

Abstract
Designing low-phytate crops without affecting the developmental process in plants had led to the identification of ABCC5 gene in soybean. The GmABCC5 gene was identified and a partial gene sequence was cloned from popular Indian soybean genotype Pusa16. Conserved domains and motifs unique to ABC transporters were identified in the 30 homologous sequences retrieved by BLASTP analysis. The homologs were analyzed for their evolutionary relationship and physiochemical properties. Conserved domains, transmembrane architecture and secondary structure of GmABCC5 were predicted with the aid of computational tools. Analysis identified 53 alpha helices and 31 beta strands, predicting 60% residues in alpha conformation. A three-dimensional (3D) model for GmABCC5 was developed based on 5twv.1.B (Homo sapiens) template homology to gain better insight into its molecular mechanism of transport and sequestration. Spatio-temporal real-time PCR analysis identified mid-to-late seed developmental stages as the time window for the maximum GmABCC5 gene expression, a potential target stage for phytate reduction. Results of this study provide valuable insights into the structural and functional characteristics of GmABCC5, which may be further utilized for the development of nutritionally enriched low-phytate soybean with improved mineral bioavailability.
Keywords: ABC transporters, Low-phytate crop, Domains, Motifs, Protein modeling
Introduction
Soybean, an agriculturally important crop, is noted for its high contents of protein (40%), oil (20%), dietary fiber and other beneficial phytochemicals like isoflavones and vitamins. Despite the manifestation of these unique nutritional attributes, it has limited consumption, which can be accredited to the presence of an absorption inhibitor, phytic acid. Phytic acid [myo-inositol 1,2,3,4,5,6-hexakisphosphate (InsP6)] bears a strong negative charge due to the presence of six attached phosphorus groups on the myo-inositol ring, enabling chelation with many essential divalent cations like iron, zinc, calcium and potassium and consequently reduces their bioavailability (Loewus and Murthy 2000). Almost half of the phosphorus fertilizer given to the crops is lost and the remaining half gets assimilated and translocated to the seeds, where a substantial (75% of the total assimilated) amount is bound and stored as phytate in vacuoles and becomes unattainable to the plant. This is a noteworthy bottleneck in the flow of phosphorus through the world’s agrarian ecosystem. Also, the digestive tracts of non-ruminants lack enzymes for the removal of phosphates from the myo-inositol ring of phytic acid, and are therefore unable to utilize the phosphorus found in legumes and cereals. Unutilized phosphorus present as phytic acid is excreted and used as manure for the fertilization of crops. The runoff from these agricultural fields containing high amount of unutilized phosphorus can further result in environmental phosphorus pollution, eutrophication and impaired water quality (Raboy 2003; Lott et al. 2000). The serious consequences have thus sparked off numerous studies and approaches toward reduction of phytate levels in the seeds of cereals and legumes used as food and feed. Considerable efforts have been made for lowering of phytic acid in cereals and oilseeds by targeting the genes involved in its biosynthesis and generating low-phytate mutants (lpa), which accumulate more inorganic phosphate without affecting the total plant phosphorus content (Raboy 2001). The biosynthesis of phytic acid takes place in the cytosol by sequential phosphorylation of the primary substrate, myo-inositol (Greenwood and Bewley 1984; Rasmussen et al. 2010), and subsequent transport and compartmentalization is undertaken by ATP-binding cassette (ABC) transporters. The targeting of ABC transporter gene has been reported to have a far-reaching effect on phytate reduction without adversely affecting plant growth and development (Shi et al. 2007).
The ABC transporters represent one of the largest superfamily of ubiquitous membrane proteins. The plant genome is particularly rich in these transporters, where Arabidopsis and Orzya sativa are known to encode more than 120 ABC transporter proteins. The ABC transporter proteins mediate MgATP-energized transmembrane transport and also regulate other transporters. These transporters in plants have been designated into different subfamilies based on the identity of their nucleotide-binding domain (NBD) and the transmembrane domains (TMD) and further prediction of function based on subfamily membership. The plant members of various subclasses have been implicated in a plethora of processes viz, lipid catabolism, polar auxin transport, xenobiotic detoxification, stomatal function, disease resistance and the transport of mineral ions and lipids. The ABC transporters of subfamily C compartmentalize phytate into protein storage vacuoles coupled with ATP hydrolysis (Raboy 2002; Xu et al. 2009; Regvar et al. 2011). The role of plant ABC transporter family proteins in phytic acid transport has been well documented (Nagy et al. 2009; Dorsch et al. 2003; Panzeri et al. 2011; Xu et al. 2009; Shi et al. 2007), but reports on its expression profile, physiochemical and structural properties in soybean are sparse. Similarly, the elucidation of the complete molecular and functional mechanisms of transmembrane phytate transport requires deep insight into the proteins’ structural features. Therefore, in this study, efforts were made to identify the ABC transporter gene implicated as a potential candidate for developing low-phytate soybean, an agriculturally and nutritionally important crop. Its protein sequence was characterized through bio-computational tools for elucidation of biochemical, physiochemical and structural properties to gain a better insight into the biological mechanism of phytate sequestration and transport.
Materials and methods
Plant material
Field-grown soybean variety Pusa 16 from the Division of Genetics, IARI, New Delhi, was used for the study. For the spatial expression profile, samples from root, stem, leaf and flower were collected from the 30-day-old plants. Developing soybean seeds were collected and sorted based on seed size into developmental stages. Tissue samples and developing seeds were harvested by flash freezing in liquid nitrogen and stored at − 80 °C for RNA isolation.
Amplification and cloning of GmABCC5 partial sequence
Total RNA was isolated from the samples using the TRIZOL reagent (Invitrogen, USA) and treated with RNase-free DNase I (Thermo Scientific, USA) for removal of any residual genomic DNA as per manufacturer’s instructions. RNA yield was quantified by spectrophotometry with Nanodrop 2000 (Thermo Scientific, USA) to achieve uniform concentration for cDNA synthesis, and RNA integrity was checked by separation on 1.2% agarose gel. 1 µg of total RNA was reverse transcribed for cDNA synthesis using the RevertAidTM H Minus First strand cDNA Synthesis Kit (Thermo Scientific, USA) according to the manufacturer’s protocol. The primer pair designed, GmABCC5F [5′-ATG AAG CCA TAG AGG CCA TGG ATA TCC CTA C-3′] and GmABCC5R [5′-TAG CTT CTG TGC AGC TGC AAG ACC AAA C-3′], were used for the amplification of GmABCC5 partial gene sequence (521 bp) from the previously reported genomic sequence of GmABCC5 deposited in NCBI GenBank (accession number XM_003554257.3), implicated in lowering of phytic acid in soybean (Maroof et al. 2009). The sequence amplified from cDNA was cloned in the pGEM®-T Easy vector (Promega, USA). The recombinant plasmids were verified by restriction analyses and sequenced using universal primers (SP6 and T7) on an automated sequencer (ABI 3730xl DNA Analyzer, USA). The nucleotide sequence data were submitted to the GenBank (accession number KF183505). To analyze the exon/intron organization of GmABCC5 (XM_003554257.3), the genomic and CDS sequences of the gene were downloaded from the NCBI database and compared in the Gene Structure Display Server 2.0 (Hu et al. 2015) program.
