

Significance
Histone deacetylase 6 (HDAC6) is a zinc metalloenzyme that serves as the tubulin deacetylase in the cell cytosol. Inhibition of tubulin deacetylase activity leads to suppression of microtubule dynamics, causing cell cycle arrest and apoptosis, which is a validated strategy for cancer chemotherapy. Selective inhibition of HDAC6 is clinically preferable, since unwanted inhibition of other HDAC isozymes can lead to undesirable off-target effects. Here, X-ray crystal structures of HDAC6-inhibitor complexes reveal molecular features responsible for the isozyme selectivity measured in inhibition assays, including an unusual monodentate hydroxamate-Zn2+ coordination mode as well as intermolecular interactions of bulky inhibitor substituents. These observations will inform the design of HDAC6-selective inhibitors with improved properties.
Keywords: protein crystallography, metalloenzyme, enzyme inhibitor, drug discovery, cancer chemotherapy
Abstract
Histone deacetylases (HDACs) regulate myriad cellular processes by catalyzing the hydrolysis of acetyl–l-lysine residues in histone and nonhistone proteins. The Zn2+-dependent class IIb enzyme HDAC6 regulates microtubule function by deacetylating α-tubulin, which suppresses microtubule dynamics and leads to cell cycle arrest and apoptosis. Accordingly, HDAC6 is a target for the development of selective inhibitors that might be useful in new therapeutic approaches for the treatment of cancer, neurodegenerative diseases, and other disorders. Here, we present high-resolution structures of catalytic domain 2 from Danio rerio HDAC6 (henceforth simply “HDAC6”) complexed with compounds that selectively inhibit HDAC6 while maintaining nanomolar inhibitory potency: N-hydroxy-4-[(N(2-hydroxyethyl)-2-phenylacetamido)methyl)-benzamide)] (HPB), ACY-1215 (Ricolinostat), and ACY-1083. These structures reveal that an unusual monodentate Zn2+ coordination mode is exploited by sterically bulky HDAC6-selective phenylhydroxamate inhibitors. We additionally report the ultrahigh-resolution structure of the HDAC6–trichostatin A complex, which reveals two Zn2+-binding conformers for the inhibitor: a major conformer (70%) with canonical bidentate hydroxamate-Zn2+ coordination geometry and a minor conformer (30%) with monodentate hydroxamate-Zn2+ coordination geometry, reflecting a free energy difference of only 0.5 kcal/mol. The minor conformer is not visible in lower resolution structure determinations. Structural comparisons of HDAC6-inhibitor complexes with class I HDACs suggest active site features that contribute to the isozyme selectivity observed in biochemical assays.
Reversible lysine acetylation is a vital cellular strategy for the regulation of protein function (1). Since the landmark discovery of histone acetylation more than 50 y ago by Allfrey et al. (2), over 35,000 acetylation sites have been characterized in histone and nonhistone proteins involved in diverse biological processes such as the cell cycle, central carbon metabolism, and cellular signaling (3–7). The acetylation of a protein l-lysine side chain is catalyzed by a histone acetyltransferase using acetyl CoA as a cosubstrate to yield acetyl–l-lysine (8, 9), and this modification is reversed by a histone deacetylase (HDAC) to yield l-lysine and acetate (10–12).
Aberrant up-regulation of HDAC activity is associated with tumorigenesis, developmental disorders, and neurodegenerative disease, so there is significant clinical interest in selectively blocking the activity of specific HDAC isozymes associated with a particular disease phenotype (13–17). Human HDAC isozymes are classified into four groups based on phylogenetic analysis (18): class I HDACs (1–3, 8); class IIa HDACs (4, 5, 7, 9) and class IIb HDACs (6, 10); class III HDACs, better known as sirtuins; and the sole class IV enzyme, HDAC11. Class I, II, and IV HDACs are Zn2+- or Fe2+-dependent enzymes (19) that adopt the arginase-deacetylase fold (11, 20–22), while the sirtuins exhibit an unrelated fold and use cofactor NAD+ (23).
Among the HDAC isozymes, HDAC6 is distinct by virtue of its localization in the cytoplasm, as signaled by a serine/glutamate-rich repeat (24), as well as its biological function (25, 26). Additionally, HDAC6 is the only isozyme that contains two catalytic domains, CD1 and CD2, the structures of which have recently been solved (27, 28). One of these domains, CD2, catalyzes the deacetylation of K40 of α-tubulin in the lumen of the microtubule (29, 30). Consequently, inhibition of HDAC6 results in microtubule hyperacetylation and suppression of microtubule dynamics, leading to cell cycle arrest and apoptosis (30, 31). HDAC6 is thus a critical target for the design of isozyme-selective inhibitors for use in cancer chemotherapy (32, 33).
