

Abstract
This report describes cultivation-dependent diversity, phylogeny and enzymatic potential of the haloalkaliphilic bacteria isolated from the unvegetated desert soil of yet unexplored, saline desert of Little Rann of Kutch (LRK), India. The LRK is a unique ecosystem displaying a combination of Dry Rann and Wet Rann. A total of 25 bacteria were isolated and characterized on the basis of colony morphology, biochemical profile, sugar utilization, secretion of the extracellular enzymes and antibiotic sensitivity. Further, the identification and phylogenetic relatedness of 23 bacteria were established by the analysis of 16S rRNA gene sequences. The phylogenetic analysis indicated that the isolates belong to the phylum Firmicutes, comprising low G + C, Gram-positive bacteria, with different genera: Bacillus (~ 39%), Staphylococcus (~ 30%), Halobacillus (~ 13%), Virgibacillus (~ 13%), Oceanobacillus (~ 4%). Majority of the bacterial isolates produced proteases (30% isolates) followed by cellulases (24% isolates), CMCases (24% isolates) and amylases (20% isolates). Halobacillus, Virgibacillus and Bacillus predominantly produced hydrolases, while many produced multiple enzymes at high salinity and alkaline pH. Highest antibiotic resistance was observed against Ampicillin and Penicillin (32%) followed by Cefaclor (20%); Colistin, Cefoperazone and Cefotaxime (16%); Cefuroxime (12%); Gentamycin and Cefixime (8%); Erythromycin, Cefadroxil, Azithromycin, Co-trimoxazole, Amoxycillin, Norfloxacin, Cefpodoxime, Amikacin and Augmentin (4%). KJ1-10-99 and KJ1-10-93 representing < 97% of 16S rRNA gene sequence similarity belong to a novel lineage within the family Bacillaceae. Comparison of the phenogram and phylogram revealed the contradiction of the phenogram pattern and the phylogenetic placement of the isolates. The isolates belonging to same species have shown considerable phenotypic variation. The study on the cultivable haloalkaliphilic bacteria of an unexplored enigmatic niche reflects ecological and biotechnological significance.
Electronic supplementary material
The online version of this article (10.1007/s13205-017-1075-0) contains supplementary material, which is available to authorized users.
Keywords: Haloalkaliphilic bacteria, Extracellular enzymes, Phenogram, Coastal saline desert, Bacterial diversity, 16S rRNA gene sequencing, Little Rann of Kutch, Novel lineage
Introduction
Arid regions comprise nearly 30% of the Earth’s terrestrial surface. Deserts represent multiple extremities of temperature, UV radiation, nutrient and water scarcity, salinity and alkalinity making the environment unfavorable for microorganisms. Till now, majority of the halophiles and haloalkaliphiles have been studied from the Soda lakes, Dead Sea, solar salterns and sea water (Duckworth et al. 1996; Arahal et al. 1996; Demergasso et al. 2004; Foti et al. 2008; Kim et al. 2013). However, the microbial diversity and biotechnological potential of the desert ecosystem remains largely unattended.
In this study, we focused on the bacterial diversity of the unexplored and unusual habitat of the Little Rann of Kutch, a salt marsh with high salinity (Gupta and Ansari 2014) of large area of 4953.7 km2 (Ishnava et al. 2011). Major region of the desert consist of 60% clay unlike of other deserts (Gupta and Ansari 2014). It is also distinct as the north head of the Gulf of the Kutch adjoins this desert. Thus, there is a regular flow of water in the Rann during the tides or through the water drifted by the south–west winds making it a saline coastal desert. Rainfall is fairly low, thus the water recedes and evaporates, leaving a crust of the halite and gypsum crystals which grow in the clay and sands. Understanding the microbial diversity of this unique habitat would help analyze the biogeochemical cycles. It would further explore the biotechnological opportunities.
Microorganisms, their distribution and identity in ecosystems play important role (Falkowski et al. 2008; Langenheder et al. 2010). The microorganisms are the key players in biomineralization, weathering of the rocks and biodegradation of various pollutants. Microbial activity is vital in the biogeochemistry of many ecosystems (Madsen 2005) and microbial diversity under extreme conditions remains a fundamental issue in microbial ecology (Torsvik and Øvreas 2002). Therefore, it is important to study the bacterial diversity and its function in desert ecosystem.
The cultivable bacteria are highly significant from ecological prospective. It is well known that not only large animals and plants but microorganisms, regardless of their size, extensively contribute to the ecosystem (Horner-Devine et al. 2003). Microbial communities have been analyzed in different desert habitats, such as Atacama Desert, Chile (Rasuk et al. 2016), Monegros Desert, Spain (Casamayor et al. 2013), Negev desert, Israel (Saul-Tcherkas et al. 2013), Gobi desert, Mongolia and Taklamakan desert, China (An et al. 2013), Sahara desert, Africa (Essoussi et al. 2012), Sonoran desert, United States of America (Andrew et al. 2012), Namib desert, Southern Africa (Scola et al. 2018) and Thar desert, India (Rao et al. 2016). Much of these studies have focused on the diversity of the microbial community of the vegetated soil of the desert crust (Raeid et al. 2010; Li et al. 2013), endolithic communities of translucent stones and gypsum deposits (Dong et al. 2007) and Shrubs (Saul-Tcherkas et al. 2013).
The microflora of the saline ecosystems has attracted a great deal of attention during the last couple of decades. These microbes possess adaptation mechanisms to thrive under the dual extremities of high salinity and alkaline pH (Margesin and Schinner 2001). From a biotechnological stand point, enzymes withstanding multitude of the extremity from the haloalkaliphilic bacteria can be quite useful in various applications (Horikoshi 1999; Joshi et al. 2009; Purohit and singh 2011; Pandey et al. 2012; Raval et al. 2014).
Little Rann of Kutch nominated as a “Biosphere Reserve” is characterized as terrestrial and coastal ecosystems (UNESCO’s Man and Biosphere (MAB) program, http://whc.unesco.org/en/tentativelists/2105/). Despite being a unique enigmatic terrain of ecological significance, only limited studies are carried out on its bacterial diversity. In the present study, we focus on the bacterial diversity of un-vegetated, desert soils to determine the phylogenetic and metabolic diversity. The phenotypic traits and 16S rRNA gene sequence based phylogenetic analyses were considered for the diversity analysis under the umbrella of the Bi-Phasic approach.
Materials and methods
Site description and sample collection
Little Rann of Kutch (LRK), roughly a triangular in shape, is located between 22°55″–24°35″N and 70°30″–71°45″E near the Great Rann of Kutch, Gujarat, covering an area of 4953.7 km2. It has an average annual rainfall of less than 400 mm (Ishnava et al. 2011) with a mean annual temperature of 26 °C, ranging 10–30 °C. In summer, the temperature reaches to 48 °C. LRK represents, both a dry and wet land depending the period of the year, representing a wide variation in soil composition and salt deposition. Soil sample, designated as KJ1, was collected from the LRK desert near Jogad (23°08″, 33.412N, 71°12″, 35.503E) at a depth of 10–15 cm, avoiding rhizosphere of any plant. The samples collected into sterile polythene bags were transported to laboratory and stored at 4 °C until further analysis.
Soil analysis
Physicochemical properties of the soil sample were analyzed. The soil pH and salinity were measured in a 1:5 (wt/wt) aqueous solution. Conductivity was measured in dS/m. pH and conductivity was measured by digital conductivity meter (CON700, Eutech instruments, Singapore). Cations (Na+, Mg2+, Ca2+ and K+) and anions (Cl− and SO42−) concentrations of the soil samples were earlier reported by Thomas et al. (2012). WHC (water holding capacity), FC (field capacity), OC (organic carbon) and N (nitrogen) of the soil of the Little Rann of Kutch were based on earlier report (Pilania and Panchal 2016).