Differential gene expression by quantitative real-time PCR
To study the expression profile, target gene primers were customized from the exonic regions of previously reported complete genomic sequences of GmABCC5 (GenBank: accession number XM_003554257.3) for amplification of approximately 200 bp from the conserved region of the gene of interest (qABCC5F 5′-TCG ACG TTG TTT TCT TCT TCT TCT TC-3′ and qABCC5R 5′-CCG TTC TCC TTA CCG AAT CTA ACT-3′). The data were normalized using the soybean housekeeping gene PEP carboxylase (qPEPCoF 5′-CAT GCA CCA AAG GGT GTT TT-3′ and qPEPCoR 5′-TTT TGC GGC AGC TAT CTC TC-3′). Real-time expression analyses of the target and reference gene was carried out in PikoReal 96 Real Time PCR System (Thermo Scientific, USA). The experimentation was performed according to the standard protocol using DyNAmo Flash SYBER Green qPCR Kit (Thermo Scientific, USA). The three biological samples were run in triplicate under the following conditions: 1 cycle of 3 min at 94 °C followed by 35 cycles of 95 °C for 30 s and 30 s at 60 °C with a final extension of 30 s at 72 °C. The amplified products were subjected to melt-curve analysis and the specificity of the amplification was assessed by dissociation curve analysis. A unique peak on the dissociation curve was confirmed for the gene. Relative expression of target genes was calculated using the efficiency calibrated method (Livak and Schmittgen 2001). For the spatial expression analysis, flower tissue was considered as the calibrator, whereas for temporal analysis, expression in the representative 2–4 mm sized seeds was taken as calibrator and their expression was taken as one and the fold change was calculated. The specificity of the PCR amplification reaction was analyzed on 1.2% agarose gel.
Sequence analysis of GmABCC5 gene
The related GmABCC5 amino acid sequences from other plants were identified via Basic Local Alignment Search Tool (BLASTP) implemented on databases of the National Center for Biotechnology Information (http://blast.ncbi.nlm.nih.gov/blastp/) using the previously deduced GmABCC5 (accession number XM_003554257.3) amino acid sequence as a query. The amino acid compositions of the selected sequences were computed using PEPSTATS (http://emboss.bioinformatics.nl/cgi-bin/emboss/pepstats/) analysis tool (Rice et al. 2000). The ExPASy ProtParam tool (http://web.expasy.org/protparam/) was used to compute the physiochemical characteristics of the selected 31 homologous sequences (Gasteiger et al. 2005). The amino acid sequences were fed into MUSCLE (http://www.ebi.ac.uk/Tools/msa/muscle/) for multiple alignments and the alignment file was imported to BioEdit program (Hall 1999) to identify and shade the conserved amino acid sequences. The phylogenetic analysis was carried out using the neighbor-joining (N-J) method with 2000 bootstrap replicates implemented in MEGA (Molecular Evolutionary Genetic Analysis) version 6 program (Tamura et al. 2013), to evaluate the degree of support for the particular grouping pattern in the constructed cladogram.
Structural analysis, domain prediction and functional characterization of GmABCC5
Secondary structure elements predictions were performed using RaptorX server (http://raptorx.uchicago.edu/) SS3 (3-State Secondary Structure Prediction) (Wang et al. 2016) and PSIPREDv3.2 server (http://bioinf.cs.ucl.ac.uk/psipred/) (Buchan et al. 2013), domain prediction was done using DomPred, protein disorder was predicted using RaptorX DISO and FoldIndex (http://bioportal.weizmann.ac.il/fldbin/findex). Online prediction tools DiANNA (http://clavius.bc.edu/~clotelab/DiANNA) (Ferre and Clote 2005) and CYS-REC (http://linux1.softberry.com/berry.phtml) were utilized to determine the most likely bonding patterns involving the available cysteine residues. Solvent accessibility of the protein was estimated using RaptorX server ACC program. Membrane helix and topology prediction was done using HMMTOP, MEMSAT3 and MEMSAT-SVM programs from the PSIPRED server. The probable subcellular protein location was determined using TargetP server (http://www.cbs.dtu.dk/) (Emanuelsson et al. 2000), MEMSAT SVM and ProtComp v. 9.0 (http://www.softberry.com/berry.phtml?topic=protcomppl&group=programs&subgroup=proloc). For the probable gene term annotations, FFPred 3 program was used for prediction of all Gene Ontology domains (Biological Process, Molecular Function, Cellular Component) from PSIPRED server (http://bioinf.cs.ucl.ac.uk/psipred/).
Homology modeling and quality assessment of the predicted structure
The 3D structure of the target protein was constructed by template-based homology modeling using SWISS-MODEL software. The theoretical model was built by ProMod3 target-template alignment using 5twv.1.B (Homo sapiens) as the reference template. The stereochemical properties of the obtained model were evaluated by Ramachandran plot analysis with RAMPAGE (Lovell et al. 2003). The 3D model of GmABCC5 was fed to Pymol Molecular Graphic System (http://pymol.org/ep) to obtain the final model structure. The PDB file of the modeled GmABCC5 was analyzed on ProSA-web server for obtaining consistent values for the model generated (http://prosa.services.came.sbg.ac.at/prosa.php) (Wiederstein and Sippl 2007) and What IF (http://swift.cmbi.ru.nl/whatif/) for packing quality assessment of the theoretical model.
The GmABCC5 has a well-documented role in phytate transport and storage; hence, identification of probable binding site residues is imperative for detailed mode-of-transport analysis. No probable ligand/substrate was identified by the SWISS-MODEL software, hence RaptorX-binding prediction server (http://raptorx.uchicago.edu/BindingSite/) (Kallberg et al. 2012) was employed for identification of probable substrate and binding site residues.
Results and discussion
Previous studies had reported that a single base mutation in ABC transporter gene resulted in a low-phytate trait in soybean. A single base substitution from A (wild-type) to T (mutant) encoded a stop codon at the amino acid level, generating a loss-of-function mutant ABC transporter protein product (Maroof et al. 2009). Using the sequence-based homology search with the mutated sequence as query helped in identifying the full-length GmABCC5 gene (gene id: XM_003554257.3).
Cloning and sequencing of GmABCC5
Custom-designed primers, synthesized based on sequence data of the GmABCC5 (XM_003554257.3) available in NCBI, were used to amplify the 521 bp open reading frame (ORF) from the seeds of Pusa 16, a popular Indian soybean genotype. This partial gene sequence earmarked for cloning will be further utilized for generation of low-phytate trait soybean. The amplicon was cloned in pGEM®-T Easy vector, sequenced and submitted in NCBI as KF183505.1 (Fig. 1a, b). The coding region of GmABCC5 was determined to be governed by 11 exons and 10 introns (Fig. 2), where the first exon encodes the maximum number of sequences and the ninth exon encodes the least as predicted by Gene Structure Display Server 2.0. The partial sequence (521 bp) cloned was found to be the part of the fourth exon in the gene.
Fig. 1.

Cloning of GmABCC5 cDNA in pGEMT easy vector system. a PCR amplification of 521 bp fragment isolated from Glycine max cv. Pusa 16 using gene-specific primers and separated on 1% agarose gel along with 100 bp molecular weight marker, b GmABCC5 sequence submitted in NCBI (KF183505.1)
Fig. 2.

Exon/intron organization of GmABCC5. The GmABCC5 genomic sequence and CDS sequences were downloaded and the sequences were compared in Gene Structure Display Server Program to infer the exon/intron organization. The targeted 521 bp sequence lies in the fourth exon of GmABCC5 gene
Expression pattern of GmABCC5
Analysis of the abundance of the GmABCC5 gene transcripts revealed differential expression pattern across the set of tissue samples displayed in Fig. 3. The maximum expression of GmABCC5 gene was found in mature soybean seeds followed by the stem, leaf, root and flower (Fig. 3), respectively. It was observed that the relative expression in seeds was 52-fold higher in comparison to the flowers (taken as the control). In other tissues like the flower, leaf, root and stem, the relative expression was less than fivefold, given that seeds are the major production site and thus necessitating maximum transporter gene activity in seeds for phytic acid transport and compartmentalization (Raboy 1997). Phytic acid is required for several vital functions in plants and is essential for their survival; hence, a basic level of phytate is observed in all tissues, coinciding with the relative abundance of the gene. In the temporal expression profiles, the maximum relative transcript accumulation was observed in the 8–10 mm seed stage. The maximum relative transcript levels observed were in the 8- to 10-mm-sized seeds almost 52-fold higher in comparison to the 2- to 4-mm-sized seeds which were used as the control (Fig. 4). Overall, the expression of GmABBC5 in seeds was found to be higher during the later developmental stages in comparison to the initial stages as observed in the relative abundance profile, which varied from 46-fold in 10–12 mm, followed by 45- and 43-fold in 14–16 and 12–14 mm, respectively, 19-fold in 6–8 mm, and less than 6-fold in 4–6 mm and 0–2 mm seed sizes. The PEPCo gene was used as the internal control of reference for assessing the relative expression pattern of the gene. The pattern of variation in the transcript abundance indicates well toward the probable major role of GmABCC5 during the later stages of seed development. In soybean after anthesis, the initial stages of seed development are devoted to the production of phosphorus compounds needed for growth and development (nucleic acids, membrane phospholipids and signaling molecules), and the synthesis of phosphorus reserve, phytic acid is minimal (Raboy and Dickinson 1987). The gene was highly expressed in the late stages of seed development as compared to the early stages. This coincides evenly with the biosynthesis of phytic acid during seed development. Temporal phytate accumulation has been studied in various crops such as Glycine max (Raboy and Dickinson 1987; Israel et al. 2011); Brassica napus (Dong et al. 2012); Pisum sativum (Shunmugam et al. 2015) and Hordeum vulgare (Hatzack et al. 2001), which revealed that the flux of phosphorus is mainly channeled toward phytic acid biosynthesis from around 21 days after flowering until seed maturity. In spite of early initiation of phytic acid biosynthesis in developing seeds (0.24 g 100 g−1 in 0–2 mm), its major accumulation occurs during middle (1.85–2.87 g 100 g−1 in 4–6 to 8–10 mm) and later stages (3.35 g 100 g−1 in 12–14 mm) of seed development (Pandey et al. 2016).