Four HDAC inhibitors are currently approved by the US Food and Drug Administration for cancer chemotherapy: the hydroxamic acids vorinostat, belinostat, and panobinostat and the cyclic depsipeptide romidepsin (34, 35). These compounds generally inhibit all HDAC isozymes potently; that is, they are “pan-HDAC” inhibitors, so off-target effects are associated with their use due to inhibition of multiple metal-dependent HDACs (13). However, significant advances have been made in the development of HDAC6-selective inhibitors, with examples being Tubastatin (36), N-hydroxy-4-(2-[(2-hydroxyethyl)(phenyl)amino]-2-oxoethyl)benzamide (HPOB) (37), N-hydroxy-4-[(N(2-hydroxyethyl)-2-phenylacetamido)methyl)-benzamide)] (HPB) (38), ACY-1215 (Ricolinostat) (39), and ACY-1083 (40) (Table 1). These inhibitors exhibit HDAC6 selectivities ranging from 12-fold to greater than 1,000-fold relative to class I enzymes such as HDAC1. Each of these inhibitors targets Zn2+ coordination with a hydroxamate group; HDAC6 selectivity is thought to be dictated by bulky capping or linker groups compared with pan-HDAC inhibitors such as suberoylanilide hydroxamic acid (SAHA) or trichostatin A (TSA) (Table 1).
Table 1.
Hydroxamate HDAC inhibitors
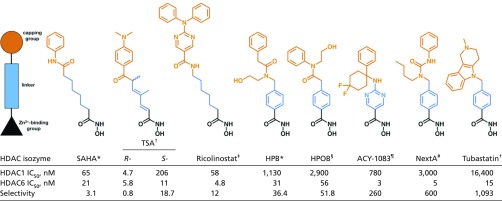 |
To determine molecular features responsible for HDAC6-selective inhibition, we now report crystal structures of inhibitor complexes with the CD2 domain of Danio rerio (zebrafish) HDAC6, henceforth referred to simply as “HDAC6.” Our recent structural and functional studies demonstrate that zebrafish HDAC6 is a validated and more readily crystallized surrogate of human HDAC6, the actual drug target (27). Here, crystal structures of HDAC6 complexed with the HDAC6-selective inhibitors HPB and ACY-1083 reveal a monodentate hydroxamate-Zn2+–binding mode similar to that recently observed in the crystal structure of the HDAC6–HPOB complex (27). Thus, this unusual binding mode is a signature of selectivity for the binding of phenylhydroxamate inhibitors with certain bulky substituents to HDAC6. In contrast, the crystal structure of the HDAC6 complex with Ricolinostat reveals a canonical bidentate hydroxamate-Zn2+ chelate complex, so the isozyme selectivity of this inhibitor is rooted solely in the interactions of its bulky capping group. Intriguingly, the 1.05 Å-resolution structure of the HDAC6 complex with the R-stereoisomer of TSA reveals two Zn2+-binding modes for this pan-HDAC inhibitor: the major binding mode corresponds to the canonical bidentate hydroxamate-Zn2+ complex observed in the previously determined 1.59 Å-resolution structure (27), and the minor binding mode corresponds to the unusual monodentate Zn2+ complex observed exclusively for HPB, HPOB, and ACY-1083.
Results
HDAC6–HPB Complex.
The 1.82 Å-resolution structure of the HDAC6–HPB complex does not reveal any significant conformational changes relative to unliganded HDAC6 [Protein Data Bank (PDB) ID code 5EEM], and the root-mean-square deviation (rmsd) between unliganded and inhibitor-bound structures is 0.14 Å for 284 Cα atoms. The hydroxamate group of HPB coordinates to the active site zinc ion only through its N–O− group (Zn2+—O distance = 1.9 Å), which binds at the coordination site that would be occupied by the substrate carbonyl in catalysis (Fig. 1A and Fig. S1). The catalytic zinc-bound water molecule remains in place and donates a hydrogen bond to the hydroxamate C=O group (O—O distance = 2.6 Å). This water molecule also forms hydrogen bonds with H573 and H574. Additionally, the Y745 hydroxyl group interacts with the hydroxamate NH (O—N distance = 2.6 Å) and O− (O—O distance = 2.7 Å) groups.
Fig. 1.

(A) Simulated annealing omit map (green, contoured at 2.0σ) for cis (yellow) and trans (orange) conformations of HPB bound to HDAC6. Omit density is also shown for the water molecule (red sphere) bound to the Zn2+ ion (gray sphere). Metal coordination and hydrogen bond interactions are indicated by solid and dashed black lines, respectively. A stereoview appears in Fig. S1. (B) Molecular surface of the HDAC6 active site showing the aromatic ring of the phenylhydroxamate nestled between F583 and F643.
