

Abstract
Background
CRTC2 is highly expressed in lung cancer and contributes to lung cancer pathogenesis; however, whether CRTC2 promotes lung cancer metastasis remains unknown. In the present study, we investigated the role of CRTC2 in lung cancer metastasis in vitro.
Methods
CRTC2 stable knockdown of lung cancer cell A549 was generated with small hairpin RNA and confirmed by quantitative reverse transcription‐PCR and Western blot. Wound healing and invasion transwell assays were performed to explore migration and invasion activity, and Western blot was conducted to detect the expression of related proteins.
Results
Suppression of CRTC2 significantly inhibited A549 cell migration and invasion in vitro. Mechanistic studies showed that knockdown of CRTC2 greatly downregulated MMP2 and MMP9 expression. CRTC2 silencing remarkably suppressed epithelial‐mesenchymal transition by modulating the expression of E‐cadherin and vimentin. Furthermore, suppression of CRTC2 expression significantly reduced MAPK/c‐Jun N‐terminal kinase activity.
Conclusion
CRTC2 may promote A549 migration and invasion by modulation of c‐Jun N‐terminal kinase‐mediated epithelial‐mesenchymal transition and matrix metalloproteinase expression.
Keywords: CRTC2, epithelial‐mesenchymal transition, lung cancer, metastasis, MMPs
Introduction
As one of the most common causes of cancer‐related mortality worldwide, lung cancer severely endangers health and life of human.1 The main types of lung cancer are small cell lung cancer (SCLC) and non‐small cell lung cancer (NSCLC). NSCLC accounts for approximately 85% of lung cancers and less than 15% of diagnosed patients will survive longer than five years.2 Despite decades of research, the prognosis for patients with lung cancer remains dismal. Although rapid advances in surgery, chemotherapy, radiotherapy and immunotherapy have been achieved, the prognosis of NSCLC patients has not significantly improved.3 Therefore, exploration of novel effective diagnostic and therapeutic targets is important. In order to develop more effective diagnostic and therapeutic technologies, a better understanding of the genetic alterations responsible for the molecular biological changes contributing to NSCLC carcinogenesis is urgently required.
Activating invasion and metastasis is one of the hallmarks of cancer, which mainly includes a series of processes: local invasion, intravasation into nearby blood and lymphatic vessels, transit of cancer cells through the lymphatic and hematogenous systems, escape of cancer cells from the lumina of such vessels into the parenchyma of distant tissues (extravasation), formation of small nodules of cancer cells (micrometastasis), and the growth of micrometastatic lesions into macroscopic tumors (colonization).4, 5 Accumulating evidence has suggested that epithelial‐mesenchymal transition (EMT) is crucial for cancer cell invasion and metastasis, during which cancer cells acquire the capacity to invade and resist apoptosis. E‐cadherin, a key cell‐to‐cell adhesion molecule, is a key suppressor of cancer cell invasion and metastasis.6, 7 E‐cadherin and vimentin are widely used markers for epithelial and mesenchymal cells, respectively.8, 9 In the early stage of metastasis, matrix metalloproteinases (MMPs) are activated and released to penetrate the extracellular matrix.10
CRTC2 belongs to the CREB‐regulated transcription coactivator (CRTC) family. CRTC2 enhances CREB transcriptional activity by associating with the leucine zipper DNA‐binding region of the CREB. CREB/CRTC2 complexes directly regulate the expression of a number of critical genes involved in proliferation and apoptosis.11, 12 The role of CRTC2 and its downstream targets in regulating hormonal and metabolic signal pathways has been well documented.13 Additionally, CRTC2 upregulates aromatase expression and may participate in the regulation of the estrogen receptor‐aromatase pathway.14
Recently, the role of CRTC2 in cancer has attracted much attention. The Catalogue of Somatic Mutations in Cancer revealed that CRTC2 is frequently mutated in several types of cancer and is downregulated in multiple cancers.15, 16 Recently, Fang et al. reported that CRTC2 acts as a tumor suppressor in T cell lymphomas by promoting transcription of MMR genes to maintain genome integrity.17 We recently identified and confirmed novel mutations of the CRTC2 gene in NSCLC patients, in which CRTC2 was highly expressed in lung cancer tissues compared to adjacent normal lung tissues.18, 19 Moreover, high CRTC2 expression was correlated with lymph node metastasis and CRTC2 played an important part in lung cancer pathogenesis in vitro and in vivo.