Fig. 3.

Real-time spatial expression analysis of GmABCC5 gene in soybean tissues (Pusa 16), PEPCo gene was used as the internal control for normalizing the relative expression. The mean value ± SE was calculated from data pooled from three experimental replicates
Fig. 4.

Real-time temporal expression analysis of GmABCC5 gene in soybean (Pusa 16) developing seeds ranging from initial 0–2 to 14–16 mm (full maturity) seed size. PEPCo gene was used as the internal control for normalizing the relative expression. The mean value ± SE was calculated from data pooled from three experimental replicates
Since the ABC transporter protein has a role in the transport and storage of phytic acid, the gene is expressed actively after a threshold level of phytic acid accumulates in the cytoplasm during seed development. The spatial expression profile identifies seed as the major tissue for its expression and the temporal profile suggests that the maximum relative expression occurs in late seed development stages; thus, the relative gene expression observed in seeds coincides with the accumulation of a large amount of phytic acid during the late development stages (Pandey et al. 2016). Thus, the present study illustrates that the initial intermediates of phytic acid biosynthesis, like myo-inositol and other phosphates, are synthesized earlier in seed developmental process, while the accumulation of the final product, phytic acid, occurs more in the later stages. Targeting the GmABBC5 gene expression during late seed development stages may provide a route for the creation of low-phytate soybean with enhanced nutritional value without affecting the intermediates, imperative for normal plant growth and development.
Physicochemical and structural features
GmABCC5 sequence similarity search using BLASTP analysis revealed homology to protein sequences of Glycine soja (KHN02460.1), Vigna radiata var. radiate (XP_014495750.1) and Phaseolus vulgaris (CBX25010.1) with 99, 93 and 92% of identity, respectively. Table 1 shows the selected 30 GmABCC5 homologs retrieved from NCBI BLASTP using GmABCC5 as the query sequence. ABC transporter gene sequences identified with the role in phytate transport were also included from viz., Zea mays (Shi et al. 2007), Arabidopsis thaliana (Nagy et al. 2009) and Triticum aestivum (Bhati et al. 2016) for better relevance.
Table 1.
ABC transporter protein sequences retrieved from the NCBI database (http://www.ncbi.nlm.nih.gov/)
| S. no. | Gene Id | Plant species | Sequence length (amino acid residue) |
|---|---|---|---|
| 1 | XP_003554305.1 | Glycine max | 1537 |
| 2 | KHN02460.1 | Glycine soja | 1690 |
| 3 | XP_003521316.1 | Glycine max | 1539 |
| 4 | XP_014495750.1 | Vigna radiate var. radiata | 1537 |
| 5 | XP_007162606.1 | Phaseolus vulgaris | 1538 |
| 6 | XP_017411139.1 | Vigna angularis | 1538 |
| 7 | XP_019455530.1 | Lupinus angustifolius | 1533 |
| 8 | XP_012569380.1 | Cicer arietinum | 1532 |
| 9 | XP_003541373.1 | Glycine max | 1517 |
| 10 | XP_014514170.1 | Vigna radiate var. radiata | 1513 |
| 11 | KHN48949.1 | Glycine soja | 1495 |
| 12 | XP_006443721.1 | Citrus clementina | 1536 |
| 13 | XP_019427850.1 | Lupinus angustifolius | 1539 |
| 14 | EOX94300.1 | Theobroma cacao | 1539 |
| 15 | XP_015901176.1 | Ziziphus jujuba | 1531 |
| 16 | XP_003625394.2 | Medicago trunculata | 1514 |
| 17 | EOX94301.1 | Theobroma cacao | 1535 |
| 18 | XP_015948969.1 | Arachis duranensis | 1531 |
| 19 | XP_013468420.1 | Medicago trunculata | 1530 |
| 20 | XP_007144409.1 | Phaseolus vulgaris | 1513 |
| 21 | XP_011024496.1 | Populus euphratica | 1532 |
| 22 | XP_017604352.1 | Gossypium arboreum | 1540 |
| 23 | OAY34676.1 | Manihot esculenta | 1531 |
| 24 | OAY32700.1 | Manihot esculenta | 1531 |
| 25 | XP_017649043.1 | Gossypium arboreum | 1543 |
| 26 | XP_012484085.1 | Gossypium raimondii | 1540 |
| 27 | NP_001106060.1 | Zea mays | 1510 |
| 28 | AIK23242.1 | Triticum aestivum | 1227 |
| 29 | NP_171908.1 | Arabidopsis thaliana | 1514 |
| 30 | XP_002468528.1 | Sorghum bicolor | 1512 |
ExPASy ProtParam and PEPSTATS tools were used for the primary structural analysis of the selected GmABCC5 homologs by computing different parameters and are tabulated in Table 2. The analysis revealed Leu as the most abundant amino acid for all the ABCC5 proteins under study, accounting for about 12% in all the protein’s primary structure, followed by Ser and Val; Cys and His were the least abundant amino acid residues encountered. The predicted average molecular weight (MW) determined was 169,358 g mol−1 (Table 2). The calculated average pI of the ABC transport proteins sequences was 7.47 (range 6.06–8.59), indicating that this enzyme is likely to precipitate in basic buffers although eight proteins have pI less than 7, signifying that in those species protein will supposedly precipitate in acidic buffers (Table 2). This information will be of much use in the development of buffer systems for the purification of recombinant ABC transport proteins by isoelectric focussing. The longer proteins are typically composed of more charged amino acids, thus much better to buffer the effect of fluctuation in their composition and can keep near neutral pI (close to 7.4), whereas extreme pI values are particular for smaller proteins (Schwartz et al. 2001). There was more variation in the pI (range 6.06–8.59) of the ABC transport proteins than their size (range 136,696–187,880 g mol−1, partial sequence from Triticum aestivum), concurrent with reports that the size of orthologous proteins found in directly related organisms is much more conserved than their pI (Nandi et al. 2005). The proteins localized to membrane contain more basic residues which are located generally on either side of the membrane-spanning region, essential for the stability of protein in the membrane (Schwartz et al. 2001). Hence, membrane proteins are basic in nature as observed here. The in vivo half-life (T1/2) of the selected transporters was computed via the instability index (Ii). It has been reported in previous studies that proteins having Ii > 40 have a T1/2 of less than 5 h, while those having Ii < 40 have a longer T1/2 of 16 h (Rogers et al. 1986). Our study showed that 13 proteins were stable, and the other ABC proteins including GmABCC5 were thermally unstable. Although the positional or interaction effects of the adjacent residues were not considered for analyzing the stability, the study provides some indication about the physical state of the protein. GRAVY indices of ABC transporter sequences ranged from 0.094 to 0.274 (Table 2), reflecting the hydrophobicity of the amino acids (Kyte and Doolittle 1982) and grand average hydropathicity. These results thus confirm that all ABC transporters tend to be hydrophobic in nature, which endorses its multifaceted role in cellular membrane transport. Aliphatic index (Ai), another parameter that measures the relative volume occupied by the aliphatic side chains of the amino acids such as Ala, Val, Leu and Ile, serves as a measure of the thermostability of proteins (Gupta et al. 2012). In this study, the Ai values of all the transporters under consideration ranged from 106.62 to 111.24. All the ABC transporter protein sequences analyzed had a high Ai (103.15–111.24) value, an indicator of protein stability over a wide temperature range, thus indicating that all the proteins are thermostable. The extinction coefficient (EC), which indicates the light absorbed by the protein at a wavelength of 280 nm, was found to be in a range of 1.19–1.36 M−1 cm−1 estimated with respect to the concentration of Cys, Trp, and Tyr, was less as Cys and Trp were the least abundant amino acids.