The phenyl linker group of HPB is sandwiched between F583 and F643 (Fig. 1B). The peptoid capping group is observed in both the trans and cis conformations with respective occupancies of 0.66 and 0.34, corresponding to ΔG = 0.4 kcal/mol. This is within the range of 0.3–0.6 kcal/mol measured for peptoid trans-cis isomerization (41), so the enzyme does not exhibit a clear preference for one conformation or the other. The phenyl group of the cis conformer makes a van der Waals contact with L1 loop residue P464; the energetically preferred trans conformation of the peptoid moiety allows the phenyl capping group to make a quadrupole-charge interaction with the side chain of R601 in an adjacent monomer. The hydroxyl moiety of the capping group forms a water-mediated hydrogen bond with S531 on the L2 loop (Fig. 1A). The side chain of S531 accepts a hydrogen bond from the backbone NH group of the acetyl–l-lysine substrate, an interaction that is unique to the HDAC6 active site (27).
HDAC6–ACY-1083 Complex.
The 1.75 Å-resolution structure of the HDAC6–ACY-1083 complex does not reveal any major structural rearrangements caused by inhibitor binding, and the rmsd is 0.16 Å for 300 Cα atoms between the inhibitor-bound and unliganded enzymes. The hydroxamate moiety adopts monodentate Zn2+ coordination geometry identical to that observed in the HPB complex: The hydroxamate N–O− group coordinates to Zn2+ (Zn2+—O distance = 1.9 Å) and interacts with Y745 (O—O distance = 2.5 Å) (Fig. 2A and Fig. S2). The hydroxamate C=O group accepts a hydrogen bond from the Zn2+-bound water molecule (O—O distance = 2.7 Å); this water molecule also hydrogen-bonds with H573 and H574. The hydroxamate NH group interacts with the side chain of Y745 (O—N distance = 2.7 Å).
Fig. 2.

(A) Simulated annealing omit map (green, contoured at 2.5σ) for ACY-1083 (orange) and the water molecule (red sphere) bound to the Zn2+ ion (large gray sphere) in the active site of HDAC6. Metal coordination and hydrogen bond interactions are indicated by solid and dashed black lines, respectively. A stereoview appears in Fig. S2. (B) Molecular surface of the HDAC6 active site showing the heteroaromatic ring of the inhibitor nestled between F583 and F643. The phenyl group of the capping group lies adjacent to F583 and also P464 (not shown in this view).
The aromatic ring of the aminopyrimidine linker is sandwiched between F583 and F643 in a similar manner to that observed for the phenyl linker of HPB (Fig. 2B). The hydroxyl side chain of S531 on the L2 loop accepts a hydrogen bond from the secondary amino group of the inhibitor (O—N distance = 3.1 Å) (Fig. 2A). The difluorocyclohexyl capping group adopts a chair conformation, and the equatorial fluorine atom packs against the edge of the F643 side chain (F—C distance = 3.6 Å). The phenyl capping group of the inhibitor makes van der Waals interactions with the side chains of P464 and F583.
HDAC6–Ricolinostat Complex.
The 1.70 Å-resolution structure of the HDAC6–Ricolinostat (ACY-1215) complex reveals that no major conformational changes are triggered by inhibitor binding, and the rmsd is 0.65 Å for 336 Cα atoms between the inhibitor-bound and unliganded enzymes. The hydroxamate moiety of Ricolinostat coordinates to Zn2+ in bidentate fashion (Fig. 3 and Fig. S3), forming a canonical five-membered chelate complex with Zn2+—O distances of 2.0 Å and 2.4 Å for the N–O− and C=O groups, respectively. The side chain of Y745 donates a hydrogen bond to the hydroxamate C=O group (O—O distance = 2.7 Å), H573 donates a hydrogen bond to the hydroxamate N–O− group (N—O distance = 2.7 Å), and H574 accepts a hydrogen bond from the hydroxamate NH group (N—N distance = 2.9 Å). Hydrogen bond interactions with the tandem histidine residues suggest that H573 is in the positively charged imidazolium state and H574 is in the neutral imidazole state. These protonation states are consistent with the assignment of the second histidine in this tandem pair as a single general base-general acid in catalysis, as recently demonstrated for HDAC8 (42, 43).
Fig. 3.

Simulated annealing omit map (green, contoured at 2.5σ) for Ricolinostat bound to HDAC6 (monomer A). Two conformations for the capping group are shown with different shades of purple, the darker of which indicates the sole conformation observed in monomer B. The Zn2+ ion is shown as a large gray sphere, and water molecules are shown as smaller red spheres. Metal coordination and hydrogen bond interactions are indicated by solid and dashed black lines, respectively. A stereoview appears in Fig. S3.
Two monomers reside in the asymmetric unit of this crystalline enzyme-inhibitor complex. In one monomer, the inhibitor capping group adopts two mutually exclusive conformations (Fig. 3). In one conformation, the amide carbonyl of the inhibitor capping group forms a water-mediated hydrogen bond with S531 on the L1 loop. The aminopyrimidinyl ring nitrogen also forms a water-mediated hydrogen bond with D460. In the second conformation, a water-mediated hydrogen bond is formed between the amide carbonyl of the inhibitor capping group and H614 (one of the Zn2+ ligands). The aminopyrimidinyl ring nitrogen also forms a water-mediated hydrogen bond with the carbonyl of P711.