Enrichment and isolation
A complex medium broth (CMB) was used for enrichment and isolation of haloalkaliphilic bacteria. CMB contained (g/l) glucose, 10; peptone, 5; yeast extract, 5; KH2PO4, 5 and with two concentrations of NaCl 5% (medium designated as CMB-5) and 10%, wt/vol (medium designated as CMB-10) at pH 9.0. The pH of the medium was checked after autoclaving, and, if required, adjusted to pH 9 with a sterile solution of Na2CO3 (20%, wt/vol). To initiate enrichment cultures, 2.0 g of the soil sample was inoculated into 100 ml CMB and the medium incubated at 37 °C in a rotator shaker at 180 rpm. After 7 days incubation the CMB enrichments in the 5 and 10% NaCl were serially diluted to 10−7 and 0.1 ml of an appropriately diluted enrichment was spread on to CMB medium that had been amended with agar (3% w/v) and incubated at 37 °C. After 72 h incubation, colonies with distinctly different morphologies and/or pigmentation were picked and pure cultures obtained by several rounds of streaking on CMB agar plates containing the respective NaCl concentrations before being regarded as pure. Cultures were designated using the following code: soil sample number-the sodium chloride concentration-pH with a unique number. 15 isolates were isolated from CMB-5 agar medium and 10 from CMB-10 agar medium.
Morphological, physiological and biochemical characterization of the bacterial isolates
The bacterial cultures were streaked on the CMB agar plates of the same conditions of NaCl and pH as enrichment. Gram reaction, cell morphology, cell size and cell arrangement were monitored. The isolates were further differentiated for their biochemical and metabolic activities. The biochemical tests included the production of catalase, oxidase, H2S, indole, hydrolysis of urea, reduction of nitrate and fermentation of the sugars such as; fructose, sucrose, maltose, lactose, galactose, raffinose, arabinose and xylose. All the test tubes were incubated at 37 °C and the results were recorded at 24, 48, and 72 h, unless stated otherwise. The biochemical media and their test reagents were prepared as mentioned by Cappuccino and Sherman (2004). Due to the haloalkaliphilic nature of the organisms, all the biochemical media were supplemented with respective NaCl concentrations and pH was adjusted to 9. The individual isolates were inoculated into the respective biochemical medium and incubated at 37 °C for 24–48 h and results were subsequently observed.
Secretion of the extracellular enzymes
Actively growing cultures in the complex medium broth (CMB) with varying concentrations of NaCl (5 and 10%, w/v) at constant pH 9 were used as mother culture for the qualitative assay of the alkaline protease (Raval et al. 2014), amylase (Amoozegar et al. 2003), CMCase and cellulase (Kasana et al. 2008). The pH of the medium was adjusted to 9, by adding separately autoclaved 20% Na2CO3 (w/v).
Antibiotic sensitivity profile
For the detection of the antibiotic sensitivity/resistance profile of the isolates, Kirby–Bauer test was performed using Dodeca discs (Hi-Media make). All the isolates were tested against 3 different universal antibiotic discs on the CMB agar plates (5 and 10%, w/v, pH 9). One ml of 24–48 h activated cultures was spread over the CMB agar plate surface, followed by placing an antibiotic impregnated disc on the agar surface. The antibiotic sensitivity was detected by measuring the zone of the inhibition around the individual antibiotic disc.
Cluster analysis and generation of phenetic dendrogram
Phenotypic characters such as biochemical tests, sugar utilization and antibiotic resistance profile were used to construct phenogram (Supplementary Table S3). For cluster analysis, the data were converted into a binary matrix, where the digit 1 represents the presence of a phenotypic character, while the digit 0 its absence. The Jaccard similarity measure was used to build a tree with the unweighted pair group mean averages (UPGMA) algorithm. Analysis of the phenotypic data was performed using the software PAST (Hammer et al. 2001).
16S rRNA gene sequencing and phylogenetic analysis
16S rRNA gene was amplified using forward and reverse primer pairs—27F (5′AGAGTTTGATCMTGGCTCAG 3′) and 1492R (5′ TACGGYTACCTTGTTACGACTT 3′), respectively. 16S rRNA gene sequencing was carried out using an automated DNA sequencer ABI 3100 (Applied Biosystems) with BigDye Terminator Kit v. 3.1 (Applied Biosystems). Gene sequences of the bacterial reference strains, type strains and closest phylogenetic relatives were selected from GenBank by subjecting the nucleotide sequences of bacterial isolates to similarity searches using BLASTn (http://www.ncbi.nlm.nih.gov/blast). Multiple sequence alignment was achieved with Clustal W of MEGA 6. The phylogenetic tree was constructed using the maximum likelihood method of MEGA 6 (Tamura et al. 2011). Distances were calculated using the Kimura correction in a pairwise deletion manner (Kimura 1980). Phylogenetic tree was constructed displaying the relatedness between all 23 isolates with their phylogenetic relatives including type species chosen from the blast analysis and EZtaxon analysis (Fig. 1). The 16S rRNA gene sequences of 23 isolates are deposited in the NCBI GenBank (Accession No. KC989937–KC989945 and KJ623591–KJ623604).
Fig. 1.
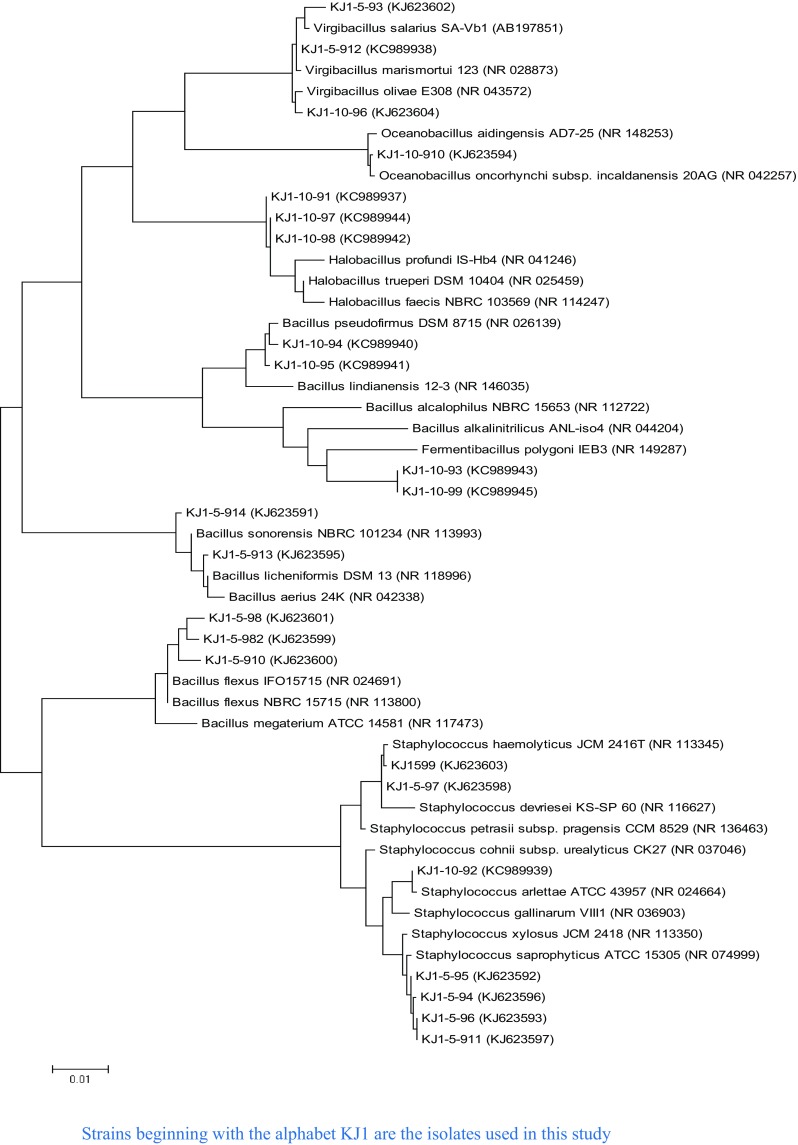
Phylogenetic tree based on the 16S ribosomal RNA gene sequences of the isolates of Jogad, Little Rann of Kutch and some of their closest phylogenetic relatives. The tree was reconstructed by the maximum likelihood method using MEGA 6 software. GenBank accession numbers are shown in parentheses. Bar, 1 nt substitution per 100 nt
Phylogenetic analysis and the identification of the novel lineage
Due to very low sequence similarity of KJ1-10-99 and KJ1-10-93 with their phylogenetic relatives, 16S rRNA gene sequences of these isolates were analyzed using EzTaxon server (Kim et al. 2012). EzTaxon based phylogenetic tree suggested novel lineage. The CLUSTAL W algorithm of MEGA 6 was used for sequence alignments and MEGA 6 (Tamura et al. 2011) software used for phylogenetic analysis of the individual sequences. Distances were calculated using Kimura correction in a pairwise deletion manner (Kimura 1980). Neighbor-joining (NJ) method in the MEGA 6 software was used to construct phylogenetic tree. Percentage support values were obtained using a bootstrap procedure based on 1000 replications.