Table 2.
Parameters of inositol phosphate kinases (plants) calculated using the ProtParam program: molecular weight (MW) (g/mol); isoelectric point (pI); extinction coefficient (EC) (M−1 cm−1); instability index (Ii); aliphatic index (Ai); grand average hydropathy (GRAVY); number of negative residues (− R); number of positive residues (+R)
| S. no. | Plant species | MW | PI | EC | II | AI | GRAVY | R − | R + |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Glycine max | 170,554 | 7.0 | 1.28 | 41.57 | 111.24 | 0.274 | 154 | 153 |
| 2 | Glycine soja | 187,880 | 7.61 | 1.27 | 42.32 | 107.62 | 0.186 | 172 | 174 |
| 3 | Glycine max | 170,953 | 6.64 | 1.25 | 40.90 | 110.60 | 0.252 | 158 | 154 |
| 3 | Vigna radiate var. radiata | 170,429 | 6.85 | 1.31 | 40.62 | 107.25 | 0.221 | 156 | 154 |
| 4 | Phaseolus vulgaris | 171,203 | 7.03 | 1.36 | 40.85 | 107.14 | 0.229 | 156 | 155 |
| 5 | Vigna angularis | 170,622 | 7.43 | 1.30 | 40.29 | 106.62 | 0.213 | 155 | 156 |
| 6 | Phaseolus vulgaris | 171,191 | 7.03 | 1.36 | 40.75 | 106.89 | 0.226 | 155 | 156 |
| 7 | Lupinus angustifolius | 170,888 | 7.26 | 1.27 | 40.59 | 107.10 | 0.188 | 155 | 155 |
| 8 | Cicer arietinum | 170,790 | 6.06 | 1.30 | 41.81 | 108.69 | 0.236 | 160 | 147 |
| 9 | Glycine max | 168,332 | 6.42 | 1.26 | 39.00 | 106.49 | 0.216 | 157 | 150 |
| 10 | Vigna radiate var. radiata | 168,698 | 7.47 | 1.32 | 39.57 | 106.50 | 0.201 | 151 | 152 |
| 11 | Glycine soja | 165,946 | 6.48 | 1.27 | 39.09 | 106.23 | 0.202 | 155 | 149 |
| 12 | Citrus clementina | 170,591 | 8.44 | 1.20 | 37.24 | 108.62 | 0.241 | 150 | 161 |
| 13 | Lupinus angustifolius | 171,123 | 7.85 | 1.31 | 40.36 | 108.25 | 0.176 | 151 | 154 |
| 14 | Theobroma cacao | 170,738 | 8.09 | 1.20 | 39.17 | 109.03 | 0.246 | 150 | 155 |
| 15 | Ziziphus jujuba | 170,310 | 8.59 | 1.20 | 41.38 | 107.20 | 0.193 | 150 | 163 |
| 16 | Medicago trunculata | 168,549 | 6.44 | 1.25 | 40.28 | 107.67 | 0.223 | 157 | 151 |
| 17 | Theobroma cacao | 170,234 | 7.97 | 1.20 | 39.32 | 109.06 | 0.249 | 150 | 154 |
| 18 | Arachis duranensis | 169,911 | 7.85 | 1.19 | 36.75 | 107.43 | 0.218 | 151 | 154 |
| 19 | Medicago trunculata | 170,057 | 6.66 | 1.27 | 39.74 | 108.82 | 0.239 | 155 | 151 |
| 20 | Phaseolus vulgaris | 168,932 | 6.71 | 1.31 | 39.26 | 106.95 | 0.213 | 156 | 152 |
| 21 | Populus euphratica | 170,369 | 8.13 | 1.17 | 41.00 | 108.53 | 0.240 | 149 | 155 |
| 22 | Gossypium arboreum | 170,660 | 7.06 | 1.211 | 39.49 | 108.77 | 0.251 | 155 | 154 |
| 23 | Manihot esculenta | 170,411 | 8.44 | 1.21 | 38.87 | 108.21 | 0.224 | 147 | 157 |
| 24 | Manihot esculenta | 170,418 | 8.20 | 1.23 | 40.33 | 109.12 | 0.234 | 147 | 153 |
| 25 | Gossypium arboreum | 171,089 | 7.82 | 1.20 | 39.70 | 107.67 | 0.223 | 154 | 157 |
| 26 | Gossypium raimondii | 170,615 | 7.47 | 1.21 | 39.86 | 109.21 | 0.252 | 153 | 154 |
| 27 | Zea mays | 166,790 | 8.44 | 1.28 | 46.28 | 107.19 | 0.190 | 144 | 155 |
| 28 | Triticum aestivum | 136,696 | 8.18 | 1.19 | 43.47 | 103.15 | 0.094 | 127 | 132 |
| 29 | Arabidopsis thaliana | 168,574 | 7.64 | 1.23 | 40.45 | 108.37 | 0.209 | 159 | 161 |
| 30 | Sorghum bicolor | 166,545 | 8.31 | 1.27 | 44.67 | 107.31 | 0.203 | 146 | 154 |
Domain identification and evolutionary analysis
Multiple alignment of the selected amino acid sequences of the identified GmABCC5 and 30 different homologs was performed using ClustalW server and the sequences were imported to BioEdit software. The significantly conserved amino acids are shown in Fig. 5. Most of the amino acids in the ABC_MRP_domains (accession: cd03250 and cd03244) are well conserved in all the homologs as indicated by low E values (Fig. 6) identified by the CDD program. The ABC_MRP domain is the nucleotide-binding fold/domain (NBD) encompassing highly conserved amino acid motifs which are involved in energy generation and hence power the transport mechanism (Kerr 2002). Two additional ATP-binding motifs were identified within the NBD: the Walker A motif (GXXGXGK(S/T)) and the Walker B motif (hhhhDE, where h is a hydrophobic amino acid) (Fig. 5). The Walker A motif is involved in establishing extensive interactions with the phosphate group of an ATP molecule; the Asp of the Walker B motif coordinates water and Mg2+ at the catalytic site while the catalytic glutamate is vital for ATP hydrolysis (Vetter and Wittinghofer 1999; Zaitseva et al. 2005; Jones and George 2002). A conserved Gln identified at the Q-loop, located between the Walker sequences, performs a similar role in ATP molecule coordination. Two additional motifs, conserved D-loop and H (or switch)-loop, were identified downstream of the Walker B motif containing a conserved Asp and His, respectively, involved in the coordination of the γ-phosphate either through interaction with a water molecule (D-loop) or through direct hydrogen bonding (H-loop) (Zaitseva et al. 2006; Jones and George 2012). An ABC signature motif, a helical subdomain, was identified located in between the Walker sequences of each NBD containing the conserved amino acid sequence LSGGQ, also involved in ATP binding (Schmees et al. 1999). The presence of ABC_membrane superfamily domain (accession: cl00549), representing hydrophobic transmembrane helices, was also identified as conserved by the low E values in all the homologs, which is an essential feature of ABC transport proteins, mandatory for transmembrane transport of molecules (Fig. 6) (Schulz and Kolukisaoglum 2006). It has been reported that NBDs tend to have high sequence similarity, but the transmembrane domains from related transporters vary from having a high sequence identity to probably no similarity, thus making phylogenetic analyses desirable to ascertain the similarity and evolutionary link between the ABC transporter proteins.