HDAC6–TSA Complex.
The 1.05 Å-resolution structure of HDAC6 complexed with the R-stereoisomer of TSA reveals significantly more detail in comparison to the previously reported 1.59 Å-resolution structure of this complex (PDB ID code 5EEK) (27). Although the polypeptide backbones of HDAC6 in the ultrahigh- and high-resolution structures are essentially identical (rmsd = 0.064 Å for 315 Cα atoms), the ultrahigh-resolution structure reveals major and minor conformers for the hydroxamate group of TSA (Fig. 4A). The minor conformation is not observable in crystal structures determined at lower resolution (27, 28).
Fig. 4.

(A) Simulated annealing omit map (green, contoured at 2.5σ) of the R-stereoisomer of TSA bound to HDAC6, showing the binding of major (purple) and minor (orange) inhibitor conformations. (B) Omit map of the major TSA conformer (70% occupancy) reveals the canonical bidentate hydroxamate-Zn2+ coordination geometry. (C) Omit map of the minor TSA conformer (30% occupancy) reveals unusual monodentate hydroxamate-Zn2+ coordination geometry. The Zn2+ ion appears as a large gray sphere; metal coordination and hydrogen bond interactions are shown as solid and dashed black lines, respectively.
The major conformer of TSA (70% occupancy) corresponds to that described in the 1.59 Å-resolution structure, in which the canonical bidentate hydroxamate-Zn2+ coordination mode is observed with O—Zn2+ distances of 2.2 Å and 2.0 Å for the hydroxamate C=O and N–O− groups, respectively. Additionally, Y745 donates a hydrogen bond to the hydroxamate C=O, while H573 and H574 form hydrogen bonds with the N–O− group (Fig. 4B).
The minor conformer of TSA (30% occupancy; Fig. 4C) corresponds to the monodentate hydroxamate-Zn2+ coordination mode observed for sterically bulky HDAC6-selective inhibitors such as HPOB, HPB, and ACY-1083. The hydroxamate group of TSA coordinates to Zn2+ only through its N–O− group (Zn2+—O distance = 1.8 Å), which binds at the coordination site that would be occupied by the substrate carbonyl in catalysis. Weak electron density is observed for the Zn2+-bound water molecule, which donates a hydrogen bond to the hydroxamate C=O group (O—O distance = 2.8 Å). Additionally, the hydroxyl group of Y745 is within hydrogen-bonding distance to the hydroxamate NH and N–O− groups with O—N and O—O distances of 2.7 Å and 2.8 Å, respectively.
Apart from differences in the conformation and orientation of the hydroxamate group, no other structural features distinguish the major and minor conformers of TSA. The dimethylheptadiene linker and p-dimethylaminophenyl capping group bind in identical fashion in high- and low-occupancy conformations. Based on the 70:30 ratio observed for bidentate/monodentate Zn2+ coordination modes for the hydroxamate group of TSA, the monodentate hydroxamate-Zn2+–binding mode is only 0.5 kcal/mol higher in energy than the canonical bidentate hydroxamate-Zn2+–binding mode.
Discussion
Structural Aspects of HDAC6-Inhibitor Selectivity.
Each component of an HDAC inhibitor (the Zn2+-binding group, the linker, and the capping group) contributes to the selectivity of inhibitor binding measured in biochemical assays. The key finding of the present study is that monodentate hydroxamate-Zn2+ coordination can be exploited by sterically bulky phenylhydroxamate inhibitors in the active site of HDAC6. The inhibitors HPB, ACY-1083, and HPOB each exhibit monodentate hydroxamate-Zn2+ coordination through their hydroxamate N–O− groups, with their hydroxamate C=O groups hydrogen-bonded to a Zn2+-bound water molecule (Fig. 5). The pan-HDAC inhibitor TSA (R-stereoisomer) engages in both bidentate and monodentate hydroxamate-Zn2+ coordination, as enabled by its comparatively slender linker group. The bidentate/monodentate ratio of 70:30 indicates a free energy difference of only 0.5 kcal/mol, so the monodentate hydroxamate-Zn2+–binding mode observed exclusively for sterically bulky phenylhydroxamate inhibitors does not significantly compromise enzyme-inhibitor affinity. The energetically accessible monodentate binding mode can be exploited by inhibitors that are too bulky to bind more deeply in the HDAC6 active site, as would be required for bidentate coordination.
Fig. 5.

Representation of the bidentate (A) and monodentate (B) Zn2+-binding modes observed for hydroxamate HDAC inhibitors.