Results
Soil analysis
The collected soil sample was dry, brown in color and rich in sand particles. The pH, electrical conductivity and salinity were 8.7, 5.7 ds/cm and 3.1 parts per thousand, respectively. According to Thomas et al. (2012) Na+, K+ and Mg2+ are 65, 2.8 and 15.8 g/l, while Cl− and SO42− are 119 and 32 g/l, respectively. Deposition of calcium (Ca2+) could not be detected. WHC (water holding capacity), FC (field capacity), OC (organic carbon) and N (nitrogen) were 29.683, 24.065, 0.726 and 0.062%, respectively, as reported earlier (Pilania and Panchal 2016).
Enrichment and isolation
A total of 25 haloalkaliphilic bacteria were isolated from the desert soil using two different enrichment conditions of salt in CMB media i.e., 5 and 10% (w/v), at constant pH 9. While 15 bacteria were obtained with the enrichment at 5% NaCl (w/v), 10 were from the enrichment at 10% NaCl (w/v). Thus, with increasing salt concentrations, the number of bacterial isolates decreased.
Phenotypic characterization of the bacterial isolates
The haloalkaliphilic bacteria were primarily diversified on the basis of their colony characteristics. Majority of them shared common features of round shape, raised elevation and opaqueness of the colonies. Yellow and cream-white pigmented colonies were observed with majority of the isolates, while some undergo a series of color change with time. In few cases, initially the colonies were off-white, later on changed to yellow followed by orange color after 48 h. KJ1-5-913 and KJ1-5-914 produced extracellular polymeric substances, while one of the isolates had raised flower shape colony filled with gummy liquid. Although all the isolates were Gram positive; KJ1-10-91, KJ1-10-93, KJ1-10-94, KJ1-10-96, KJ1-10-97 and KJ1-10-99 belonging to Halobacillus, Virgibacillus and Bacillus, consistently stained as gram negative and contradicted their phylogenetic placement as gram positive. The rod shape bacteria ranged from 2.11 to 4.86 μm in length and 0.63 to 1.44 μm in width. While 65% of the isolates were rod shape, the others represented spherical shapes and few adapted “V”, “Y” and “T” shape of the cell arrangement (Supplementary Table S2).
All isolates were oxidase positive and majority displayed catalase positive test followed by the H2S production (60% isolates), Nitrate reduction (36% isolates) and Indole production (8% isolates). None of the isolates hydrolyzed urea (Supplementary Table S3). The patterns of the sugar utilization varied among the isolates, and none produced gas in the Durham’s tube. Galactose, xylose, sucrose, lactose and raffinose were most preferred carbon sources, while maltose and fructose were least favored. About 75% of the isolates utilized galactose, a most favored carbon source (Supplementary Table S3).
Bacteria were screened for the extracellular protease, amylase, cellulase and CMCase. Protease production was observed in 30% of the isolates followed by cellulase (24%), CMCase (24%) and amylases (20%). Many isolates produced more than one of these enzymes. For instance, two hydrolytic activities were represented by ~ 35% of the isolates, while 4% produced three and four hydrolytic enzymes. Two enzymes were produced by Bacillus pseudofirmus KJ1-10-94, Halobacillus trueperi (KJ1-10-91 and KJ1-10-97), Bacillus flexus (KJ1-10-98 and KJ1-10-982), Bacillus licheniformis KJ1-5-913 and Bacillus sp. KJ1-5-914. On the other hand, three hydrolytic enzymes produced by Staphylococcus saprophyticus KJ1-5-911 and four by Virgibacillus salarius KJ1-5-912 (Table 1). It appears that the isolates obtained by the enrichment at lower salt concentration were potent cellulase, CMCase and amylase producers, while those enriched at the higher salt concentration were protease producers. The enzyme production was highly pH and salt dependent. Nearly half of the isolates did not secrete any enzymes.
Table 1.
Extracellular enzyme secretion profile of the isolates
| Isolates | Hydrolytic activities | Total enzyme activities | |||
|---|---|---|---|---|---|
| Protease | Amylase | Cellulase | CMCase | ||
| KJ1-5-93 | + | − | − | − | 1 |
| KJ1-5-98 | − | + | + | − | 2 |
| KJ1-5-982 | − | + | + | − | 2 |
| KJ1-5-911 | + | − | + | + | 3 |
| KJ1-5-912 | + | + | + | + | 4 |
| KJ1-5-913 | − | − | + | + | 2 |
| KJ1-5-914 | − | − | + | + | 2 |
| KJ1-10-91 | + | − | − | + | 2 |
| KJ1-10-94 | + | + | − | − | 2 |
| KJ1-10-96 | + | − | − | − | 1 |
| KJ1-10-97 | + | + | − | − | 2 |
| KJ1-10-99 | − | − | − | + | 1 |
No protease, amylase, cellulase or CMCase activities were detected in isolates; KJ1-5-94, KJ1-5-95, KJ1-5-96, KJ1-5-97, KJ1-5-99, KJ1-5-910, KJ1-10-92, KJ1-10-93, KJ1-10-95, KJ1-10-98 and KJ1-10-910
The antibiotic sensitivity profile varied among the isolates. The isolates were highly sensitive against Co-trimoxazole (except KJ1-10-94 and KJ1-10-95), Rifampicin, Clindamycin, Chloramphenicol, Norfloxacin (except KJ1-5-94, KJ1-5-97, KJ1-5-99, KJ1-10-95, KJ1-10-94), Ciprofloxacin (except KJ1-5-97, KJ1-5-99, KJ1-10-94 and KJ1-10-95) and Ceftriaxone (Supplementary Table S3). The antibiotics; Colistin, Cefaclor, Ampicillin, Penicillin and Cefadroxil were comparatively less effective. It appears that the isolates obtained by the enrichment at high salt were in general more sensitive against the antibiotics as compared to those enriched at low salt enrichment conditions. However, KJ1-10-92 was an exception, as it resisted a range of the antibiotics. Most of the isolates exhibited maximum resistance against Ampicillin and Penicillin (32%) followed by Cefaclor (20%); Colistin, Cefoperazone and Cefotaxime (16%); Cefuroxime (12%); Gentamycin and Cefixime (8%); Erythromycin, Cefadroxil, Azithromycin, Co-trimoxazole, Amoxycillin, Norfloxacin, Cefpodoxime, Amikacin and Augmentin (4%) (Supplementary Figure S1). On the other hand, the strains KJ1-5-97, KJ1-5-913 and KJ1-5-914 exhibited resistance against maximum number of the antibiotics (8), followed by KJ1-10-92 (7); KJ1-5-99 (5); KJ1-5-94 (3); KJ1-5-98, KJ1-5-910 and KJ1-5-982 (2); KJ1-5-94, KJ1-5-912, KJ1-5-95, KJ1-10-96 (1) (Supplementary Figure S2).