Fig. 5.

a, b Schematic representation of multiple sequence alignment of GmABCC5 homologs by ClustalW and image generated by BioEdit program for identification of conserved domains and motifs
Fig. 6.

Domain analysis of GmABCC5 and its homologs as analyzed by BLASTP and CDD program
Sequence conservation across different species is an important indicator of functionality and evolution. (Baum 2008). Phylogenetic analysis provides a useful framework to understand the relationship of different homologs and their evolution from a common ancestor. We constructed a neighbor-joining phylogenetic tree of GmABCC5 and its 30 homologs using (Fig. 7) MEGA 6.0 software. The proteins formed four distinct clades, each comprising evolutionarily related members from the selected homologs and the branching patterns were supported by high bootstrap values. Pattern branching of the phylogenetic tree revealed that the ABC transport proteins of plants belonging to Fabaceae formed a monophyletic cluster with the proteins from citrus, coffee, cotton, etc., indicating close evolutionary relationship and suggesting a common ancestor. The Fabaceae family formed a separate clade with two sub-clades comprising members from soybean, chickpea, beans, etc., and the proteins from citrus, coffee, cotton etc. formed another subgroup in the second clade of phylogeny. Out of the four ABC transport proteins having a well-identified role in phytate transport, the protein from Arabidopsis formed a separate third clade, and the three ABC transporters from the Graminae family formed another well-delineated fourth clade. The phylogram thus illustrated a high degree of divergence among the legumes, cereals, and the horticultural crops despite having high sequence similarity.
Fig. 7.

Cladogram of the selected 30 ABC transport proteins based on their amino acid sequences as outlined in Table 1 using Mega 6.0 software with an outgroup. The tree was inferred based on the neighbor-joining algorithm
Secondary structure prediction
Structural analyses are prized sources of information on shapes and domain structure, protein classification, function prediction and interactions with other macromolecules. The structure provides the first framework to understand the interactions of transporter protein with the ligands/substrates at the molecular level. The secondary structure information in addition aids in the prediction of homology-based protein models. The secondary structure of the GmABCC5 protein is dominated by alpha helix (60%), whereas beta sheets and random coils contribute to 7 and 32%, respectively, as revealed by three-state secondary structure prediction (SS3) by RaptorX server. PSIPRED secondary structure prediction determined a total of 53 alpha helices and 31 beta strands in the protein’s structure. The secondary structures are more conserved than the nucleotide sequences, which help in understanding the molecular evolution (Reehana et al. 2013). Protein sequence analysis by RaptorX predicted disorder in 22% of GmABCC5 residues, further confirmed by FoldIndex server (Wang et al. 2016; Prilusky et al. 2005). The 1537 residues of GmABCC5 revealed 0.326 unfoldability, 0.001 charge and 0.530 phobic values. There were 6 predicted disordered regions having a total of 171 disordered residues. Predicted disordered segments were [290]–[297] length: 8 score: − 0.02 ± 0.03, [299]–[307] length: 9 score: − 0.03 ± 0.02, [505]–[533] length: 29 score: –0.13 ± 0.06, [864]–[879] length: 16 score: − 0.06 ± 0.03, [892]–[956] length: 65 score: − 0.15 ± 0.09 (longest disordered region) and [1373]–[1416] length: 44 score: − 0.13 ± 0.06. Protein intrinsic disorder since long has been linked to molecular recognition, binding activities and regulatory processes in the light of both experimental and computational investigations and hence is functionally important (Cozzetto and Jones 2013; Wright and Dyson 2015).
Disulfide predictions using DiANNA server revealed the presence of 29 Cys residues in GmABCC5 and the most probable bonds as predicted by CYS-REC were 82–1313, 158–896, 163–1480 and 654–905. Solvent access was determined by RaptorX (Wang et al. 2016) revealing residues as exposed, intermediate and buried, 30, 42 and 27%, respectively, based on relative solvent accessibility (RSA), where exposed had 40–100%, intermediate 10–40% and buried 0–10% RSA. HMMTOP, MEMSAT SVM and MEMSAT 3 are transmembrane protein topology prediction programs where MEMSAT SVM is a support vector machines (SVM) based predictor, a highly reliable program which can also identify pore-lining residues. These programs predicted 16 transmembrane helices (Table 3) with the total entropy of the model 17.0221. The transmembrane helices illustrated were rich in hydrophobic amino acids, which is well documented by the Kyte and Dolittle mean hydrophobicity profile, and the final annotated topology of this transmembrane protein was visualized by MEMSAT SVM (Fig. 8a, b) (Nugent and Jones 2009). It also identified three pore-lining helices, featuring residues from 323 to 353, 587 to 610 and 1085 to 1108 with a pore stoichiometry of 1. It has been reported that the transmembrane domain region of the transporter proteins, which forms the transport channel, exclusively consists several membrane-spanning alpha-helices, along with kinks and bends, providing considerable structural variability essential for its function (Dahl et al. 2004; Moussatova et al. 2008). Thus, the presence of transmembrane and hydrophobic residues identified its membrane-embedded nature and role in transmembrane transport which can be further validated by structural and functional characterization.
Table 3.
Total number of transmembrane helices and regions involved in the formation of transmembrane helix as predicted by MEMSAT SVM
| Helix no. | Residues |
|---|---|
| 1 | 37–61 |
| 2 | 98–122 |
| 3 | 141–156 |
| 4 | 168–187 |
| 5 | 205–233 |
| 6 | 323–353 |
| 7 | 370–388 |
| 8 | 445–465 |
| 9 | 469–491 |
| 10 | 549–577 |
| 11 | 587–610 |
| 12 | 963–990 |
| 13 | 1012–1041 |
| 14 | 1085–1108 |
| 15 | 1112–1127 |
| 16 | 1188–1218 |
Fig. 8.

a Schematic diagram for topology analysis prediction of the GmABCC5 query sequence by MEMSAT SVM and MEMSAT3. b Topology analysis cartoon diagram for GmABCC5 by MEMSAT SVM
Structural and functional characterization
Computational protein localization and targeting predictions aid in in silico protein function characterization as well as genome annotation and is therefore a significant study. Signal peptides and transmembrane helices provide useful hints about the protein’s subcellular localization and transmembrane transporter activities. Protein sequence analyses based on TargetP scores (cTP: 0.315, mTP: 0.014, SP: 0.216, other: 0.702) suggested GmABCC5 to be located anywhere in the cell besides mitochondria, with less chance of presence in chloroplast as well. Also, the signal peptide (SP) score reinforced its membranous nature. Results compared to predictions obtained from MEMSAT-SVM server identified a 32-residue long N-terminal signal region positioned at 1–32, indicating that the protein was guided through a secretary pathway to the cellular membrane. ProtComp server identified protein’s location to the plasma membrane and vacuole, and also a probable GPI anchor at the 1501 residue. The GPI anchors are prevalent in membrane proteins and have a confirmed role in providing membrane-attached proteins a stable membrane-anchoring device, and are resistant to most extracellular proteases and lipases (Low 1989). FFPred is an SVM-based program, where a large suite of protein’s physicochemical property predictors cover many features such as signal peptides, membrane helices, secondary structure and disorder terms for prediction of gene ontology (Cozzetto et al. 2016). Gene ontology prediction by FFPred predicted biological function as transmembrane transport and ion transport with 0.940 and 0.907 probabilities, respectively. Molecular function prediction determined anion/substrate-specific transmembrane transporter activity with 0.981 probability and cellular component prediction as integral component of membrane with 1.0 probability. Domain analysis, structural information and GO prediction results, together, strongly recommend GmABCC5 protein’s membrane localization and role in transmembrane transport of substrates, particularly phytate which needs further evidence from 3D structure study.