Interestingly, the secondary amino linker group of ACY-1083 donates a hydrogen bond directly to the hydroxyl group of S531 (Fig. 2). S531 plays a key role in HDAC6-substrate recognition by accepting a hydrogen bond from the backbone NH group of the scissile acetyl–l-lysine substrate (27). In class I enzymes such as HDAC8, D101 serves this role and accepts hydrogen bonds from both backbone NH groups flanking the scissile acetyl–l-lysine residue (44, 45). While HPB lacks comparable functionality to make a direct interaction with S531, the hydroxyl group of its peptoid capping group forms a water-mediated hydrogen bond with S531 (Fig. 1). Thus, direct or water-mediated hydrogen bonding with S531 is an interaction unique to the HDAC6 active site that confers some measure of isozyme selectivity.
The capping group of each inhibitor significantly contributes to HDAC6 affinity and selectivity. The capping group binds at the mouth of the active site cleft, and this region exhibits significant structural differences among the HDAC isozymes. In HDAC6, the L1 (H455-E465), L2 (M517-N536), and L7 (A706–Q716) loops can interact with inhibitor capping groups. The aromatic capping groups of HPB, ACY-1083, and HPOB pack primarily against the L1 loop; in addition, the capping group of Ricolinostat is sufficiently large to interact with the L7 loop as well.
The L1 loops of HDAC6 and class I HDACs adopt different conformations that significantly influence inhibitor binding (Fig. 6). The phenyl group of the 260-fold selective inhibitor ACY-1083 packs tightly against the side chains of H463 and P464 in the L1 loop. Superposition of this complex with the structures of HDAC1, HDAC2, and HDAC3 shows that the L1 loops of class I HDACs adopt different conformations that constrict their active sites relative to HDAC6 (46–48). This would create a steric clash with the binding conformation of ACY-1083. Thus, ACY-1083 binding to class I HDACs appears to be disfavored due to clashes between the large capping group and the L1 loop.
Fig. 6.

Superposition of HDAC6 CD2 (pale blue) complexed with ACY-1083 (orange), HDAC1 (PDB ID code 4BKX), HDAC2 (PDB ID code 4LY1), and HDAC3 (PDB ID code 4A69). All class I HDACs are shown in shades of gray. The L1 and L2 loops are shown in shades of red, orange, and yellow for HDAC1, HDAC2, and HDAC3, respectively, and shades of blue for HDAC6. Corepressor proteins complexed with HDAC1 and HDAC3 (MTA1 and NCOR2, respectively) are shown in green and appear to stabilize the L1 loop conformation. The inositol tetraphosphate (IP4) bound at the HDAC3–NCOR2 interface is also shown. A conserved proline residue is shown on each L1 loop to highlight the steric crowding that would occur upon ACY-1083 binding to the class I HDACs. The L1 loop is set back by ca. 1 Å in HDAC6 relative to the class I HDACs. The Zn2+ ion of HDAC6 is shown as a lavender sphere.
Ricolinostat has the largest capping group of the inhibitors shown in Table 1, and this cap binds in a cleft between the L1 and L7 loops of HDAC6. Superposition with HDAC3 shows an ∼1 Å difference in these loop conformations, resulting in a narrowed cleft that would be less ideal for Ricolinostat binding. The 12-fold HDAC6 selectivity of Ricolinostat arises solely from this capping group, since the flexible aliphatic linker and bidentate hydroxamate-Zn2+–binding mode are otherwise identical to those of the pan-HDAC inhibitor SAHA.
HDAC6–Nexturastat A Complex.
The 1.99 Å-resolution crystal structure of HDAC6 CD2 complexed with the HDAC6-selective inhibitor Nexturastat A (NextA) (49) (Table 1 and Fig. S4A) was recently reported (28). Curiously, despite its structural resemblance to HPOB and HPOB as a bulky phenylhydroxamate derivative, NextA is reported to bind with an alternative monodentate hydroxamate-Zn2+ coordination geometry in which the hydroxamate carbonyl oxygen coordinates to Zn2+ with a Zn2+—O distance of 2.6 Å (Fig. S4B). This separation is rather long for inner-sphere metal coordination. However, we inspected the electron density map generated with structure factor amplitudes and phases calculated from the final model of the enzyme-inhibitor complex deposited in the PDB (PDB ID code 5G0I); we suggest that the map better accommodates monodentate hydroxamate-Zn2+ coordination by the hydroxamate N–O− group (Fig. S4C). This binding mode refines with a Zn2+—O distance of 2.2 Å, which is more typical for an inner-sphere metal coordination interaction. Additionally, the hydroxamate C=O group accepts a hydrogen bond from the Zn2+-bound water molecule, as observed for HPOB (27), HPB (Fig. 1), ACY-1083 (Fig. 2), as well as the minor conformer of TSA (Fig. 4). Thus, we suggest that Fig. S4C illustrates the preferred binding mode of NextA.