Phylogenetic analysis
The isolates were identified and analyzed on the basis of 16S rRNA gene sequence homology. The sequences analyzed to find the closest phylogenetic relatives. The isolates were classified in the kingdom bacteria and phylum Firmicutes, which included Low G + C Gram-positive bacteria related to the families of Bacillaceae and Staphylococcaceae. The isolates were distributed among the five different genera with an uneven distribution: Bacillus, Halobacillus, Oceanobacillus, Virgibacillus and Staphylococcus. The relatively abundant genera were Bacillus (9 isolates: 39%), Staphylococcus (7 isolates: 30%), Halobacillus (3 isolates: 13%), Virgibacillus (3 isolates: 13%), Oceanobacillus (1 isolate: 4%).
The BLAST analysis suggested homology among the isolates from 97 to 100% with the reference strains of the NCBI database. Among all, Bacillus genus was most abundant, representing 39% of the isolates exhibiting 97–99% identity with the reference strains in NCBI database. Strains of this genus were allocated into different species; B. licheniformis, Bacillus sp., B. pseudofirmus and B. flexus. The second most abundant genus was Staphylococcus with 3 different species; Staphylococcus haemolyticus, S. saprophyticus and Staphylococcus arlettae.
Interestingly, KJ1-5-912 has shown equally high sequence similarity and placed close to both, V. salarius and Virgibacillus marismortui in the phylogenetic tree (Fig. 1). A similar pattern was reflected by KJ1-5-95, which has shown 99% similarity with S. saprophyticus, Staphylococcus xylosus and Staphylococcus succinus subsp. succinus. These two phylotypes could not be assigned to any particular species, as their 16S rRNA gene sequences were identical to two different species. Either they belong to one of these species or may represent a new species. Moreover, isolates KJ1-10-91, KJ1-10-97 and KJ1-10-98 formed separate clade from their closest phylogenetic neighbours (Fig. 1). These isolates might represent novel species. However, polyphasic characterization is required to validate their taxonomic identity.
KJ1-10-93 and KJ1-10-99 have shown low 16S rRNA gene sequence similarity with the neighboring taxa Bacillus (97.1% similarity) and Anaerobacillus (96.3% similarity) and thus could not be assigned to an existing genus. They either belong to one of these genera or alternatively may represent new genera.
Phylogenetic analysis and identification of novel lineage
To analyze KJ1-10-99 and KJ1-10-93 as a novel lineage, their16S rRNA gene sequences were further analyzed by EzTaxon program. Both strains display highest sequence similarity with Fermentibacillus polygoni IEB3T (96.90%) followed by B. alkalinitrilicus ANL-iso4T (96.3%), B. nanhaiisediminis NH3T (96.3%) and Anaerobacillus arseniciselenatis DSM 15340T. Therefore, phylogenetic tree was prepared using 16S rRNA gene sequences of both strains and neighbouring type species, retrieved from NCBI. The phylogenetic analysis of the 16S rRNA gene sequences suggested that the two novel isolates clustered in a separate clade (Fig. 3), indicating that these two isolates might represent a novel species in a new genus. Later, polyphasic taxonomic analysis of these isolates confirmed their status as a novel genus in Bacillaceae family (Bhatt et al. 2017).
Fig. 3.

Phylogenetic tree showing the relationships among strains KJ1-10-99T and KJ1-10-93 and closely related species of genera within the family Bacillaceae. The tree was reconstructed by the neighbour-joining method using MEGA 6 software and rooted using Paenibacillus polymyxa DSM 36T as the out group. Numbers at nodes represent bootstrap values (based on 1000 resamplings); GenBank accession numbers are shown in parentheses. Bar, 1 nt substitution per 100 nt
Numerical taxonomy and cluster analysis of the phenotypic characteristics
A set of 44 tests was used to construct a phenotype profile. The number of positive tests displayed by the isolates ranged from 1 to 19. The phenotypic profiles were compared amongst the organisms and a similarity dendrogram was constructed (Fig. 2). The similarity comparison was based on the number of the positive tests and the tests which were positive between the organisms.
Fig. 2.

Phenogram of the 23 isolates generated from 44 phenetic characteristics using Jaccard similarity coefficient and UPGMA algorithm. A value of 60% similarity was used to assign isolates to phenons (A, B, C, D and E)
The phenotype profile of each isolate was compared against the other 22 isolates. Overall, all the isolates displayed phenotype profiles that were approximately 30% similar. On the basis of 44 phenotypic characteristics, 2 major clusters (X and Y) at ~ 30% similarity level and 5 sub-clusters (A, B, C, D and E) at 60% similarity level were generated (Fig. 2). Seven isolates had very low phenotypic similarity and did not cluster with other isolates. In the cluster pattern, the significant effect of the salt was quite apparent. Clusters D and E consisted of the isolates obtained at 5% NaCl, while Cluster A included the strains obtained at 10% NaCl. Conversely, the Clusters B and C included those strains which were isolated at both, 5 and 10% NaCl enrichment.
The term ‘Phenon’ is used to describe the clusters in phenogram. The members of all the phenons produced catalase and oxidase. As shown in Fig. 3, the members of the Phenon E (on average 83.33%) displayed maximum positive biochemical tests, while Phenon D (on average 33.33%) exhibited least. Phenon C isolates exhibited maximum sugar utilization (on average 75%), followed by Phenon E (on average 50%), Phenon B (on average 46.87%), Phenon D (on average 33.32%) and Phenon A (on average 18.75%). Antibiotic resistance was highest in phenon E (on average 26.66%) followed by Phenon D (on average 6.66%), Phenon B (on average 1.66%), Phenon C (on average 0.66%) and Phenon A (on average 0%).
Comparison of phenogram and phylogram
Overall, the patterns of phenotypic diversity corresponded with the phylogenetic diversity. Phenons A, D and E, corresponded the trends reflected in the phylogram (Figs. 1, 2). Phenon A comprised of KJ1-10-91 and KJ1-10-98, phylogenetically identified as H. trueperi, clustered together and displayed high phenotypic similarity of 60%. Similarly, Phenon D included KJ1-5-98, KJ1-5-982 and KJ1-5-910, taxonomically identified as B. flexus with 86% similarity. Among them, KJ1-5-982 and KJ1-5-910 shared maximum phenotypic similarity of 100%. KJ1-5-913 and KJ1-5-914 of Phenon E were phylogenetically identical with a high phenotypic similarity of 70%.
Conversely, in few cases, phenotypic diversity did not correspond with the phylogenetic relatedness. For instance, the phylogram exhibited H. trueperi (KJ1-10-91, KJ1-10-97 and KJ1-10-98) identical to each other, while in phenogram only two H. trueperi isolates (KJ1-10-91 and KJ1-10-98) were identical while KJ1-10-97, although belonged to the same species, clustered in different segment (Figs. 1, 2). Similarly, KJ1-5-93, KJ1-5-912 and KJ1-10-96 displayed identity with V. salarius and placed with Oceanobacillus sp. (KJ1-10-910) in the phylogenetic tree. However, as revealed in the phenogram, only two isolates belonging to V. salarius (KJ1-5-93 and KJ1-10-96) clustered with Oceanobacillus sp. (KJ1-10-910), while KJ1-5-912, being phenotypically different, placed far apart in different phenon.
