Panel C. S3U respiration (Figure 1A) normalized to CS activity is shown in Panel D. For data presented panel A, n=6 (sham) and n=5 (burn), respectively. For data presented in panels B–D, n=11 (sham) and n=10 (burn), respectively. All values are group means ± SE, **P<0.01.
Effect of burn injury on mitochondrial protein abundance in the liver
Western blot analysis of ETC proteins is shown in Figure 3A. Burn injury did not increase the abundance of NADH oxidase (Complex I) or succinate dehydrogenase (Complex II) (Figure 3B). Burn injury acutely increased cytochrome bc oxido-reductase (Complex III) by 17% and cytochrome C oxidase (Complex IV) protein abundance by 14%, (Figure 3B). ATP synthase (Complex V) protein abundance in the liver was not altered by acute burn trauma (Figure 3B).
Figure 3.
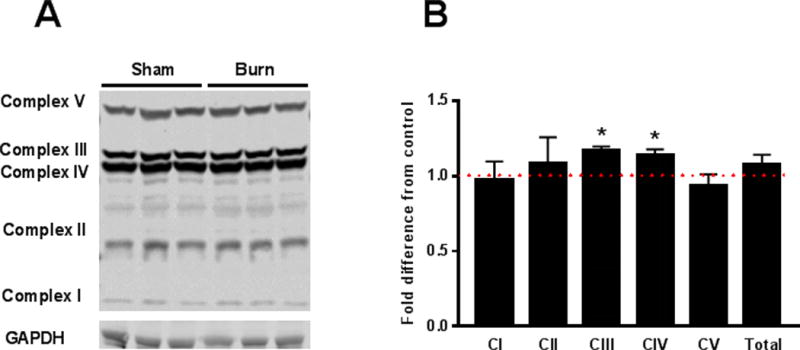
A representative micro-film depicting the detection of electron transport chain proteins in liver homogenates of sham and burn treated mice is shown in panel A. Quantification of western blot data for of electron transport chain proteins is shown in panel B. For data presented panel B, n=6 (sham) and n=6 (burn), respectively. Values are group means ± SE, *P<0.05.
Circulating factors induced by burn trauma acutely alter energy sensing proteins in hepatocytes
AMP-activated protein kinase alpha (AMPKα) abundance was significantly reduced in hepatocytes following acute (24h) exposure to serum collected from scalded rats (Figure 4A; P=0.007). This reduction was attributable to a ~3-fold increase in the abundance of phosphorylated AMPKα (pAMPKα) (Figure 4B; P=0.02) in hepatocytes acutely exposed to serum from burn injured rats. Subsequently, there was a significant shift in the pAMPK:totalAMPK ratio in hepatocytes exposed to burn serum in vitro (Figure 4C; P<0.001). NAD-dependent deacetylase sirtuin-1 (SIRT1) protein abundance was acutely elevated in hepatocytes exposed to serum collected from burned rats (Figure 4D; P=0.003), as was the expression of the peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) (Figure 4E; P=0.01), and nuclear respiratory factor 2 (NRF2) (Figure 4F; P<0.001).
Figure 4.