Summary and Conclusions
Canonical bidentate hydroxamate-Zn2+ coordination was first observed for inhibitor binding to thermolysin (50), and exceptions are rarely observed (51). Here, we outline a hydroxamate-Zn2+–binding mode that can be exploited by phenylhydroxamates with bulky substituents in the active site of HDAC6: These inhibitors coordinate to Zn2+ through a monodentate hydroxamate N–O− group, while the hydroxamate C=O group accepts a hydrogen bond from Zn2+-bound water. A six-membered ring hydroxamate-Zn2+–H2O complex results, as summarized in Fig. 5. The free energy of this Zn2+-binding mode is just 0.5 kcal/mol higher than that of the canonical bidentate hydroxamate-Zn2+ complex. This alternative Zn2+-binding mode can be exploited by bulky phenylhydroxamate inhibitors in the active site of HDAC6, even though the binding of these inhibitors would be disfavored in the more sterically constricted active sites of class I HDACs. Intriguingly, this alternative binding mode would disfavor the potential for the Zn2+-dependent Lossen rearrangement that could convert the hydroxamate moiety into a mutagenic isocyanate derivative (52).
The linker groups of HDAC6-selective inhibitors can make direct or water-mediated hydrogen bonds with S531. This residue accepts a hydrogen bond from the backbone NH group of the acetyl–l-lysine substrate (27), and this interaction is unique to the HDAC6 active site. Thus, an inhibitor that targets this interaction will exhibit selectivity for binding to HDAC6.
Finally, interactions of inhibitor capping groups at the mouth of the HDAC6 active site reveal that structural differences in the L1 loop also contribute to isozyme selectivity. Thus, specific interactions of the Zn2+-binding group, linker, and capping group together contribute to selectivity for HDAC6 binding.
Materials and Methods
HDAC6 CD2 from D. rerio (HDAC6) was recombinantly expressed using the MBP-tobacco etch virus-z6CD2-pET28a(+) vector and purified as previously described with minor modifications (27), and crystalline enzyme-inhibitor complexes were prepared by cocrystallization in sitting drops. Crystals diffracted to 1.05–1.82 Å-resolution using synchrotron radiation, and each structure was solved by molecular replacement using the structure of unliganded HDAC6 (PDB 5EEM) (27) as a search probe for rotation and translation function calculations. Each structure was refined to convergence with Rwork values ranging from 0.113–0.184 and Rfree values ranging from 0.132–0.209. Full experimental details are outlined in Supporting Information, and data collection and refinement statistics are provided in Table S1.
Supplementary Material
Acknowledgments
We thank Drs. John Van Duzer and Matthew Jarpe, formerly of Acetylon (now Celgene), for the generous gifts of Ricolinostat and ACY-1083, and we thank Dr. Stephen Shinsky for helpful discussions. We also thank synchrotron beamline staff for assistance with data collection. Specifically, we thank Dr. Raj Rajashankar and Dr. Narayansami Sukumar at the Northeastern Collaborative Access Team beamlines (supported by NIH Grants P41 GM103403 and S10 RR029205) at the Advanced Photon Source, a US Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract DE-AC02-06CH11357. We also thank Dr. Jay Nix at the Advanced Light Source (University of California, Berkeley), which is a DOE Office of Science User Facility under Contract DE-AC02-05CH11231. Finally, we thank Dr. Tzanko Doukov and Dr. Clyde Smith at the Stanford Synchrotron Radiation Lightsource (SSRL), SLAC National Accelerator Laboratory, a facility supported by the DOE Office of Science, Office of Basic Energy Sciences, under Contract DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the NIH/National Institute of General Medical Sciences (including Grant P41GM103393). This research was supported by NIH Grant GM49758. N.J.P. received financial support from NIH through Chemistry-Biology Interface Training Grant T32 GM071339. This paper is dedicated to the memory of R.B., our collaborator, mentor, and friend.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates and crystallographic structure factors for HDAC6 complexes with the inhibitors trichostatin A, N-hydroxy-4-[(N(2-hydroxyethyl)-2-phenylacetamido)methyl)-benzamide)], Ricolinostat, and ACY-1083 have been deposited in the Protein Data Bank (www.rcsb.org) with accession codes 5WGI, 5WGK, 5WGL, and 5WGM, respectively.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1718823114/-/DCSupplemental.