Phenon C partially corroborates the phylogram, as only KJ1-10-93 and KJ1-10-99 of 5 members exhibited high phenotypic similarity of 0.92 (92%). While KJ1-10-95 (Bacillus sp.) and KJ1-10-97 (Halobacillus sp.), although belonged to different species/genus, had high phenotypic similarity of 88% and clustered together in phenogram.
Among the Staphylococcus genus, KJ1-5-94, KJ1-5-95, KJ1-5-96 and KJ1-5-911, despite being phylogenetically identical with S. saprophyticus, were scattered in the phenogram. While KJ1-5-97 and KJ1-5-99 being phylogenetically identified as S. haemolyticus, clustered together in the phenogram.
Discussion
Deserts are the extreme environment with respect to water scarcity and factors limiting growth and survival of the organisms. It is identified with different extreme challenges of nutrient availability, temperature fluctuation, UV radiation, alkalinity and salinity. Studies of the microorganisms of such habitats will help to understand the interactions among the living organisms, eventually leading to the discovery of new organisms with distinctive survival strategies, physiological processes or secondary metabolites.
The Little Rann of Kutch is a typical ecosystem of the saline desert climate harboring unique flora and fauna. It has a different demography from the rest of the desert as it is located near sea and low-lying areas, through which marine water enters into the desert, making it an admixture of the saline, marshy and coastal desert.
The intermediate disturbance hypothesis suggests that highly stable or highly unstable environments harbor only limited diversity as compared to the moderately unstable environments (Petraitis et al. 1989). In LRK, during monsoon storm tides aided by the wind forces, water of the Gulf of Kutch flows towards the flat surface of the Rann, while during the rest of the time the desert is dry. Therefore, this desert reflects intermediate level of disturbance and hence expected to harbor greater microbial diversity. To explore the microbial diversity of this unique desert, various enrichment conditions were employed to assess the diversity and enumeration of the bacterial isolates. The bacteria were phylogenetically related to those reported from the aquatic, hypersaline and desert habitats, representing a diverse distribution pattern.
Phenotypic analysis
The colony characteristics of the haloalkaliphilic bacteria indicated their diverse nature. Yellow pigmented colonies were represented by a large number of the isolates, while several reflected color changes from cream white to yellow to yellowish red with the increasing age of the culture, a trend earlier reported for the bacteria of hypersaline environment (Phillips et al. 2012). All the isolates were Gram positive, while some reflected Gram negative behavior despite of their phylogenetic placement as gram-positive bacteria. Filobacillus milosensis and Halobacillus sp. from the Great Salt Plains of Oklahoma having unusual cell wall have been reported to display this character of Gram reaction (Schlesner et al. 2001; Caton et al. 2004).
Majority represented rods and cocci shape with some exceptions of unusual cell arrangements. Cells might aggregate due to certain stress conditions of the desert climate, such as desiccation and high temperature (Monier and Lindow 2003). The EPS (exopolymeric substance) production in few isolates arguably suggest towards a protection strategy against desiccation (Schnider-Keel et al. 2001; Monier and Lindow 2003).
In view of the significance of the metabolic and physiological characters in diversifying the microorganisms, the isolates were assessed for various such properties. All the isolates were catalase and oxidase positive indicating their aerobic nature. The extent of these enzymes, however, significantly varied, suggesting variation in O2 requirements and oxidative enzyme systems (Nowlan et al. 2006; Taprig et al. 2013). The lack of utilization of urea and tryptophan was consistent with some earlier reports on moderate halophilic and alkaliphilic bacteria (Romano et al. 2005). Detection of H2S production and nitrate reduction in some members indicated the availability of protein rich contents and utilization of sulfur-containing amino acids in the habitat. The isolates were highly variable in their sugar utilization pattern.
The extracellular enzymes of potential applications have been reported in recent years in halophiles and haloalkaliphiles (Rohban et al. 2009; Purohit and Singh 2011; Sahay et al. 2012; Raval et al. 2014). Many bacteria in this study have shown hydrolytic activities at high pH and NaCl concentrations. Bacillus, Piscibacillus and Halobacillus from Howz Soltan Lake of Iran are earlier described as the predominant gram-positive bacteria producing hydrolases (Rohban et al. 2009). However, in the present study, Halobacillus, Virgibacillus and Bacillus predominantly produced hydrolases. Isolates belonging to Virgibacillus genus were predominant protease producers followed by Halobacillus, Staphylococcus and Bacillus. Amylase production was maximum in the genus Bacillus followed by Virgibacillus and Halobacillus. CMCase production was maximally detected in the members of Bacillus genus followed by Virgibacillus, Staphylococcus and Halobacillus, while cellulase was predominant in Bacillus followed by Staphylococcus and Virgibacillus.
The antibiotic sensitivity profiling has emerged as an effective tool for judging the microbial diversity (Litzner et al. 2006). The isolates obtained from high salt concentration enrichment were more sensitive to different antibiotics as compared to those enriched at low salt, suggesting the repression of certain specific genes at higher salt concentrations. While the bacterial growth was not significantly affected by the antibiotics that inhibit cell wall synthesis, the antibiotics affecting the protein synthesis were more effective. Some of the isolates were sensitive against the antibiotics inhibiting RNA synthesis and DNA replication. Many isolates were resistant against penicillin and cephalosporin, arguably due to the synthesis of the beta-lactamase. Cefpodoxime is an antibiotic which is stable with beta-lactamase enzymes. The resistant nature of KJ-5-913 against cefpodoxime suggests the production of the extended spectrum of the beta-lactamase by this isolate.
The phylogenetics
The bacterial isolates based on the phylogenetic analysis were related to the Phylum Firmicutes, Gram-positive bacteria belonging to the families of Bacillaceae and Staphylococcaceae with the genera: Bacillus, Halobacillus, Oceanobacillus, Virgibacillus and Staphylococcus. These organisms were phylogenetically identical to those from the hypersaline soil or desert environment. The earlier reports of the hypersaline habitats have suggested the dominance of the halotolerant bacteria that include Flavobacterium, Pseudomonas, Alcaligenes, Micrococcus, Acinetobacter, Arthrobacter, Planococcus, and Bacillus (Quesada et al. 1985; Su et al. 2004). Firmicutes are dominant among the members of the cultivable bacterial community in a variety of hyper saline habitats (Yeon et al. 2005; Hedi et al. 2009; Baati et al. 2010). The dominance of Bacillaceae and Staphylococcaceae has recently been reported in the saline desert of Kutch (Great Rann) (Subrahmanyam et al. 2014).
The bacterial communities of the hypersaline soil (Caton et al. 2004) or saltern (Quesada et al. 1985) relates to the species reported from the aquatic environment. Since salterns are formed by flooding the lands with seawater, the bacterial community in such environment may resemble to those from the aquatic habitats. Our findings in this report also suggest the similar trend.
Archaea dominates the microbial community of the hypersaline soil over bacteria (Hacĕne et al. 2004). However, recent studies have suggested huge bacterial diversity with the dominance of Gram-stain-negative Betaproteobacteria, Gammaproteobacteria, and Deltaproteobacteria groups and a lower representation of the Gram-positive bacteria in saline soils (Baati et al. 2010). Our studies, however, have suggested the dominance of the Gram-stain-positive bacteria in the saline desert. Dominance of the Gram-positive bacteria in deep-sea hypersaline lake anoxic sediments has been reported (Sass et al. 2008). Further, high occurrence of the Gram-positive bacteria of the genera Halobacillus, Virgibacillus, Bacillus, Oceanobacillus, Gracilibacillus, Paenibacillus, and Pontibacillus are evident in the Arid Natural Saline Systems of the Southern Tunisian Sahara (Guesmi et al. 2013). In an alkaline Lonar Lake of India, endospore forming bacteria represented by Paenibacillus, Bacillus, and Alkalibacillus genera are reported (Joshi et al. 2008). A wide distribution of Bacillus genus in various stressful environments suggests their physiological and metabolic competence.