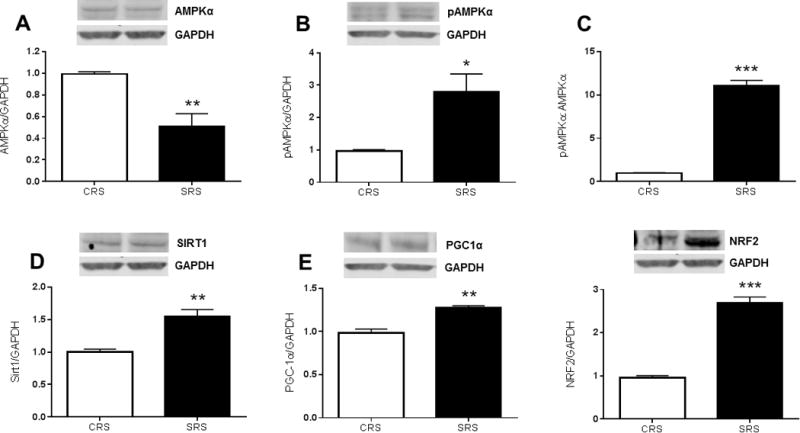
Serum from rats with a 60% TBSA scald burn alters metabolic signaling in cultured hepatocytes. The impact of acute (24h) exposure to hepatocytes to serum from sham or burned rats on metabolic proteins is shown in Figure 4. Panel A shows the acute effect of burn trauma on AMPKα abundance in hepatocytes, while panel B depicts the acute effect of burn trauma on the abundance of AMPKα in its phosphorylated form. Panel C shows the ratio of phosphorylated to total AMPKα. SIRT2 protein levels in hepatocytes exposed to serum from burned rats is shown in panel D, the response of PGC1α and NRF1 to acute burn trauma are depicted in panel E and panel F, respectively. All values are group means ± SE (n=4 experimental observations per group), *P<0.05, **P<0.01 and ***P<0.001, respectively.
Discussion
Because of the role of the liver in the stress response to burns, we hypothesized that liver mitochondrial respiration would be acutely augmented following burn trauma. Further, we theorized that increased ATP turnover would mediate this response, meaning that respiratory control, i.e., coupling of respiration to oxidative phosphorylation, would be augmented in the liver after severe burn trauma. Indeed, our results show that, in the acute response to burn injury, liver mitochondria undergo an adaptive response whereby respiratory capacity and function are increased. Specifically, respiratory capacity and respiration coupled to ATP production significantly increased after burn. In addition, mitochondrial coupling control was elevated in the liver of burned animals, evidenced by a greater respiratory response to ADP and a better matching between ATP production and respiratory capacity. Thus, both respiratory capacity of the mitochondrial network and the intrinsic function of mitochondrion appear to be acutely augmented in the liver in response to severe burn trauma, expanding capacity for ATP production.
Burn trauma results in a prolonged pathophysiological stress response that is unrivaled in terms of its magnitude and persistence. Hypermetabolism, an increase in resting metabolic rate, is a hallmark of this stress response. Indeed, patients with severe burns have significantly elevated whole-body O2 consumption rates (18, 19), which remain elevated for up to 2 years post injury (20). This hypermetabolic phenotype is associated with greater substrate turnover (12, 21–25), a greater degree of cachexia (26), and poorer clinical outcomes (20). Approximately 50–60% of hypermetabolism in burned individuals is accounted for by increased ATP turnover (3). The liver is thought to be the organ responsible for a significant portion of this increased ATP consumption post burn (3), given its important role in the acute-phase response (24, 25) and in intermediary metabolism (3, 11, 20, 22, 26, 27). Surprisingly, very little is known about how burn trauma affects cellular energetics in the liver.
In healthy humans, the liver accounts for ~20% of whole-body O2 consumption (27). Following a major burn covering >40% of the TBSA, the oxygen consumption rate of the liver can double (11), where is has been estimated that the liver accounts for ~25% of the increase in metabolic rate in response to severe burn trauma (9). Clearly then, the liver is one of the most important organs mediating burn-induced hypermetabolism. This increase in O2 consumption likely supports the acute-phase response to burns. Indeed, others have previously reported large increases in the production of acute-phase proteins in response to severe burn trauma (28–31). Since protein synthesis accounts for ~25% of whole-body ATP turnover in vivo (2), the large increase in liver O2 consumption post burn may support increased ATP production required for protein synthesis.