References
- 1.Choudhary C, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 2.Allfrey VG, Faulkner R, Mirsky AE. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc Natl Acad Sci USA. 1964;51:786–794. doi: 10.1073/pnas.51.5.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hornbeck PV, et al. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015;43:D512–D520. doi: 10.1093/nar/gku1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao S, et al. Regulation of cellular metabolism by protein lysine acetylation. Science. 2010;327:1000–1004. doi: 10.1126/science.1179689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang Q, et al. Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science. 2010;327:1004–1007. doi: 10.1126/science.1179687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choudhary C, Weinert BT, Nishida Y, Verdin E, Mann M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat Rev Mol Cell Biol. 2014;15:536–550. doi: 10.1038/nrm3841. [DOI] [PubMed] [Google Scholar]
- 7.Kouzarides T. Acetylation: A regulatory modification to rival phosphorylation? EMBO J. 2000;19:1176–1179. doi: 10.1093/emboj/19.6.1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sterner DE, Berger SL. Acetylation of histones and transcription-related factors. Microbiol Mol Biol Rev. 2000;64:435–459. doi: 10.1128/mmbr.64.2.435-459.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Friedmann DR, Marmorstein R. Structure and mechanism of non-histone protein acetyltransferase enzymes. FEBS J. 2013;280:5570–5581. doi: 10.1111/febs.12373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem J. 2003;370:737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lombardi PM, Cole KE, Dowling DP, Christianson DW. Structure, mechanism, and inhibition of histone deacetylases and related metalloenzymes. Curr Opin Struct Biol. 2011;21:735–743. doi: 10.1016/j.sbi.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.López JE, Sullivan ED, Fierke CA. Metal-dependent deacetylases: Cancer and epigenetic regulators. ACS Chem Biol. 2016;11:706–716. doi: 10.1021/acschembio.5b01067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov. 2014;13:673–691. doi: 10.1038/nrd4360. [DOI] [PubMed] [Google Scholar]
- 14.Penney J, Tsai LH. Histone deacetylases in memory and cognition. Sci Signal. 2014;7:re12. doi: 10.1126/scisignal.aaa0069. [DOI] [PubMed] [Google Scholar]
- 15.Dokmanovic M, Clarke C, Marks PA. Histone deacetylase inhibitors: Overview and perspectives. Mol Cancer Res. 2007;5:981–989. doi: 10.1158/1541-7786.MCR-07-0324. [DOI] [PubMed] [Google Scholar]
- 16.Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. Epigenetic protein families: A new frontier for drug discovery. Nat Rev Drug Discov. 2012;11:384–400. doi: 10.1038/nrd3674. [DOI] [PubMed] [Google Scholar]
- 17.Ganai SA, Ramadoss M, Mahadevan V. Histone deacetylase (HDAC) inhibitors–Emerging roles in neuronal memory, learning, synaptic plasticity and neural regeneration. Curr Neuropharmacol. 2016;14:55–71. doi: 10.2174/1570159X13666151021111609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gregoretti IV, Lee YM, Goodson HV. Molecular evolution of the histone deacetylase family: Functional implications of phylogenetic analysis. J Mol Biol. 2004;338:17–31. doi: 10.1016/j.jmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 19.Gantt SL, Gattis SG, Fierke CA. Catalytic activity and inhibition of human histone deacetylase 8 is dependent on the identity of the active site metal ion. Biochemistry. 2006;45:6170–6178. doi: 10.1021/bi060212u. [DOI] [PubMed] [Google Scholar]
- 20.Finnin MS, et al. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature. 1999;401:188–193. doi: 10.1038/43710. [DOI] [PubMed] [Google Scholar]
- 21.Kanyo ZF, Scolnick LR, Ash DE, Christianson DW. Structure of a unique binuclear manganese cluster in arginase. Nature. 1996;383:554–557. doi: 10.1038/383554a0. [DOI] [PubMed] [Google Scholar]
- 22.Ash DE, Cox JD, Christianson DW. Arginase: A binuclear manganese metalloenzyme. Met Ions Biol Syst. 2000;37:407–428. [PubMed] [Google Scholar]
- 23.Yuan H, Marmorstein R. Structural basis for sirtuin activity and inhibition. J Biol Chem. 2012;287:42428–42435. doi: 10.1074/jbc.R112.372300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bertos NR, et al. Role of the tetradecapeptide repeat domain of human histone deacetylase 6 in cytoplasmic retention. J Biol Chem. 2004;279:48246–48254. doi: 10.1074/jbc.M408583200. [DOI] [PubMed] [Google Scholar]
- 25.Grozinger CM, Hassig CA, Schreiber SL. Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc Natl Acad Sci USA. 1999;96:4868–4873. doi: 10.1073/pnas.96.9.4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Verdel A, Khochbin S. Identification of a new family of higher eukaryotic histone deacetylases. Coordinate expression of differentiation-dependent chromatin modifiers. J Biol Chem. 1999;274:2440–2445. doi: 10.1074/jbc.274.4.2440. [DOI] [PubMed] [Google Scholar]
- 27.Hai Y, Christianson DW. Histone deacetylase 6 structure and molecular basis of catalysis and inhibition. Nat Chem Biol. 2016;12:741–747. doi: 10.1038/nchembio.2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miyake Y, et al. Structural insights into HDAC6 tubulin deacetylation and its selective inhibition. Nat Chem Biol. 2016;12:748–754. doi: 10.1038/nchembio.2140. [DOI] [PubMed] [Google Scholar]