In this study, isolates including Halobacillus sp. (KJ1-10-91, KJ1-10-97 and KJ-10-98), Bacillus sp. (KJ1-10-95), Oceanobacillus sp. (KJ1-10-910) and Virgibacillus sp. (KJ1-5-912) displayed phylogenetic relatedness with Halobacillus sp. GSP 35, Bacillus sp. GSP77, Oceanobacillus sp. GSP73 and V. marismortui strain GSP17 of the Great Salt plains of Oklahoma (Caton et al. 2004).
Novel lineage identification
Detailed analyses of the 16S rRNA sequences of the strains KJ1-10-99 and KJ1-10-93 with the neighbouring type species led to the identification of a novel lineage. In BLAST analysis, both isolates were detected as Bacillus sp. with the nearest homolog Bacillus sp. ISO_06 kulunda of only 97% identity. Further, 16S rRNA gene sequence comparison in Eztaxon revealed that both isolates share 96.90% similarity with F. polygoni IEB3T. Sequence similarity greater than or equal to 97% is widely considered for the genus level match, while a species level match is based on a similarity greater than or equal to 99% (Guesmi et al. 2013). Based on the polyphasic taxonomic analysis, the two phylotypes were identified as the type species of novel genus recently described as Desertibacillus haloalkaliphilus KJ1-10-99T (Bhatt et al. 2017).
Comparison of the diversity based on the phylogenetic and phenotypic approaches
The genotypic changes constantly occur in organisms irrespective of the phenotypic variations. In other words, the evolution is quasi-independent of the phenotypic characters. Nevertheless, the phenotypic characteristics play significant role, and thus cannot be ignored (Bhatt and Singh 2016). While some phenotypic change may lead to a short term adaptation of the organism to the environment, a long term change might be due to the changes at the genetic level.
The Phenogram supported the phylogenetic tree based on the 16S rRNA gene sequences. For instance, isolates KJ1-5-913 and KJ1-5-914 were in close proximity and clustered together as Phenon E in the phenogram and were also placed together in phylogram. On the other hand, in few cases the phenogram contradicted the trend reflected in phylogram. For instance, on comparing the phylogram and phenogram, the phylogram (Fig. 1) exhibited all the three isolates KJ1-10-91, KJ1-10-97 and KJ1-10-98 identical to each other, while in phenogram (Fig. 2) only two isolates, KJ1-10-91 and KJ1-10-98 were identical being in same cluster (Phenon A), whereas KJ1-10-97 although belonged to same species have shown only ~ 44% phenotypic similarity with their phylogenetic relatives and placed in different Phenon C. Therefore, the observations clearly indicated that although the isolates had high similarity in the 16S rRNA gene sequences, they demonstrated considerable phenotypic heterogeneity.
Earlier, its reported that the strains belonging to the same species based on the 16S rRNA gene sequences have quite distinct phenotypic characteristics (Caton et al. 2004; Litzner et al. 2006; Lima-Bittencourt et al. 2007; Phillips et al. 2012; Bhatt and Singh 2016). Thus, the phenotypic diversity can be quite useful where phylogenetic diversity fails to discriminate strains of the same species or two different species sharing high 16S rRNA gene sequence similarity. Moreover, the presence and extent of the phenotypic characteristics of the bacterial communities reflect their role in biogeochemical cycles and nutrient composition of the soil.
Conclusion
This study is the first culture-dependent report on the bacterial diversity in the saline desert of the Little Rann of Kutch. The novel lineage found in this study will add to our quest for analyzing bacterial community structure, the adaptation strategies and mining of novel genes. Being enigmatic terrain, it is highly possible that this habitat might possess many such novel lineages. Besides identifying new lineage of haloalkaliphilic bacteria, the analysis provided insights into the yet-to-culture bacteria in this saline desert. The application of the Bi-phasic approach led to greater understanding of the bacterial diversity. The cultivable approach holds its significance in exploring the catalytic and metabolic potential of the organisms and their role in the biogeochemical cycles.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
The authors are grateful to University Grants Commission (UGC), India and Saurashtra University for the infrastructural and financial support. Support under the UGC-Centre of Advanced Study and DST-FIST programs is acknowledged. Award of Research Associateship to SDG by CSIR, New Delhi is duly acknowledged. HB is grateful to the UGC, New Delhi for Meritorious Fellowship under BSR Scheme.
Compliance with ethical standards
Conflict of interest
The authors have declared no conflict of interest.
Footnotes
Electronic supplementary material
The online version of this article (10.1007/s13205-017-1075-0) contains supplementary material, which is available to authorized users.
References
- Amoozegar MA, Malekzadeh F, Malik KA. Production of amylase by newly isolated moderate halophile, Halobacillus sp. strain MA-2. J Microbiol Methods. 2003;52:353–359. doi: 10.1016/S0167-7012(02)00191-4. [DOI] [PubMed] [Google Scholar]
- An S, Couteau C, Luo F, Neveu J, DuBow MS. Bacterial diversity of surface sand samples from the Gobi and Taklamaken deserts. Microb Ecol. 2013;66:850–860. doi: 10.1007/s00248-013-0276-2. [DOI] [PubMed] [Google Scholar]
- Andrew DR, Fitak RR, Munguia-Vega A, Racolta A, Martinson VG, Dontsova K. Abiotic factors shape microbial diversity in Sonoran desert soils. Appl Environ Microbiol. 2012;78:7527. doi: 10.1128/AEM.01459-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arahal DR, Dewhirst FE, Paster BJ, Volcani BE, Ventosa A. Phylogenetic analyses of some extremely halophilic archaea isolated from Dead Sea water, determined on the basis of their 16S rRNA sequences. Appl Environ Microbiol. 1996;62:3779–3786. doi: 10.1128/aem.62.10.3779-3786.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baati H, Amdouni R, Gharsallah N, Sghir A, Ammar E. Isolation and characterisation of moderately halophilic bacteria from Tunisia solar saltern. Curr Microbiol. 2010;60:157–161. doi: 10.1007/s00284-009-9516-6. [DOI] [PubMed] [Google Scholar]
- Bhatt HB, Singh SP. Phylogenetic and phenogram based diversity of haloalkaliphilic bacteria from the saline desert. In: Bhukya B, Tangutur AD, editors. Microbial biotechnology-technological challenges and developmental trends. Oakville: Apple Academic Press; 2016. pp. 373–386. [Google Scholar]
- Bhatt HB, Begum AM, Chintalapati S, Chintalapati VR, Singh SP. Desertibacillus haloalkaliphilus gen. nov. sp. nov., isolated from a saline desert. Int J Syst Evol Microbiol. 2017;67(11):4435–4442. doi: 10.1099/ijsem.0.002310. [DOI] [PubMed] [Google Scholar]