However, until now, the role of CRTC2 in metastasis remains elusive. In the present study, therefore, we investigated whether CRTC2 was involved in lung cancer metastasis in vitro. We demonstrated that CRTC2 promoted lung cancer migration and invasion by regulating EMT and MMP expression in a c‐Jun N‐terminal kinase (JNK)‐dependent manner.
Methods
Reagents
Anti‐E‐cadherin (P12830), MMP2 (P08253) and MMP9 (P14780) were obtained from Affinity (Cincinatti, OH, USA). Antibodies for vimentin (sc‐6260) and glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) were from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). The antibody against CRTC2 (ab109081) was purchased from Abcam (Cambridge, UK). Anti‐phospho‐JNK (2679369) and phospho‐MAPK1/2 (Erk1/2) were obtained from Millipore (Billerica, MA, USA). The antibody for phospho‐p38 MAPK (4511) was obtained from Cell Signaling Technology (Danvers, MA, USA).
Cell culture
Lung cancer cell line A549 was obtained from American Type Culture Collection (Manassas, VA, USA) and grown in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), 2 mM L‐glutamine, 100 U penicillin, and 100 μg/mL streptomycin. All cells were cultured in standard incubator conditions at 37°C with 5% CO2. RPMI 1640 medium without glucose and FBS were purchased from Gibco (Grand Island, NY, USA).
Generation of CRTC2 stable knockdown non‐small cell lung cancer cell lines
Small hairpin RNA (shRNA) were constructed for the human CRTC2 lentivirus gene transfer vector to select shRNA‐CRTC2‐positive NSCLC cells. Sequences for targeting the CRTC2 gene (GenBank accession number NM_181715.2) using the BLOCK‐iT RNA interference (RNAi) Designer were selected. Complementary DNA of the shRNA was inserted into the lentivirus gene transfer vector. The double stranded shRNA oligonucleotide was ligated to the pGCsil‐GFP lentivirus vector (Neuron Biotech, Shanghai, China) using the Age I and EcoRI restriction enzyme sites. A control shRNA that was unrelated to the sequence of CRTC2 (5′‐TTCTC CGAAC GTGTCAC GT‐3′) was transferred to the same vector as a negative control (NC). The viral particles were then produced, the recombinant lentivector was used to infect the target cells, and shRNA CRTC2‐positive NSCLC cells were selected by fluorescence‐activated cell sorting, as previously described.20 The stable silencing effect of CRTC2 cell lines was confirmed by quantitative reverse transcription‐PCR (q‐RT‐PCR) and Western blotting.
Migration wound healing assay
A549 wild‐type (WT) and CRTC2 stable knockdown (KD) cells were grown to 90% confluency in RPMI 1640 medium with 10% FBS and were then incubated with medium containing 2% FBS overnight. The cultured cells were then scratched using a 200 μL pipette tip and rinsed twice with phosphate buffered saline to remove free floating cells and debris. After incubation with medium containing 10% FBS, the cells were visualized by light microscopy at 0 and 16 hours after scratching. All experiments were repeated three times.
Invasion transwell assay
In vitro invasion assays were performed in Matrigel‐coated transwells. A549 WT and CRTC2 KD cells (5 × 104 in 200 μL serum‐free medium) were placed in the top chamber, while the lower chamber was filled with 600 μL of medium with 10% FBS as chemoattractant. After 24 hours, cells that had not invaded the lower chamber were wiped away from the upper surface of the transwell membrane with a cotton swab. Invaded cells on the lower membrane surface were fixed, stained, photographed, and counted. The invasion index was calculated by taking the invaded cell number of the control sample as 1.
Reverse transcription and quantitative PCR analysis
Total RNA was extracted from cultured cells using TRIzol Reagent (Invitrogen, Carlsbad, CA, USA) and treated with RNase‐free DNase I according to the manufacturer's instructions. qRT‐PCR using SYBR premix EX Taq (TaKaRa BIO, Japan) and SYBR green I was used to compare the relative expression levels of the gene messenger RNAs. β‐actin was used as an internal control. Primer sequences for real‐time PCR are shown in Table 1.