Our current data support the assertion that liver hypermetabolism in response to burn trauma supports ATP production. We found that, in burned mice, increased respiration in liver mitochondria was brought about by both an increase in respiratory capacity and improvements in mitochondrial coupling control. These data are in line with a previous report of increased ATP synthase activity in hepatocytes as early as 30 min post burn (32). Furthermore, Hu and colleagues (33) showed that, compared to mitochondria from sham-treated mice, those from burned mice had greater coupled (ATP-producing) respiration and a greater respiratory response to ADP. Our current data extend these findings by showing that increased mitochondrial respiratory capacity, as well as an increased respiratory response to ADP, underlie the acute increase in ATP-producing capacity of liver mitochondria post burn. Further, in the current study, we provide additional evidence of altered mitochondrial flux control in the liver following burn trauma. Specifically, per mitochondrion, increased ATP production was accompanied by a reduction in reserve respiratory capacity, meaning that in response to burn trauma, liver mitochondria use a greater proportion of their respiratory capacity for oxidative phosphorylation.
To further investigate whether burn injury induced alterations in mitochondrial respirometry measures by increasing mitochondrial volume density and/or altering mitochondrial function, we normalized mitochondrial respiratory fluxes to CS activity. Activity of this key citric acid cycle enzyme is reflective of mitochondrial protein abundance (15). CS activity was not altered in the liver by acute burn trauma. Accordingly, when normalized to CS activity, both maximal respiration and respiration coupled to ATP production remained higher in the livers of burned mice than in those of controls, adding further support to the assertion that the acute increase in liver mitochondrial respiratory capacity in response to burns was not brought about by a gross increase in mitochondrial protein abundance.
To further study the acute effect of burn trauma on hepatic mitochondrial protein levels, we quantified the abundance of ETC proteins in liver homogenates from burn and sham-treated mice. We found no significant differences in the abundance of Complex I, II, or V of the ETC between burn and control groups. However, we did observe a significant, albeit modest (~15%), increase in the abundance of Complex III and IV in liver homogenates of burned mice. Nevertheless, total ETC protein abundance was comparable in the livers of burn and sham-treated mice, further suggesting that there are no acute gross changes in liver mitochondrial protein abundance post burn. While we did not observe a global increase in mitochondrial protein levels, increases in the ETC proteins distal of ubiquinone, the point of electron convergence in the ETC, suggest that increased Complex III and IV protein abundance may be a specific adaptation to augment the electron transfer and respiratory capacity of liver mitochondria in response to burn trauma.
Our current data further suggest a role for the mitochondrion in the hypermetabolic response to burns. Interestingly, in contrast to our data generated in skeletal muscle (5, 14, 34) and both white (4, 6) and brown adipose tissue (35), which primarily suggest a loss in mitochondrial coupling control and thus increased mitochondrial thermogenesis, the current data suggest that liver mitochondria contribute to burn-induced hypermetabolism through increase ATP turnover. Further, while we observed modest changes in the abundance of a select few electron transport chain proteins, CS activity, a robust marker of mitochondrial volume density was not altered in the liver acutely post burn, suggesting that altered mitochondrial function underlies increased capacity for oxidative phosphorylation in the liver acutely post burn. This is in contrast to our data white adipose tissue (6), where changes in mitochondrial energy transduction (i.e., increased thermogenesis) are accompanied by altered mitochondrial volume density, evidenced by decreased CS activity in muscle and increased CS in fat of burn patients. However, we should note that CS activity increases in white adipose tissue of burned rodents (4) or humans (6) over several days to weeks, so it may be that mitochondrial volume density does change overtime in the liver post injury.