- Cappuccino JG, Sherman N. Microbiology, a laboratory manual. 6. Singapore: Pearson education; 2004. [Google Scholar]
- Casamayor EO, Triadó-Margarit X, Castañeda C. Microbial biodiversity in saline shallow lakes of the Monegros Desert, Spain. FEMS Microbiol Ecol. 2013;85:503–518. doi: 10.1111/1574-6941.12139. [DOI] [PubMed] [Google Scholar]
- Caton TM, Witte LR, Ngyuen HD, Buchheim JA, Buchheim MA, Schneegurt MA. Halotolerant aerobic heterotrophic bacteria from the Great Salt Plains of Oklahoma. Microb Ecol. 2004;48:449–462. doi: 10.1007/s00248-004-0211-7. [DOI] [PubMed] [Google Scholar]
- Demergasso C, Casamayor EO, Chong G, Galleguillos P, Escudero L, Pedrós-Alió C. Distribution of prokaryotic genetic diversity in athalassohaline lakes of the Atacama Desert, Northern Chile. FEMS Microbiol Ecol. 2004;48:57–69. doi: 10.1016/j.femsec.2003.12.013. [DOI] [PubMed] [Google Scholar]
- Dong HL, Rech JA, Jiang HC, Sun H, Buck BJ. Endolithic cyanobacteria in soil gypsum: occurrences in Atacama (Chile), Mojave (United States), and Al-Jafr Basin (Jordan) deserts. J Geophys Res Biogeosci. 2007;112:2005–2012. [Google Scholar]
- Duckworth AW, Grant WD, Jones BE, Steenbergen RV. Phylogenetic diversity of Soda lake alkaliphiles. FEMS Microbiol Ecol. 1996;19:181–191. doi: 10.1111/j.1574-6941.1996.tb00211.x. [DOI] [Google Scholar]
- Essoussi I, Boujmil R, Nouioui I, Abbassi-Ghozzi I, Hamza A, Boudabous A, Gtari M. Genetic diversity and esterase-profiling of actinobacteria isolated from Sahara desert stones and monuments. Geomicrobiol J. 2012;29:23–28. doi: 10.1080/01490451.2010.521367. [DOI] [Google Scholar]
- Falkowski PG, Fenchel T, Delong EF. The microbial engines that drive Earth’s biogeochemical cycles. Science. 2008;320:1034–1039. doi: 10.1126/science.1153213. [DOI] [PubMed] [Google Scholar]
- Foti MJ, Sorokin DY, Zacharova EE, Pimenov NV, Kuenen JG, Muyzer G. Bacterial diversity and activity along a salinity gradient in soda lakes of the Kulunda steppe (Altai, Russia) Extremophiles. 2008;12:133–145. doi: 10.1007/s00792-007-0117-7. [DOI] [PubMed] [Google Scholar]
- Guesmi A, Ettoumi B, Hidri DE, Essanaa J, Cherif H, Mapelli F, Marasco R, Rolli E, Boudabous A, Cherif A. Uneven distribution of Halobacillus trueperi species in arid natural saline systems of Southern Tunisian Sahara. Microb Ecol. 2013;66:831–839. doi: 10.1007/s00248-013-0274-4. [DOI] [PubMed] [Google Scholar]
- Gupta V, Ansari AA. Geomorphic portrait of the Little Rann of Kutch. Arab J Geosci. 2014;1:527–536. doi: 10.1007/s12517-012-0743-y. [DOI] [Google Scholar]
- Hacĕne H, Rafa F, Chebhouni N, Boutiba S, Bhatnagar T, Baratti JC, Ollivier B. Biodiversity of prokaryotic microflora in El GoleaSalt, Algerian Sahara. J Arid Environ. 2004;58:273–284. doi: 10.1016/j.jaridenv.2003.08.006. [DOI] [Google Scholar]
- Hammer Ø, Harper DAT, Ryan PD. PAST: paleontological statistics software package for education and data analysis. Palaeontol Electron. 2001;4:1–9. [Google Scholar]
- Hedi A, Sadfi N, Fardeau M-L, Rebib H, Cayol J-L, Ollivier B, Boudabous A. Studies on the biodiversity of halophilic microorganisms isolated from El-Djerid Salt Lake (Tunisia) under aerobic conditions. Int J Microbiol. 2009;2009:1–17. doi: 10.1155/2009/731786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horikoshi K. Alkaliphiles: some applications of their products for biotechnology. Microbiol Mol Biol Rev. 1999;63:735–750. doi: 10.1128/mmbr.63.4.735-750.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horner-Devine MC, Carney KM, Bohannan BJM. An ecological perspective on bacterial diversity. Proc R Soc Lond Ser B Biol Sci. 2003;271:113–122. doi: 10.1098/rspb.2003.2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishnava K, Ramarao V, Mohan JSS, Kothari IL. Ecologically important and life supporting plants of little Rann of Kachchh, Gujarat. J Ecol Nat Environ. 2011;3:33–38. [Google Scholar]
- Joshi AA, Kanekar PP, Kelkar AS, Shouche YS, Vani AA, Borgave SB, Sarnaik SA. Cultivable bacterial diversity of alkaline Lonar Lake, India. Microb Ecol. 2008;55:163–172. doi: 10.1007/s00248-007-9264-8. [DOI] [PubMed] [Google Scholar]
- Joshi RH, Dodia MS, Singh SP. Production and optimization of a commercially viable alkaline protease from a haloalkaliphilic bacterium. Biotechnol Bioprocess Eng. 2009;13:552–559. doi: 10.1007/s12257-007-0211-9. [DOI] [Google Scholar]
- Kasana RC, Salwan R, Dhar H, Dutt S, Gulati A. A rapid and easy method for the detection of microbial cellulases on agar plates using Gram’s iodine. Curr Microbiol. 2008;57:503–507. doi: 10.1007/s00284-008-9276-8. [DOI] [PubMed] [Google Scholar]
- Kim OS, Cho YJ, Lee K, Yoon SH, Kim M, Na H, Park SC, Jeon YS, Lee JH, Yi H, Won S. Introducing EzTaxon-e: a prokaryotic 16S rRNA gene sequence database with phylotypes that represent uncultured species. Int J Syst Evol Microbiol. 2012;62:716–721. doi: 10.1099/ijs.0.038075-0. [DOI] [PubMed] [Google Scholar]
- Kim TY, Kim SJ, Park SJ, Kim JG, Cha IT, Jung MY, Lee SA, Roh SW, Yim KJ, Itoh T, Rhee SK. Natronomonas gomsonensis sp. nov. isolated from a solar saltern. Antonie Van Leeuwenhoek. 2013;104:627–635. doi: 10.1007/s10482-013-9970-9. [DOI] [PubMed] [Google Scholar]
- Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980;16:111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- Langenheder S, Bulling MT, Solan M, Prosser JI. Bacterial biodiversity-ecosystem functioning relations are modified by environmental complexity. PLoS One. 2010;5:e10834. doi: 10.1371/journal.pone.0010834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K, Liu R, Zhang H, Yun J. The diversity and abundance of bacteria and oxygenic phototrophs in saline biological desert crusts in Xinjiang, Northwest China. Microb Ecol. 2013;66:40–48. doi: 10.1007/s00248-012-0164-1. [DOI] [PubMed] [Google Scholar]
- Lima-Bittencourt CI, Astolfi-Filho S, Chartone-Souza E, Santos FR, Nascimento AMA. Analysis of Chromobacterium sp. natural isolates from different Brazilian ecosystems. BMC Microbiol. 2007;7:58. doi: 10.1186/1471-2180-7-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litzner BR, Caton TM, Schneegurt MA. Carbon substrate utilization, antibiotic sensitivity, and numerical taxonomy of bacterial isolates from the Great Salt Plains of Oklahoma. Arch Microbiol. 2006;185:286–296. doi: 10.1007/s00203-006-0096-6. [DOI] [PubMed] [Google Scholar]
- Madsen EL. Identifying microorganisms responsible for ecologically significant biogeochemical processes. Nat Rev Microbiol. 2005;3:439–446. doi: 10.1038/nrmicro1151. [DOI] [PubMed] [Google Scholar]
- Margesin R, Schinner F. Potential of halotolerant and halophilic microorganisms for biotechnology. Extremophiles. 2001;5:73–83. doi: 10.1007/s007920100184. [DOI] [PubMed] [Google Scholar]