Table 1.
Primer sequences for real‐time PCR
| CRTC2(forward) | 5′‐CCAATTTGACCCACACCATGA‐3′ |
| CRTC2(reverse) | 5′‐CCTGGAGGTTGGGATTGCTTAG‐3′ |
| β‐Actin(forward) | 5′‐ATAGCACAGCCTGGATAGCAACGTAC‐3′ |
| β‐Actin(reverse) | 5′‐CAC CT TCTACAATGAGCTGCGTGTG −3′ |
Western blotting
Cell lysates were prepared by suspending cells in M2 buffer (20 mM Tris–HCl pH 7.6, 0.5% NP40, 250 mM NaCl, 3 mM ethylene‐diamine‐tetraacetic acid, 2 mM dithiothreitol, 0.5 mM phenylmethylsulfonyl fluoride, 20 mM β‐glycerophosphate, 1 mM sodium vanadate, and 1 μg/mL leupeptin). The samples were boiled for five minutes and then equal amounts of proteins from each cell lysate were resolved by 8% or 12% sodium dodecyl sulfate‐polyacrylamide gel electrophoresis and analyzed by Western blot. The proteins were visualized with enhanced chemiluminescence (Millipore) following the manufacturer's instructions. Each experiment was repeated at least three times and representative results are shown.21, 22 Fold changes were calculated with the control taken as 1.
Results
CRTC2 suppression inhibits A549 migration and invasion
Our previous study have found that CRTC2 is overexpressed in lung cancer tissues compared to normal lung tissues, CRTC2 expression is correlated with metastasis, and knockdown of CRTC2 expression significantly inhibits lung cancer cell proliferation and promotes apoptosis in vitro and in vivo (data not shown). In the present study, we continued to investigate whether CRTC2 was involved in lung cancer metastasis in vitro. CRTC2 stable knockdown A549 cells were generated by shRNA transfection, confirmed by q‐RT‐PCR and Western blot (Fig 1a,b). Wound healing and invasion transwell assays were then performed to investigate the migration and invasion capacity of A549 WT and CRTC2 KD cells. The wound healing results suggested that A549‐WT cells had higher migration potential than CRTC KD cells (Fig 1c). To substantiate this observation, an invasion transwell assay was performed, which consistently showed that A549‐WT cells exhibited more migrating cells through the membrane (Fig 1d,e). These results indicate that CRTC2 KD suppressed A549 migration and invasion capacity in vitro.
Figure 1.
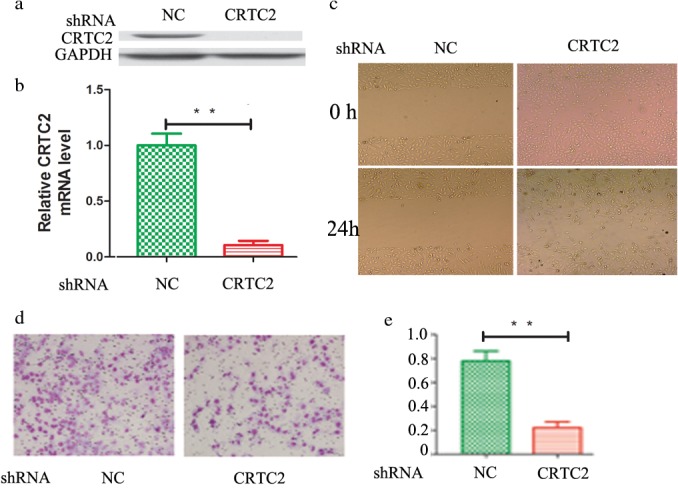
Suppression of CRTC2 expression inhibits A549 migration and invasion. (a,b) Silence of CRTC2 was confirmed by quantitative reverse transcription‐PCR and Western blot. (c) Wound healing assay. Cell cultures in confluence photographed immediately after scratching the wound (0 hours) or 16 hours post scratching. Representative images are shown. (d,e) Transwell invasion assay. A549 wild‐type or CRTC2 knockdown cells (5 × 104 in 200 μL serum‐free medium) were seeded in the top chamber containing a layer of Matrigel; the lower chamber contained medium with 10% fetal bovine serum as a chemoattractant. After incubation for 24 hours, invaded cells on the lower membrane surface were detected. Representative images of migrated A549 wild‐type and CRTC2 cells are shown. Quantification of cell migration of each line is shown (right). Data shown are mean ± standard deviation. *P < 0.05; **P < 0.01. GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; mRNA, messenger RNA; NC, negative control; shRNA, small hairpin RNA.