The scald burn model used in the current study was a cutaneous injury that was not complicated by additional conditions such as inhalation injury or sepsis. Since the liver is not directly injured in this model, systemic factors must mediate any alterations in liver mitochondrial function that were observed. To confirm this supposition, we used an in vitro approach to investigate the signaling events underpinning this response. After culturing HepG2 hepatocytes in serum collected from either burn or sham-treated rats for 24 h (serum was collected from rats 24 h post sham or burn treatment), we found that there was a robust alteration in the phosphorylation status of AMPKα, where AMPKα was phosphorylated and thus activated. AMPKα is an energy-sensing regulatory protein that can sense subtle changes in the ratio of AMP to ATP. In response to energy deprivation, i.e., an increase in the AMP to ATP ratio, AMPK activation can modulate metabolic activity within the cell, switching off energy-dependent processes such as protein and fatty acid synthesis, while augmenting glucose uptake and fatty acid oxidation (36). Thus, these data suggest that systemic factors induce an energy deficient state in the liver after burn injury by increasing ATP turnover. In addition to observing AMPK phosphorylation, we observed an increase in the abundance of SIRT1 in hepatocytes cultured in media containing the serum of burned rats. This NAD+-dependent acetylase is known to play critical metabolic roles in the liver, including altering fuel selection and mitochondrial biogenesis (37). Like AMPK, SIRT1 is sensitive to cellular energy state since it requires NAD+ to be active. Indeed, burn-induced activation of SIRT1 in hepatocytes may reflect increased NADH oxidation and thus greater NAD+ levels.
Activation of both AMPK and SIRT following cellular energy deprivation is capable of increasing the expression of PGC-1α (36, 37). PGC-1α is considered an important nuclear regulator of mitochondrial biogenesis. In conjunction with Nrf2, PGC-1α promotes the expression of mitochondrial genes encoded by both nuclear and mitochondrial genomes, ultimately increasing organelle density and function (38). In the current in vitro model of burn trauma, we observed concurrent elevations in PGC-1α and Nrf2 protein levels in HEPG2 cells, suggesting that acute burn trauma stimulates mitochondrial biogenesis in the liver. In the long-term, this response would likely increase mitochondrial volume/density, increasing cellular capacity for ATP production.
While the current data provides novel information on the acute response of hepatic bioenergetics to burn trauma, we should note the potential limitations of our experimental approach. As is typical with most rodent models of burn trauma, no wound excision or grafting was performed post injury, which is in contrast to how patients with burns would be treated. Further, animals were housed at ~22°C leading up to and during the experiments. While this is temperature is normal and in line with our local IACUC and national guidelines on housing for rodents in biomedical research, this ambient temperature potentially pose two issues. One, 22°C represents mild cold exposure to a mouse (~30°C represents thermoneutrality for most common laboratory mouse strains), and thus may alter energy expenditure. Two, animals were not kept in a warmer environment post burn, which again is in contrast to who most burn patients would be cared for. With that said, whether, these potential limitations impact liver bioenergetics is unclear, it is of course feasible that the second insult of major skin excision could further augment the acute stress response to burns, and research addressing this question would be welcomed. With regards to temperature animals were house at, this is certainly likely to affect metabolic rate and potentially the mitochondrial response to burns. Indeed, we see more profound browning of white adipose tissue in rodents (4) compared to humans (6) following severe burns. However, since mitochondrial adaptions in the liver post burn seen to reflect increase capacity for ATP turnovers and not thermogenesis, we are of the opinion that the housing temperature of mice in the current study did not have a significant bearing on results.
To summarize, these data underscore the importance of the liver in the hypermetabolic stress response to severe burn trauma. Specifically, we have shown that severe burn trauma markedly alters the respiratory capacity and function of liver mitochondria. Collectively, these adaptations increase liver capacity for ATP production, likely providing greater metabolic flexibility in the context of the hypermetabolic response to burns. We have also show that systemic factors that increase ATP and NADH turnover in hepatocytes may mediate this response by activating regulatory signaling proteins responsible for the integration of fuel metabolism and mitochondrial biogenesis. Further studies defining the long-term alterations in hepatic bioenergetics will likely shed new light on the role of the liver in the hypermetabolic stress response to burns.
Acknowledgments
This work was funded by National Institutes of Health (P50 GM060338, R01 GM056687, R01 GM112936, and T32 GM008256) and Shriners Hospitals for Children (84080, 85410, and 84090) research grants. This work was also supported by the Department of Surgery at the University of Texas Medical Branch.
Footnotes
Disclosure
The authors have no affiliations or involvement in any organization or entity with any financial or non-financial interest in the subject matter or materials discussed in this manuscript.