Homology modeling and model validation
Atomic resolution crystal structures of several soluble proteins have been reported, but similar progress has not been made for the transporter proteins, where the major obstacle encountered is the extreme difficulty in their crystallization owing to conformational flexibility. Hence, the development of homology-based protein model is a relevant alternative (Dahl et al. 2004). Pymol Molecular Graphic System was used to visualize the final 3D structure of GmABCC5 (Fig. 9a), generated through template-based homology model with SWISS-MODEL software. Molecular modeling methods have been routinely used to analyze the structure, dynamics, surface properties and thermodynamics of inorganic, biological and polymeric systems. The 5twv.1.B template [ATP-binding cassette subfamily C member 8, channel conductance-controlling ATPase (cystic fibrosis transmembrane conductance regulator from Homo sapiens)] with a sequence identity score of 35.17% was identified as the top template for homology-based modeling for GmABCC5. In the context of QMEAN global scores, QMEAN-Z score was observed as − 4.96, indicating relatively good model quality (Fig. 9b) and the bio-computed model was further assessed by the ProSA server. The Z score value is a determinant of model quality that predicts the total energy of the structure (Wiederstein and Sippl 2007). As shown in Fig. 9c, the obtained Z score value of GmABCC5 was − 10.47, which is within the range typically found for comparable protein chains of PDB, indicating reliability of the structure. The energy plot (Fig. 9d) shows the local model quality by plotting energies as a function of amino acid sequence position and, in general, positive values correspond to problematic or erroneous parts of a model. The residues with the negative energies further confirmed the consistency of this predicted model (Kulkarni and Devarumath 2014).
Fig. 9.

a Homology model of GmABCC5 protein generated using Swiss-model and rendered using PyMOL. b Plot showing normalized Q mean score of the model. c Z score value of the model generated. d Ramachandran plot validating backbone dihedral angels of the energy-minimized model
The packing environment for residues of the modeled GmABCC5 was compared with the experimental structures by WHAT IF. Poor structural packing is specified by a score of less than or equal to − 5.0 (Singh and Pandey 2015). Analysis reveals that the generated structure has similar packing scores compared to the template X-ray structure with very few residues having poor packing as reflected by their scores being less than − 5.0. Ramachandran plot assessment of GmABCC5 for stereochemical and energetic properties by RAMPAGE server revealed 92% of residues (1188) in the most favored zone, 6.3% of residues (81) in the allowed region and 1.7% of residues (22) in the disallowed/outlier region (Fig. 9d). The statistics in the favored and allowed region and relative low percentage in the outlier suggest that the Ramachandran plot for GmABCC5 is acceptable. The goodness factor (G-factor) from the PROCHECK results showed relevant information between covalent and overall bond angle distances. Analysis of G-factor of the modeled GmABCC5 was − 0.32, which revealed the quality of the predicted model as very good. The overall Ramachandran plot attributes and the G-factor assured the quality of the GmABCC5 structure (Table 4). ERRAT analysis of GmABCC5 showed an overall quality factor of 79.935. The results obtained from various quality evaluation servers certify the quality of the 3D protein model developed in this study. Although GmABCC5 has a well-documented role in ATP-coupled phytate transport, where ATP and phytate are obvious substrates, no substrate/ligand was identified for the model developed and, hence, no binding sites were found. However, another reliable online server RaptorX-binding site prediction determined PO4 as the probable ligand and identified two binding sites governed by the residues: site 1: W655, V684, G685, S686, G687, K688, S689, Q717 (Fig. 10a); and site 2: F175, F178, W193 (Fig. 10b). The first site identified lies in the ABC_MRP domain, the nucleotide-binding fold specific for the coordination and hydrolysis of the ATP molecule (López-Marqués et al. 2015); thus, the probable ligand (PO4) can be speculated to be the part of ATP moiety, and the binding site residues might be involved in energy-coupled transport. As for the second site, no probable domain was identified, but PO4 moiety identified as ligand can either be the component of ATP or phytate salt, which requires further validation. Unraveling the complete mechanistic of phytate discrimination and transport across the membrane requires further studies on complete model, preferably developed through atomic resolution. The 3D structure thus developed will open new avenues and experimental approaches for identification of protein–ligand-/substrate-binding sites and interactions for better insights into the transport mechanism for planning of in vivo experiments.
Table 4.
PROCHECK statistics of the predicted 3D model
| G factors | ||
|---|---|---|
| Parameter | Score | Average score |
| Dihedral angles | ||
| Phi–psi distribution | − 0.12 | − 0.24 |
| Chi1–chi2 distribution | 0.22 | |
| Chi1 only | 0.08 | |
| Chi3 and chi4 | 0.73 | |
| Omega | − 1.04 | |
| Main-chain covalent forces | ||
| Main-chain bond lengths | 0.18 | − 0.26 |
| Main-chain bond angles | − 0.57 | |
| Overall average | − 0.23 | |
Fig. 10.
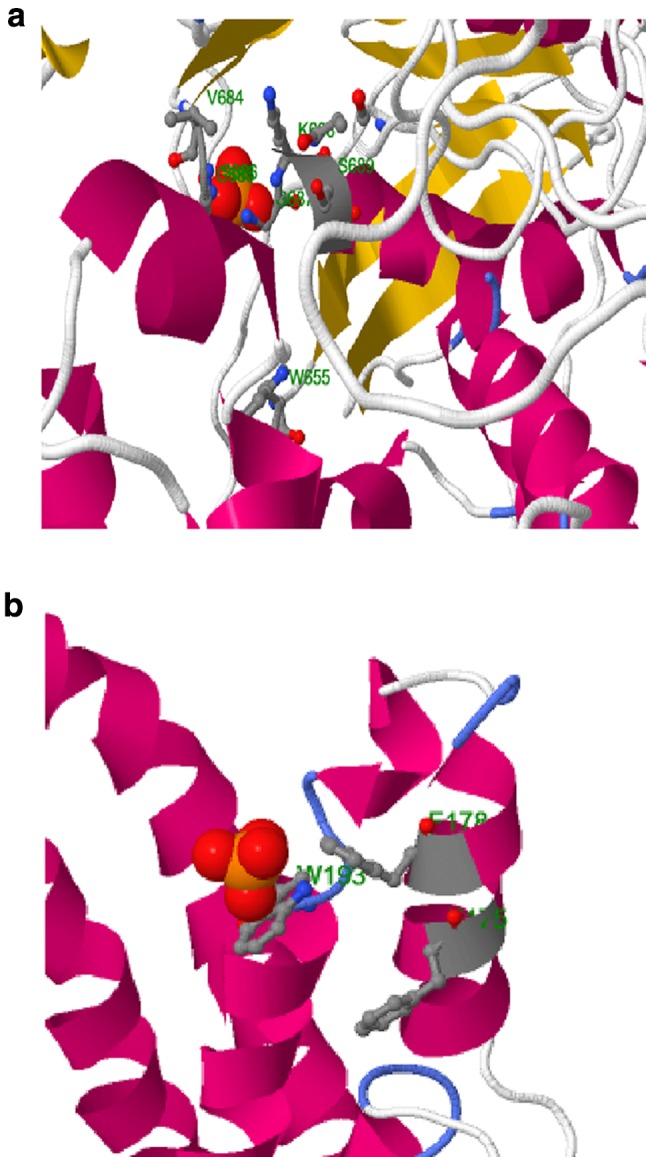
Interactions of PO4 substrate with residues in the active site of GmABCC5 protein a site 1 and b site 2, generated by RaptorX and visualized using JasMol
Conclusion
ATP-binding cassette (ABC) C family 5 transporters in soybean couple ATP hydrolysis to the sequestration and transport of phytate across the cellular membrane in protein storage vacuoles. Here, we have identified and cloned the partial gene sequence of ABCC5 from Glycine max var. Pusa16. The GmABCC5 gene was predominantly expressed in the later phase of seed development, and hence is an appropriate target window for generation of low-phytate soybean without affecting its growth and development. Analyses by computational tools shed light on its secondary structure, physicochemical features, evolutionary linkages, localization and function which can act as a starting point for experimental studies. The three-dimensional protein model, developed through template-based homology modeling, can provide insight into the functional mechanism and molecular structure and can enable the formulation of hypotheses regarding transporter structure and function, which needs further experimental validation. This study would thus be helpful in developing strategies for the development of low-phytate, nutritionally enhanced crops without affecting other plant parameters.