CRTC2 promotes A549 migration and invasion by regulation of MMP expression
Penetration of the extracellular matrix is one of the early steps in cancer cell metastasis, which mainly involves the activation and release of matrix metalloproteinases (MMPs). Previous studies have demonstrated that MMP1, MMP2, and MMP9 contribute to lung cancer cell metastasis.23, 24 We investigated whether CRTC2 regulated the expression of these MMPs. When CRTC2 expression was knocked down, MMP2 and MMP9 expression were substantially suppressed (Fig 2). These results suggest that CRTC2 regulates A549 migration and invasion by modulating MMP2 and MMP9 expression.
Figure 2.
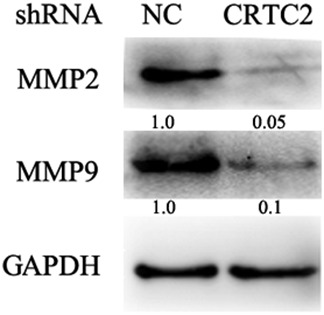
Knockdown of CRTC2 suppresses matrix metalloproteinase (MMP) expression. The indicated proteins were detected by Western blot. Glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) was detected as an input control. NC, negative control; shRNA, small hairpin RNA.
CRTC2 regulates epithelial‐mesenchymal transition by regulating E‐cadherin and vimentin expression
The role of EMT in lung cancer metastasis has been well established; therefore we continued to investigate whether CRTC2 regulated A549 migration and invasion by modulating EMT. The expression of markers for epithelial (E‐cadherin) and mesenchymal (Vimentin) cells were detected by Western blot. The results showed that CRTC2 suppression remarkably downregulated E‐cadherin and upregulated vimentin expression (Fig 3), indicating that knock down of CRTC2 expression suppressed EMT. These results indicate that CRTC2 regulates EMT by modulating the expression of E‐cadherin and vimentin.
Figure 3.
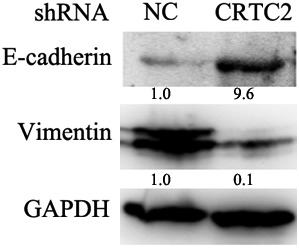
CRTC2 knockdown suppresses epithelial‐mesenchymal transition of A549. The indicated proteins were detected by Western blot. Glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) was detected as an input control. NC, negative control; shRNA, small hairpin RNA.
CRTC2 knockdown downregulates c‐Jun N‐terminal kinase activity
Previous studies have reported that MAPKs (including JNK, ERK, and p38) are involved in the regulation of the EMT and MMP expression, therefore we examined whether CRTC2 also regulated these pathways through MAPKs. Interestingly, suppression of CRTC2 expression distinctively downregulated JNK kinase activity (Fig 4a); however, it had almost no effect on ERK and p38 kinase activity (Fig 4b). Previous reports have suggested that JNK plays a crucial role in the regulation of EMT and MMPs expression, and these data, along with our results, suggest that CRTC2 promotes A549 migration and invasion through JNK‐mediated EMT and MMP expression.24
Figure 4.
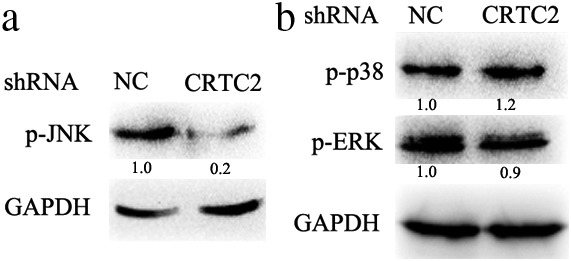
CRTC2 silencing downregulates c‐Jun N‐kinase (JNK) activity in A549 CRTC2 knockdown cells. The indicated proteins were detected by Western blot. Glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) was detected as an input control. NC, negative control; p, phosphorylated; shRNA, small hairpin RNA.