References
- 1.Porter C, Tompkins RG, Finnerty CC, Sidossis LS, Suman OE, Herndon DN. The metabolic stress response to burn trauma: Current understanding and therapies. Lancet. 2016;388:1417–1426. doi: 10.1016/S0140-6736(16)31469-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rolfe DF, Brown GC. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol Rev. 1997;77:731–758. doi: 10.1152/physrev.1997.77.3.731. [DOI] [PubMed] [Google Scholar]
- 3.Yu YM, Tompkins RG, Ryan CM, Young VR. The metabolic basis of the increase of the increase in energy expenditure in severely burned patients. JPEN J Parenter Enteral Nutr. 1999;23:160–168. doi: 10.1177/0148607199023003160. [DOI] [PubMed] [Google Scholar]
- 4.Porter C, Herndon DN, Bhattarai N, Ogunbileje JO, Szczesny B, Szabo C, Toliver-Kinsky T, Sidossis LS. Severe burn injury induces thermogenically functional mitochondria in murine white adipose tissue. Shock. 2015;44:258–264. doi: 10.1097/SHK.0000000000000410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Porter C, Herndon DN, Borscheim E, Chao T, Reidy PT, Borack M, Rasmussen BB, Chondonikola M, Saraf MK, Sidossis LS. Uncoupled skeletal muscle mitochondria contribute to hypermetabolism in severely burned adults. Am J Physiol Endocrinol Metab. 2014;307:462–467. doi: 10.1152/ajpendo.00206.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sidossis LS, Porter C, Saraf MK, Borsheim E, Radhakrishnan RS, Chao T, Ali A, Chondronikola M, Mlcak R, Finnerty CC, Hawkins HK, Toliver-Kinsky T, Herndon DN. Browning of subcutaneous white adipose tissue in humans after severe adrenergic stress. Cell Metab. 2015;22:219–227. doi: 10.1016/j.cmet.2015.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Porter C, Herndon DN, Børsheim E, Bhattarai N, Chao T, Reidy PT, Rasmussen BB, Andersen CR, Suman OE, Sidossis LS. Long-term skeletal muscle mitochondrial dysfunction is associated with hypermetabolism in severely burned children. J Burn Care Res. 2015;37:53–63. doi: 10.1097/BCR.0000000000000308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Porter C, Herndon DN, Bhattarai N, Ogunbileje JO, Szczesny B, Szabo C, Toliver-Kinsky T, Sidossis LS. Differential acute and chronic effects of burn trauma on murine skeletal muscle bioenergetics. Burns. 2016;42:112–122. doi: 10.1016/j.burns.2015.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Porter C, Chondronikola M, Sidossis L. The therapeutic potential of brown adipocytes in humans. Front Endocrinol (Lausanne) 2015;13:156. doi: 10.3389/fendo.2015.00156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Porter C, Herndon DN, Chondronikola M, Chao T, Annamalai P, Bhattarai N, Saraf MK, Capek KD, Reidy PT, Daquinag AC, Kolonin MG, Rasmussen BBB,E, Toliver-Kinsky T, Sidossis LS. Human and mouse brown adipose tissue mitochondria have comparable ucp1 function. Cell Metab. 2016;24:246–255. doi: 10.1016/j.cmet.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilmore DW, Aulick LH. Systemic responses to injury and the healing wound. JPEN J Parenter Enteral Nutr. 1980;4:147–151. doi: 10.1177/014860718000400215. [DOI] [PubMed] [Google Scholar]
- 12.Wolfe RR, Herndon DN, Jahoor F, Miyoshi H, Wolfe M. Effect of severe burn injury on substrate cycling by glucose and fatty acids. N Engl J Med. 1987;317:403–408. doi: 10.1056/NEJM198708133170702. [DOI] [PubMed] [Google Scholar]