- 29.Hubbert C, et al. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417:455–458. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- 30.Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc Natl Acad Sci USA. 2003;100:4389–4394. doi: 10.1073/pnas.0430973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Szyk A, et al. Molecular basis for age-dependent microtubule acetylation by tubulin acetyltransferase. Cell. 2014;157:1405–1415. doi: 10.1016/j.cell.2014.03.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dallavalle S, Pisano C, Zunino F. Development and therapeutic impact of HDAC6-selective inhibitors. Biochem Pharmacol. 2012;84:756–765. doi: 10.1016/j.bcp.2012.06.014. [DOI] [PubMed] [Google Scholar]
- 33.Seidel C, Schnekenburger M, Dicato M, Diederich M. Histone deacetylase 6 in health and disease. Epigenomics. 2015;7:103–118. doi: 10.2217/epi.14.69. [DOI] [PubMed] [Google Scholar]
- 34.West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest. 2014;124:30–39. doi: 10.1172/JCI69738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ma N, et al. Selective histone deacetylase inhibitors with anticancer activity. Curr Top Med Chem. 2016;16:415–426. doi: 10.2174/1568026615666150813145629. [DOI] [PubMed] [Google Scholar]
- 36.Butler KV, et al. Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, tubastatin A. J Am Chem Soc. 2010;132:10842–10846. doi: 10.1021/ja102758v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee JH, et al. Development of a histone deacetylase 6 inhibitor and its biological effects. Proc Natl Acad Sci USA. 2013;110:15704–15709. doi: 10.1073/pnas.1313893110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee JH, et al. Creation of a histone deacetylase 6 inhibitor and its biological effects [corrected] Proc Natl Acad Sci USA. 2015;112:12005–12010, and correction (2015) 112:E5899. doi: 10.1073/pnas.1515882112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Santo L, et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood. 2012;119:2579–2589. doi: 10.1182/blood-2011-10-387365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krukowski K, et al. HDAC6 inhibition effectively reverses chemotherapy-induced peripheral neuropathy. Pain. 2017;158:1126–1137. doi: 10.1097/j.pain.0000000000000893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sui Q, Borchardt D, Rabenstein DL. Kinetics and equilibria of cis/trans isomerization of backbone amide bonds in peptoids. J Am Chem Soc. 2007;129:12042–12048. doi: 10.1021/ja0740925. [DOI] [PubMed] [Google Scholar]
- 42.Gantt SL, Joseph CG, Fierke CA. Activation and inhibition of histone deacetylase 8 by monovalent cations. J Biol Chem. 2010;285:6036–6043. doi: 10.1074/jbc.M109.033399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gantt SM, et al. General base-general acid catalysis in human histone deacetylase 8. Biochemistry. 2016;55:820–832. doi: 10.1021/acs.biochem.5b01327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vannini A, et al. Substrate binding to histone deacetylases as shown by the crystal structure of the HDAC8-substrate complex. EMBO Rep. 2007;8:879–884. doi: 10.1038/sj.embor.7401047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dowling DP, Gantt SL, Gattis SG, Fierke CA, Christianson DW. Structural studies of human histone deacetylase 8 and its site-specific variants complexed with substrate and inhibitors. Biochemistry. 2008;47:13554–13563. doi: 10.1021/bi801610c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Watson PJ, et al. Insights into the activation mechanism of class I HDAC complexes by inositol phosphates. Nat Commun. 2016;7:11262. doi: 10.1038/ncomms11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Watson PJ, Fairall L, Santos GM, Schwabe JWR. Structure of HDAC3 bound to co-repressor and inositol tetraphosphate. Nature. 2012;481:335–340. doi: 10.1038/nature10728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bressi JC, et al. Exploration of the HDAC2 foot pocket: Synthesis and SAR of substituted N-(2-aminophenyl)benzamides. Bioorg Med Chem Lett. 2010;20:3142–3145. doi: 10.1016/j.bmcl.2010.03.091. [DOI] [PubMed] [Google Scholar]
- 49.Bergman JA, et al. Selective histone deacetylase 6 inhibitors bearing substituted urea linkers inhibit melanoma cell growth. J Med Chem. 2012;55:9891–9899. doi: 10.1021/jm301098e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Holmes MA, Matthews BW. Binding of hydroxamic acid inhibitors to crystalline thermolysin suggests a pentacoordinate zinc intermediate in catalysis. Biochemistry. 1981;20:6912–6920. doi: 10.1021/bi00527a026. [DOI] [PubMed] [Google Scholar]
- 51.Scolnick LR, et al. Novel binding mode of hydroxamate inhibitors to human carbonic anhydrase II. J Am Chem Soc. 1997;119:850–851. [Google Scholar]
- 52.Shen S, Kozikowski AP. Why hydroxamates may not be the best histone deacetylase inhibitors–What some may have forgotten or would rather forget? ChemMedChem. 2016;11:15–21. doi: 10.1002/cmdc.201500486. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.