Discussion
The incidence and mortality rate of lung cancer ranks highest among different types of cancer in China, and the population diagnosed with this disease has increased rapidly in recent years because of severe pollution and cigarette smoking. As one of the main hallmarks of cancer, metastasis is the cause of 90% of human cancer‐related deaths.25 It has been well established that EMT and MMPs play key roles in metastasis in various cancers. Although decades of studies have been devoted to investigate the regulation of EMT and MMPs expression, the underlying mechanisms of these pathways in metastasis remain largely unclear.
In this study, we investigated the role of CRTC2 in lung cancer cell metastasis in vitro. Firstly, we investigated the migration and invasion potential of A549 WT and CRTC2 KD cells with wound healing and invasion transwell assays. The results showed that knockdown of CRTC2 expression greatly suppressed A549 cell migration and invasion. We then further explored the mechanisms underlying CRTC2‐mediated migration and invasion. The data demonstrated that CRTC2 regulated the expression of MMPs, and moreover, CRTC2 promoted EMT by modulating E‐cadherin and vimentin expression. Furthermore, we found that CRTC2 silencing distinctively downregulated JNK activity but had no effect on ERK or p38 activity. These data imply that CRTC2 may promote lung cancer metastasis through JNK‐mediated MMPs expression and EMT.
Epithelial‐mesenchymal transition is a key step in cancer metastasis, through which cancer cells acquire mesenchymal characteristics, including the capacity to migrate and resist apoptosis.26, 27 E‐cadherin and vimentin are widely used biomarkers to investigate the process of EMT.8, 9 As a key cell‐to‐cell adhesion molecule, E‐cadherin has been well documented to suppress migration and invasion, and is often downregulated in human cancinomas.4, 6 In this study, we found that suppression of CRTC2 expression remarkably downregulated E‐cadherin and upregulated vimentin (Fig 3), confirming that CRTC2 was a novel regulator of EMT. Li et al. previously reported that JNK and ERK were required for MMPs expression in TRAIL‐resistant lung cancer cells.24 Consistently, our results showed that silencing CRTC2 expression significantly decreased JNK kinase activity (Fig 4), further supporting the role of JNK in lung cancer metastasis. However, unlike previous results that ERK was involved in lung cancer metastasis, CRTC2 knockdown did not influence ERK and p38 kinase activity, suggesting that ERK and p38 may be not required for CRTC2‐mediated migration and invasion.9 Therefore the role of MAPKs in lung cancer metastasis may be specific, especially in different settings.
In conclusion, our data provide evidence that CRTC2 promotes lung cancer cell migration and invasion in vitro, and mechanistic studies show that CRTC2 regulates the expression of MMPs and EMT in a JNK‐dependent manner. Combined with our previous study that CRTC2 promotes lung cancer pathogenesis in vivo and in vitro, CRTC2 may present a novel promising target for lung cancer diagnosis and prognosis.
Disclosure
No authors report any conflict of interest.
Acknowledgments
This study was supported in part by grants from Department of Science and Technology of Yunnan Province (2017FE468 [−151], 2017FE468 [−200]) and the Department of Education of Yunnan (2017zDX169, 2015Y182).
Contributor Information
Shaoqing Shi, Email: sqs621@yeah.net.
Lihua Zhang, Email: lihuazhang772002@163.com.
Zhuang Luo, Email: skyny4511@126.com.