- 13.Mascarenhas D, El Ayadi AS,BK, Prasai A, Hegde S, Herndon D, Finnerty C. Nephrilin peptide modulates a neuroimmune stress response in rodent models of burn trauma and sepsis. Int J Burns Trauma. 2013;3:190–200. [PMC free article] [PubMed] [Google Scholar]
- 14.Porter C, Hurren NM, Cotter M, Bhattarai N, Reidy PT, Dillon EL, Durham W, Tuvdendorj D, Sheffield-Moore M, Volpi E, Sidossis LS, Rasmussen BB, Børsheim E. Mitochondrial respiratory capacity and coupling control decline with age in human skeletal muscle. Am J Physiol Endocrinol Metab. 2015;309:224–32. doi: 10.1152/ajpendo.00125.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Larsen S, Nielsen J, Hansen CN, Nielsen LB, Wibrand F, Stride N, Schroder HD, Boushel R, Helge JW, Dela F, Hey-Mogensen M. Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J Physiol. 2012;590:3349–3360. doi: 10.1113/jphysiol.2012.230185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Srere PA. Citrate synthase. Methods Enzymol. 1969;13:3–11. [Google Scholar]
- 17.Nunez Lopez O, Bohanon F, Wang X, Ye N, Corsello T, Rojas-Khalil Y, Chen H, Zhou J, Radhakrishnan R. Stat3 inhibition suppresses hepatic stellate cell fibrogenesis: Hjc0123, a potential therapeutic agent for liver fibrosis. RSC Adv. 2016;6:100652–100663. doi: 10.1039/C6RA17459K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goran MI, Peters EJ, Herndon DN, Wolfe RR. Total energy expenditure in burned children using the doubly labeled water technique. Am J Physiol. 1990;259:576–585. doi: 10.1152/ajpendo.1990.259.4.E576. [DOI] [PubMed] [Google Scholar]
- 19.Wilmore DW, Long JM, Mason ADJ, Skreen RW, Pruitt BAJ. Catecholamines: Mediator of the hypermetabolic response to thermal injury. Ann Surg. 1974;180:653–669. doi: 10.1097/00000658-197410000-00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jeschke MG, Gauglitz GG, Kulp GA, Finnerty CC, Williams FN, Kraft R, Suman OE, Mlcak RP, Herndon DN. Long-term persistance of the pathophysiologic response to severe burn injury. PLoS One. 2011;6:e21245. doi: 10.1371/journal.pone.0021245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Biolo G, Fleming RY, Maggi SP, Nguyen TT, Herndon DN, Wolfe RR. Inverse regulation of protein turnover and amino acid transport in skeletal muscle of hypercatabolic patients. J Clin Endocrinol Metab. 2002;87:3378–3384. doi: 10.1210/jcem.87.7.8699. [DOI] [PubMed] [Google Scholar]
- 22.Wolfe RR, Durkot MJ, Allsop JR, Burke JF. Glucose metabolism in severely burned patients. Metabolism. 1979;28:1031–1039. doi: 10.1016/0026-0495(79)90007-6. [DOI] [PubMed] [Google Scholar]
- 23.Wolfe RR, Herndon DN, Peters EJ, Jahoor F, Desai MH, Holland OB. Regulation of lipolysis in severely burned children. Ann Surg. 1987;206:214–221. doi: 10.1097/00000658-198708000-00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wolfe RR, Jahoor F, Herndon DN, Miyoshi H. Isotopic evaluation of the metabolism of pyruvate and related substrates in normal adult volunteers and severely burned children: Effect of dichloroacetate and glucose infusion. Surgery. 1991;110:54–67. [PubMed] [Google Scholar]
- 25.Wolfe RR, Klein S, Herndon DN, Jahoor F. Substrate cycling in thermogenesis and amplification of net substrate flux in human volunteers and burned patients. J Trauma. 1990;30:6–9. doi: 10.1097/00005373-199012001-00004. [DOI] [PubMed] [Google Scholar]