Acknowledgements
Financial support was provided by the Department of Science and Technology, Government of India, in the form of INSPIRE fellowship (IF120064). Funding by the National Fund for Basic, Strategic and Frontier Application Research in Agriculture (RNAi 20-11), Indian Council of Agricultural Research, is duly acknowledged.
Abbreviations
- ATP
Adenosine triphosphate
- ABC
ATP-binding cassette transporter
- ABCC5
ABC transporter subfamily C member 5
- Gm
Glycine max
- PEPCo
PEP carboxylase
- PDB
Protein Data Bank
- NJ
Neighbor joining
- MW
Molecular weight
- pI
Isoelectric pH
- EC
Extinction coefficient
- Ai
Aliphatic index
- Ii
Instability index
- GRAVY
Grand average hydropathy
Compliance with ethical standards
Conflict of interest
The authors declare that there is no conflict of interest regarding the publication of this article.
References
- Baum D. Reading a phylogenetic tree: the meaning of monophyletic groups. Nat Educ. 2008;1(1):190–196. [Google Scholar]
- Bhati KK, Alok A, Kumar A, Kaur J, Tiwari S, Pandey AK. Silencing of ABCC13 transporter in wheat reveals its involvement in grain development, phytic acid accumulation and lateral root formation. J Exp Bot. 2016 doi: 10.1093/jxb/erw224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchan DWA, Minneci F, Nugent TCO, Bryson K, Jones DT. Scalable web services for the PSIPRED Protein Analysis Workbench. Nucleic Acids Res. 2013;41(W1):W340–W348. doi: 10.1093/nar/gkt381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozzetto D, Jones DT. The contribution of intrinsic disorder prediction to the elucidation of protein function. Curr Opin Struct Biol. 2013;23:467–472. doi: 10.1016/j.sbi.2013.02.001. [DOI] [PubMed] [Google Scholar]
- Cozzetto D, Minneci F, Currant H, Jones DT. FFPred 3: feature-based function prediction for all Gene Ontology domains. Sci Rep. 2016;6:31865. doi: 10.1038/srep31865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl SG, Sylte I, Aina Ravna AW. Structures and models of transporter proteins. J Pharmacol Exp Ther. 2004;309(3):853–860. doi: 10.1124/jpet.103.059972. [DOI] [PubMed] [Google Scholar]
- Dong J, Yan W, Bock C, Nokhrina K, Keller W, Georges F. Perturbing the metabolic dynamics of myo-inositol in developing Brassica napus seeds through in vivo methylation impacts its utilization as phytate precursor and affects downstream metabolic pathways. BMC Plant Biol. 2012;13:84. doi: 10.1186/1471-2229-13-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsch JA, Cook A, Young KA, Anderson JM, Bauman AT, Volkmann CJ, Murthy PPN, Raboy V. Seed phosphorus and inositol phosphate phenotype of barley low phytic acid genotypes. Phytochemistry. 2003;62:691–706. doi: 10.1016/S0031-9422(02)00610-6. [DOI] [PubMed] [Google Scholar]
- Emanuelsson O, Nielsen H, Brunakm S, Heijne GV. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J Mol Biol. 2000;300:1005–1016. doi: 10.1006/jmbi.2000.3903. [DOI] [PubMed] [Google Scholar]
- Ferre F, Clote P. DiANNA: a web server for disulfide connectivity prediction. Nucleic Acids Res. 2005;1:230–232. doi: 10.1093/nar/gki412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. Protein identification and analysis tools on the ExPASy Server. In: Walker J, editor. The proteomics protocols handbook. New York: Humana Press; 2005. pp. 571–607. [Google Scholar]
- Greenwood JS, Bewley JD. Subcellular distribution of phytin in the endosperm of developing castor bean: a possibility for its synthesis in the cytoplasm prior to deposition within protein bodies. Planta. 1984;160:113–120. doi: 10.1007/BF00392859. [DOI] [PubMed] [Google Scholar]
- Gupta SK, Rai AK, Kanwar SS, Sharma TR. Comparative analysis of zinc finger proteins involved in plant disease resistance. PLoS One. 2012;7(8):e42578. doi: 10.1371/journal.pone.0042578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Window 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95–98. [Google Scholar]
- Hatzack F, Hübel F, Zhang W, Hansen PE, Rasmussen SK. Inositol phosphates from barley low-phytate grain mutants analysed by metal-dye detection HPLC and NMR. Biochem J. 2001;354:473–480. doi: 10.1042/bj3540473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B, Jin J, Guo A, Zhang H, Luo J, Gao G. GSDS 2.0: an upgraded gene feature visualization server. Bioinformatics. 2015;31(8):1296–1297. doi: 10.1093/bioinformatics/btu817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israel DW, Talierico E, Kwanyuen P, Burton JW, Dean L. Inositol metabolism in developing seeds of low and normal phytic acid soybean lines. Crop Sci. 2011;51:282–289. doi: 10.2135/cropsci2010.03.0123. [DOI] [Google Scholar]
- Jones PM, George AM. Mechanism of ABC transporters: a molecular dynamics simulation of a well characterized nucleotide-binding subunit. Proc Natl Acad Sci USA. 2002;99:12639–12644. doi: 10.1073/pnas.152439599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PM, George AM. Role of the D-loops in allosteric control of ATP hydrolysis in an ABC transporter. J Phys Chem A. 2012;116:3004–3013. doi: 10.1021/jp211139s. [DOI] [PubMed] [Google Scholar]
- Kallberg M, Wang H, Wang S, Peng J, Wang Z, Lu H, Xu J. Template-based protein structure modeling using the RaptorX web server. Nat Protoc. 2012;7(8):1511–1522. doi: 10.1038/nprot.2012.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr ID. Structure and association of ATP-binding cassette transporter nucleotide-binding domains. Biochim Biophys Acta. 2002;1561:47–64. doi: 10.1016/S0304-4157(01)00008-9. [DOI] [PubMed] [Google Scholar]
- Kulkarni PA, Devarumath RM. In silico 3Dstructure prediction of SsMYB2R: a novel MYB transcription factor from Saccharum spontaneum. Indian J Biotech. 2014;13:437–447. [Google Scholar]
- Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157(1):105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using realtime quantitative PCR and the 2∆∆C(T) method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Loewus FA, Murthy PPN. myo-inositol metabolism in plants. Plant Sci. 2000;150:1–19. doi: 10.1016/S0168-9452(99)00150-8. [DOI] [Google Scholar]
- López-Marqués RL, Poulsen LR, Bailly A, Geisler M, Pomorski TG, Palmgren MG. Structure and mechanism of ATP-dependent phospholipid transporters. Biochim Biophys Acta. 2015;1850(3):461–475. doi: 10.1016/j.bbagen.2014.04.008. [DOI] [PubMed] [Google Scholar]
- Lott JNA, Ockenden I, Raboy V, Batten GD. Phytic acid and phosphorus in crop seeds and fruits: a global estimate. Seed Sci Res. 2000;10:11–33. [Google Scholar]
- Lovell SC, Davis IW, Arendall WB, de Bakker PI, Word JM. Structure validation by C alpha geometry: phi, psi and C beta deviation. Proteins. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- Low MG. Glycosyl-phosphatidylinositol: a versatile anchor for cell surface proteins. FASEB J. 1989;3:1600–1608. doi: 10.1096/fasebj.3.5.2522071. [DOI] [PubMed] [Google Scholar]
- Maroof M, Glover N, Biyashev R, Buss G, Grabau E. Genetic basis of the low-phytate trait in the soybean line CX1834. Crop Sci. 2009;49:69–76. doi: 10.2135/cropsci2008.06.0362. [DOI] [Google Scholar]