References
- 1. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014 (Published erratum appears in CA Cancer J Clin 2014;64:364.). CA Cancer J Clin 2014; 64: 9–29.24399786 [Google Scholar]
- 2. Katlic MR, Facktor MA, Berry SA, McKinley KE, Bothe A Jr, Steele GD Jr. ProvenCare lung cancer: A multi‐institutional improvement collaborative. CA Cancer J Clin 2011; 61: 382–96. [DOI] [PubMed] [Google Scholar]
- 3. Spira A, Ettinger DS. Multidisciplinary management of lung cancer. (Published erratum appears in N Engl J Med 2009;360:1917.). N Engl J Med 2004; 350: 379–92. [DOI] [PubMed] [Google Scholar]
- 4. Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell 2011; 144: 646–74. [DOI] [PubMed] [Google Scholar]
- 5. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100: 57–70. [DOI] [PubMed] [Google Scholar]
- 6. Berx G, van Roy F. Involvement of members of the cadherin superfamily in cancer. Cold Spring Harb Perspect Biol 2009; 1: a003129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cavallaro U, Christofori G. Cell adhesion and signalling by cadherins and Ig‐CAMs in cancer. Nat Rev Cancer 2004; 4: 118–32. [DOI] [PubMed] [Google Scholar]
- 8. Zeisberg M, Neilson EG. Biomarkers for epithelial‐mesenchymal transitions. J Clin Invest 2009; 119: 1429–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. LaGamba D, Nawshad A, Hay ED. Microarray analysis of gene expression during epithelial‐mesenchymal transformation. Dev Dyn 2005; 234: 132–42. [DOI] [PubMed] [Google Scholar]
- 10. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell 2010; 141: 39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Conkright MD, Canettieri G, Screaton R et al TORCs: Transducers of regulated CREB activity. Mol Cell 2003; 12: 413–23. [DOI] [PubMed] [Google Scholar]
- 12. Koo SH, Flechner L, Qi L et al The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature 2005; 437: 1109–11. [DOI] [PubMed] [Google Scholar]
- 13. Dentin R, Hedrick S, Xie J, Yates J III, Montminy M. Hepatic glucose sensing via the CREB coactivator CRTC2. Science 2008; 319: 1402–5. [DOI] [PubMed] [Google Scholar]
- 14. Samarajeewa NU, Docanto MM, Simpson ER, Brown KA. CREB‐regulated transcription co‐activator family stimulates promoter II‐driven aromatase expression in preadipocytes. Horm Cancer 2013; 4: 233–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Forbes SA, Bindal N, Bamford S et al COSMIC: Mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res 2011; 39: D945–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rhodes DR, Kalyana‐Sundaram S, Tomlins SA et al Molecular concepts analysis links tumors, pathways, mechanisms, and drugs. Neoplasia 2007; 9: 443–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fang M, Pak ML, Chamberlain L, Xing W, Yu H, Green MR. The CREB Coactivator CRTC2 is a lymphoma tumor suppressor that preserves genome integrity through transcription of DNA mismatch repair genes. Cell Rep 2015; 11: 1350–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. He Y, Li Y, Qiu Z et al Identification and validation of PROM1 and CRTC2 mutations in lung cancer patients. Mol Cancer 2014; 13: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li Y, He Y, Qiu Z et al CRTC2 and PROM1 expression in non‐small cell lung cancer: Analysis by western blot and immunohistochemistry. Tumour Biol 2014; 35: 11719–26. [DOI] [PubMed] [Google Scholar]
- 20. Lam P, Sian Lim K, Mei Wang S, Hui KM. A microarray study to characterize the molecular mechanism of TIMP‐3‐mediated tumor rejection. Mol Ther 2005; 12: 144–52. [DOI] [PubMed] [Google Scholar]
- 21. Wang Q, Chen W, Bai L et al Receptor‐interacting protein 1 increases chemoresistance by maintaining inhibitor of apoptosis protein levels and reducing reactive oxygen species through a microRNA‐146a‐mediated catalase pathway. J Biol Chem 2014; 289: 5654–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. He W, Wang Q, Xu J et al Attenuation of TNFSF10/TRAIL‐induced apoptosis by an autophagic survival pathway involving TRAF2‐ and RIPK1/RIP1‐mediated MAPK8/JNK activation. Autophagy 2012; 8: 1811–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ohbayashi H. Matrix metalloproteinases in lung diseases. Curr Protein Pept Sci 2002; 3: 409–21. [DOI] [PubMed] [Google Scholar]
- 24. Li Z, Xu X, Bai L, Chen W, Lin Y. Epidermal growth factor receptor‐mediated tissue transglutaminase overexpression couples acquired tumor necrosis factor‐related apoptosis‐inducing ligand resistance and migration through c‐FLIP and MMP‐9 proteins in lung cancer cells. J Biol Chem 2011; 286: 21164–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sporn MB. The war on cancer. Lancet 1996; 347: 1377–81. [DOI] [PubMed] [Google Scholar]
- 26. Yilmaz M, Christofori G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev 2009; 28: 15–33. [DOI] [PubMed] [Google Scholar]
- 27. Singh A, Settleman J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010; 29: 4741–51. [DOI] [PMC free article] [PubMed] [Google Scholar]