- Monier JM, Lindow SE. Differential survival of solitary and aggregated bacterial cells promotes aggregate formation on leaf surfaces. PNAS. 2003;100:15977–15982. doi: 10.1073/pnas.2436560100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowlan B, Dodia MS, Singh SP, Patel BKC. Bacillus okhensis sp. nov., a halotolerant and alkalitolerant bacterium from an Indian saltpan. Int J Syst Evol Microbiol. 2006;56:1073–1077. doi: 10.1099/ijs.0.63861-0. [DOI] [PubMed] [Google Scholar]
- Pandey S, Rakholiya K, Raval VH, Singh SP. Catalysis and stability of an alkaline protease from a haloalkaliphilic bacterium under non-aqueous conditions as a function of pH, salt and temperature. J Biosci Bioeng. 2012;114:251–256. doi: 10.1016/j.jbiosc.2012.03.003. [DOI] [PubMed] [Google Scholar]
- Petraitis PS, Latham RE, Niesenbaum RA. The maintenance of species diversity by disturbance. Q Rev Biol. 1989;64:393–418. doi: 10.1086/416457. [DOI] [Google Scholar]
- Phillips K, Zaidan F, Elizondo OR, Lowe KL. Phenotypic characterization and 16S rDNA identification of culturable non-obligate halophilic bacterial communities from a hypersaline lake, La Sal del Rey, in extreme South Texas (USA) Aquat Biosyst. 2012;8:5. doi: 10.1186/2046-9063-8-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilania PK, Panchal NS. Influence of sodium and calcium on vegetation at Saline Desert (Little Rann of Kutch) of Gujarat State in India. Ann Arid Zone. 2016;55(1&2):1. [Google Scholar]
- Purohit MK, Singh SP. Comparative analysis of enzymatic stability and amino acids sequences of thermostable alkaline proteases from two haloalkaliphilic bacteria isolated from coastal region of Gujarat, India. Int J Biol Macromol. 2011;49:103–112. doi: 10.1016/j.ijbiomac.2011.04.001. [DOI] [PubMed] [Google Scholar]
- Quesada E, Bejar V, Valderrama MJ, Ventosa A, Ramos-Cormenzana A. Isolation and characterization of moderately halophilic non motile rods from different saline habitats. Microbiologia. 1985;1:89–96. [PubMed] [Google Scholar]
- Raeid MM, Kharusi ASA, Schramm A, Robinson MD. Bacterial diversity, pigments and nitrogen fixation of biological desert crusts from the Sultanate of Oman. FEMS Microbiol Ecol. 2010;72:418–428. doi: 10.1111/j.1574-6941.2010.00854.x. [DOI] [PubMed] [Google Scholar]
- Rao S, Chan Y, Bugler-Lacap DC, Bhatnagar A, Bhatnagar M, Pointing SB. Microbial diversity in soil, sand dune and rock substrates of the Thar Monsoon Desert, India. Indian J Microbiol. 2016;56:35–45. doi: 10.1007/s12088-015-0549-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasuk MC, Fernández AB, Kurth D, Contreras M, Novoa F, Poiré D, Farías ME. Bacterial diversity in microbial mats and sediments from the Atacama Desert. Microb Ecol. 2016;71:44–56. doi: 10.1007/s00248-015-0649-9. [DOI] [PubMed] [Google Scholar]
- Raval VH, Pillai S, Rawal CM, Singh SP. Biochemical and structural characterization of a detergent-stable serine alkaline protease from seawater haloalkaliphilic bacteria. Process Biochem. 2014;49:955–962. doi: 10.1016/j.procbio.2014.03.014. [DOI] [Google Scholar]
- Rohban R, Amoozegar MA, Ventosa A. Screening and isolation of halophilic bacteria producing extracellular hydrolases from Howz Soltan Lake, Iran. J Ind Microbiol Biotechnol. 2009;36:333–340. doi: 10.1007/s10295-008-0500-0. [DOI] [PubMed] [Google Scholar]
- Romano I, Giordano A, Lama L, Nicolaus B, Gambacorta A. Halomonas campaniensis sp. nov. a haloalkaliphilic bacterium isolated from a mineral pool of Campania Region, Italy. Syst Appl Microbiol. 2005;28:610–618. doi: 10.1016/j.syapm.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Sahay H, Mahfooz S, Singh AK, Singh S, Kaushik R, Saxena AK, Arora DK. Exploration and characterization of agriculturally and industrially important haloalkaliphilic bacteria from environmental samples of hypersaline Sambhar Lake, India. World J Microbiol Biotechnol. 2012;28:3207–3217. doi: 10.1007/s11274-012-1131-1. [DOI] [PubMed] [Google Scholar]
- Sass AM, McKew BA, Sass H, Fichtel J, Timmis KN, McGenity TJ. Diversity of Bacillus-like organisms isolated from deep-sea hypersaline anoxic sediments. Saline Syst. 2008;4:1–11. doi: 10.1186/1746-1448-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saul-Tcherkas V, Unc A, Steinberger Y. Soil microbial diversity in the vicinity of desert shrubs. Microb Ecol. 2013;65:689–699. doi: 10.1007/s00248-012-0141-8. [DOI] [PubMed] [Google Scholar]
- Schlesner H, Lawson PA, Collins MD, Weiss N, Wehmeyer U, Volker H, Thomm M. Filobacillus milensis gen. nov., sp. nov., a new halophilic spore-forming bacterium with Orn-d-Glutypepeptidoglycan. Int J Syst Evol Microbiol. 2001;51:425–431. doi: 10.1099/00207713-51-2-425. [DOI] [PubMed] [Google Scholar]
- Schnider-Keel U, Lejbølle KB, Baehler E, Haas D, Keel C. The sigma factor AlgU (AlgT) controls exopolysaccharide production and tolerance towards desiccation and osmotic stress in the biocontrol agent pseudomonas fluorescence CHA0. Appl Environ Microbiol. 2001;67:5683–5693. doi: 10.1128/AEM.67.12.5683-5693.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scola V, Ramond JB, Frossard A, Zablocki O, Adriaenssens EM, Johnson RM, Seely M, Cowan DA. Namib Desert soil microbial community diversity, assembly, and function along a natural xeric gradient. Microb Ecol. 2018;75:193–203. doi: 10.1007/s00248-017-1009-8. [DOI] [PubMed] [Google Scholar]
- Su J, Wu Y, Ma X, Zhang G, Feng H, Zhang Y. Soil microbial counts and identification of culturable bacteria in an extreme by arid zone. Folia Microbiol. 2004;49:423–429. doi: 10.1007/BF02931604. [DOI] [PubMed] [Google Scholar]
- Subrahmanyam G, Khonde N, Maurya DM, Chamyal LS, Archana G. Microbial activity and culturable bacterial diversity in sediments of the Great Rann of Kachchh, Western India. Pedosphere. 2014;24(1):45–55. doi: 10.1016/S1002-0160(13)60079-X. [DOI] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taprig T, Akaracharanya A, Sitdhipol J, Visessanguan W, Tanasupawat S. Screening and characterization of protease-producing Virgibacillus, Halobacillus and Oceanobacillus strains from Thai fermented fish. J Appl Pharm Sci. 2013;3:025–030. [Google Scholar]
- Thomas M, Pal KK, Dey R, Saxena AK, Dave SR. A novel haloarchael lineage widely distributed in the hypersaline marshy environment of Little and Great Rann of Kutch in India. Curr Sci. 2012;10:1078–1084. [Google Scholar]
- Torsvik V, Øvreas L. Microbial diversity and function in soil: from genes to ecosystems. Curr Opin Microbiol. 2002;5:240–245. doi: 10.1016/S1369-5274(02)00324-7. [DOI] [PubMed] [Google Scholar]
- UNESCO (2006) World Heritage Centre-Nomination entry. http://whc.unesco.org/en/tentativelists/2105/. Accessed 3 June 2017
- Yeon SH, Jeong WJ, Park JS. The diversity of culturable organotrophic bacteria from local solar salterns. The Journal of Microbiology. 2005;43(1):1–10. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.