- Moussatova A, Kandt C, O’Mara ML, Tieleman DP. ATP-binding cassette transporters in Escherichia coli. Biochim Biophys Acta. 2008;1778:1757–1771. doi: 10.1016/j.bbamem.2008.06.009. [DOI] [PubMed] [Google Scholar]
- Nagy R, Grob H, Weder B, Green P, Klein M, Frelet-Barrand A, Schjoerring JK, Brearley C, Martinoia E. The Arabidopsis ATP-binding cassette protein AtMRP5/AtABCC5 is a high affinity inositol hexakisphosphate transporter involved in guard cell signalling and phytate storage. J Biol Chem. 2009;284:33614–33622. doi: 10.1074/jbc.M109.030247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandi SN, Mehra AM, Lynn Bhattacharya A. Comparison of theoretical proteomes: identification of COGs with conserved and variable pI within the multimodal pI distribution. BMC Genom. 2005;6:116. doi: 10.1186/1471-2164-6-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent T, Jones DT. Transmembrane protein topology prediction using support vector machines. BMC Bioinform. 2009;10:159. doi: 10.1186/1471-2105-10-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey V, Krishnan V, Basak N, Hada A, Punjabi M, Jolly M, Lal SK, Singh SB, Sachdev A. Phytic acid dynamics during seed development and it’s composition in yellow and black Indian soybean (Glycine max L.) genotypes through a modified HPLC method. J Plant Biochem Biot. 2016;25(4):367–374. doi: 10.1007/s13562-015-0348-0. [DOI] [Google Scholar]
- Panzeri D, Cassani E, Doria E, et al. A defective ABC transporter of the MRP family, responsible for the bean lpa1 mutation, affects the regulation of the phytic acid pathway, reduces seed myo-inositol and alters ABA sensitivity. New Phytol. 2011;191:70–83. doi: 10.1111/j.1469-8137.2011.03666.x. [DOI] [PubMed] [Google Scholar]
- Prilusky J, Felder CE, Zeev-Ben-Mordehai T, Rydberg E, Man O, Beckmann JS, Silman I, Sussman JL. FoldIndex: a simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics. 2005;21(16):3435–3438. doi: 10.1093/bioinformatics/bti537. [DOI] [PubMed] [Google Scholar]
- Raboy V (1997) Accumulation and storage of phosphate and minerals. In: BA Larkins, Vasil IK (eds) Cellular and molecular biology of plant seed development, pp 441–477
- Raboy V. Seeds for a better future: ‘low phytate’, grains help to overcome malnutrition and reduce pollution. Trends Plant Sci. 2001;6:458–462. doi: 10.1016/S1360-1385(01)02104-5. [DOI] [PubMed] [Google Scholar]
- Raboy V. Progress in breeding low phytate crops. J Nutr. 2002;132:503S–505S. doi: 10.1093/jn/132.3.503S. [DOI] [PubMed] [Google Scholar]
- Raboy V. Myo-Inositol-1,2,3,4,5,6-hexakisphosphate. Phytochemistry. 2003;64:1033–1043. doi: 10.1016/S0031-9422(03)00446-1. [DOI] [PubMed] [Google Scholar]
- Raboy V, Dickinson DB. The timing and rate of phytic acid accumulation in developing soybean seeds. Plant Physiol. 1987;85:841–844. doi: 10.1104/pp.85.3.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SR, Ingvardsen CR, Torp AM. Mutations in genes controlling the biosynthesis and accumulation of inositol phosphates in seeds. Biochem Soc Trans. 2010;38:689–694. doi: 10.1042/BST0380689. [DOI] [PubMed] [Google Scholar]
- Reehana N, Ahamed AP, Ali DM, Suresh A, Kumar RA, Thajuddin N. Structure based computational analysis and molecular phylogeny of C-Phycocyanin gene from the selected cynobacteria. Int J Biol Vet Agric Food Eng. 2013;7:47–51. [Google Scholar]
- Regvar M, Eichert D, Kaulich B, Gianoncelli A, Pongrac P, Vogel-Mikus K, Kreft I. New insights into globoids of protein storage vacuoles in wheat aleurone using synchrotron soft X-ray microscopy. J Exp Bot. 2011;62:3929–3939. doi: 10.1093/jxb/err090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice P, Longden I, Bleasby A. Emboss: the European molecular biology open software suite. Trends Genet. 2000;16(6):276–277. doi: 10.1016/S0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- Rogers S, Wells R, Rechsteiner M (1986) Amino acid sequences common to rapidly degraded proteins: the PEST hypothesis. Science 234(4774):364-368 [DOI] [PubMed]
- Schmees G, Stein A, Hunke S, Landmesser H, Schneider E. Functional consequences of mutations in the conserved ‘signature sequence’ of the ATP-binding cassette protein MalK. Eur J Biochem. 1999;266:420–430. doi: 10.1046/j.1432-1327.1999.00871.x. [DOI] [PubMed] [Google Scholar]
- Schulz B, Kolukisaoglum HU. Genomics of plant ABC transporters: the alphabet of photosynthetic life forms or just holes in membranes? FEBS Lett. 2006;580(4):1010–1016. doi: 10.1016/j.febslet.2006.01.002. [DOI] [PubMed] [Google Scholar]
- Schwartz R, Ting CS, King J. Whole proteome pI values correlate with subcellular localizations of proteins for organisms within the three domains of life. Genome Res. 2001;11(5):703–709. doi: 10.1101/gr.GR-1587R. [DOI] [PubMed] [Google Scholar]
- Shi J, Wang H, Schellin K, Li B, Faller M, Stoop JM, Meeley RB, Ert DS, Ranch JP, Glassman K. Embryo-specific silencing of a transporter reduces phytic acid content of maize and soybean seeds. Nat Biotechnol. 2007;25:930–937. doi: 10.1038/nbt1322. [DOI] [PubMed] [Google Scholar]
- Shunmugam ASK, Bock C, Arganosam GC, Georges F, Gray GR, Warkentin TD. Accumulation of phosphorus-containing compounds in developing seeds of low-phytate pea (Pisum sativum L.) Mutants Plants. 2015;4:1–26. doi: 10.3390/plants4010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Pandey PN. Molecular docking and molecular dynamics study on SmHDAC1 to identify potential lead compounds against Schistosomiasis. Mol Biol Rep. 2015;42:689–698. doi: 10.1007/s11033-014-3816-z. [DOI] [PubMed] [Google Scholar]
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol Biol Evol. 2013;30(12):2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetter IR, Wittinghofer A. Nucleoside triphosphate-binding proteins: different scaffolds to achieve phosphoryl transfer. Q Rev Biophys. 1999;32:1–56. doi: 10.1017/S0033583599003480. [DOI] [PubMed] [Google Scholar]
- Wang S, Li W, Liu S, Xu J. RaptorX-property: a web server for protein structure property prediction. Nucleic Acids Res. 2016;44:W430–W435. doi: 10.1093/nar/gkw306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35:W407–W410. doi: 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright PE, Dyson HJ. Intrinsically disordered proteins in cellular signalling and regulation. Nat Rev Mol Cell Biol. 2015;16:18–29. doi: 10.1038/nrm3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu XH, Zhao HJ, Liu QL, Frank T, Engel KH, An G, Shu QY. Mutations of the multi-drug resistance-associated protein ABC transporter gene 5 result in reduction of phytic acid in rice seeds. Theor Appl Genet. 2009;119:75–83. doi: 10.1007/s00122-009-1018-1. [DOI] [PubMed] [Google Scholar]
- Zaitseva J, Jenewein S, Jumpertz T, Holland IB, Schmitt L. H662 is the linchpin of ATP hydrolysis in the nucleotide-binding domain of the ABC transporter HlyB. EMBO J. 2005;24:1901–1910. doi: 10.1038/sj.emboj.7600657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaitseva J, Oswald C, Jumpertz T, Jenewein S, Wiedenmann A, Holland IB, Schmitt L. A structural analysis of asymmetry required for catalytic activity of an ABC-ATPase domain dimer. EMBO J. 2006;25:3432–3443. doi: 10.1038/sj.emboj.7601208. [DOI] [PMC free article] [PubMed] [Google Scholar]