- 26.Hart DW, Wolf SE, Chinkes DL, Gore DC, Mlcak RP, Beauford RB, Obeng MK, Lal S, Gold WF, Wolfe RR, Herndon DN. Determinants of skeletal muscle catabolism after severe burn. Ann Surg. 2000;232:455–465. doi: 10.1097/00000658-200010000-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gallagher D, Belmonte D, Deurenberg P, Wang Z, Krasnow N, Pi-Sunyer FX, Heymsfield SB. Organ-tissue mass measurement allows modeling of ree and metabolically active tissue mass. Am J Physiol. 1998;275:249–258. doi: 10.1152/ajpendo.1998.275.2.E249. [DOI] [PubMed] [Google Scholar]
- 28.Gauglitz GG, Song J, Herndon DN, Finnerty CC, Boehning D, Barral JM, Jeschke MG. Characterization of the inflammatory response during acute and post-acute phases after severe burn. Shock. 2008;30:503–507. doi: 10.1097/SHK.0b013e31816e3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jeschke MG, Barrow RE, Herndon DN. Extended hypermetabolic response of the liver in severely burned pediatric patients. Arch Surg. 2004;139:641–647. doi: 10.1001/archsurg.139.6.641. [DOI] [PubMed] [Google Scholar]
- 30.Jeschke MG, Chinkes DL, Finnerty CC, Kulp G, Suman OE, Norbury WB, Branski LK, Gauglitz GG, Mlcak RP, Herndon DN. Pathophysiologic response to severe burn injury. Ann Surg. 2008;248:387–401. doi: 10.1097/SLA.0b013e3181856241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jeschke MG, Micak RP, Finnerty CC, Herndon DN. Changes in liver function and size after a severe thermal injury. Shock. 2007;28:172–177. doi: 10.1097/shk.0b013e318047b9e2. [DOI] [PubMed] [Google Scholar]
- 32.Wang XM, Chen KM, Shi Y, Shi HP. Functional changes of the nadh respiratory chain in rat-liver mitochondria and the content changes of high-energy phosphate groups in rat liver and heart during the early phase of burn injury. Burns. 1990;16:377–380. doi: 10.1016/0305-4179(90)90012-l. [DOI] [PubMed] [Google Scholar]
- 33.Hu H, Greif RL, Goodwin CW. The effects of thermal injury on mitochondrial oxygen consumption and the glycerol phosphate shuttle. Metabolism. 1994;43:913–916. doi: 10.1016/0026-0495(94)90276-3. [DOI] [PubMed] [Google Scholar]
- 34.Porter C, Herndon D, Bhattarai N, Ogunbileje J, Szczesny B, Szabo C, Toliver-Kinsky T, Sidossis L. Differential acute and chronic effects of burn trauma on murine skeletal muscle bioenergetics. Burns. 2016;42:112–122. doi: 10.1016/j.burns.2015.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Porter C, Herndon D, Chondonikola M, Chao T, Annamalai P, Bhattarai N, Saraf M, Capek K, Reidy P, Daquinag A, Kolonin M, Rasmussen B, Borsheim E, Toliver-Kinsky T, Sidossis L. Human and mouse brown adipose tissue mitochoondria have similar ucp1 function. Cell Metab. 2016 doi: 10.1016/j.cmet.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Viollet B, Foretz M, Guigas B, Horman S, Dentin R, Bertrand L, Hue L, Andreelli F. Activation of amp-activated protein kinase in the liver: A new strategy for the management of metabolic hepatic disorders. J Physiol. 2006;574:42–53. doi: 10.1113/jphysiol.2006.108506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chang H, Guarente L. Sirt1 and other sirtuins in metabolism. Trends Endocrinol Metab. 2013;25:138–145. doi: 10.1016/j.tem.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scarpulla R. Metabolic control of mitochondrial biogenesis through the pgc-1 family regulatory network. Biochim Biophys Acta. 2010;1813:1269–1278. doi: 10.1016/j.bbamcr.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]