

Abstract
A previous study has reported that frequent amplifications of the TG-interacting factor (TGIF) were observed in esophageal squamous cell carcinoma. The aim of the present study was to investigate the potential role of TGIF in the proliferation and tumorigenicity of the esophageal cancer cell line EC109 and cisplatin-induced apoptosis. Stable TGIF-knockdown EC109 cell line was established by infecting short hairpin RNA (shRNA) lentiviral particles. Soft agar and tumor xenograft assays were applied in nude mice. Flow cytometry was employed to evaluate the cell cycle and apoptosis. Western blot analysis was used to detect the expression of proteins. TGIF knockdown suppressed EC109 cell proliferation, colony formation in soft agar and tumor growth in nude mice, induced cell cycle arrest in the G1 phase, and promoted cisplatin-induced apoptosis. In addition, TGIF knockdown significantly reduced the expression of phospho-Rb in EC109 cells. The reduced level of full length PARP expression and the increased level of cleaved caspase-3 expression were observed in EC109 cells with the treatment of cisplatin and TGIF knockdown. The results suggest that knockdown of TGIF attenuated the proliferation and tumorigenicity of EC109 cells, and promoted cisplatin-induced apoptosis.
Keywords: TG-interacting factor, esophageal cancer, tumorigenicity, cell cycle, apoptosis
Introduction
Esophageal cancer, the sixth leading cause of cancer-related death, has currently ranked the eighth in the malignant tumors worldwide (1). There were 455,800 new esophageal cancer cases and 400,200 deaths estimated in 2012. The incidence rates of esophageal cancer vary internationally, the highest rates are found in Eastern Asia and in Eastern and Southern Africa and the lowest rates are found in Western Africa. There are two main histological types of esophageal cancer, including squamous cell carcinoma and adenocarcinoma (2). Although, the diagnosis and treatments for esophageal cancer update constantly (3,4), the overall 5-year survival rate is only around 20% (5). Therefore, it is important and urgent to elucidate an exactly novel molecular mechanism underlying esophageal cancer formation, which may provide new strategies for the diagnosis and treatments of esophageal cancer in future healthcare.
TG-interacting factor (TGIF) belongs to the three-amino acid loop extension (TALE) subfamily of atypical homeodomain proteins (6). Heterozygous loss of TGIF gene causes holoprosencephaly in humans (7). It has been reported that TGIF is involved in at least three signaling pathways, including retinoic acid (RA) (6), transforming growth factor β (TGF-β) (8), and wnt/β-catenin signaling pathways (9). A number of studies have reported that TGIF plays an important role in the initiation, development, and progression of breast cancer (9), lung cancer (10–12), hepatocellular carcinoma (13,14), upper urinary tract urothelial carcinoma (15,16), leukemia (17–19), and oral squamous cell carcinoma (20,21). Nakakuki et al reported that frequent amplifications of TGIF were observed in esophageal squamous cell carcinoma (ESCC) (22), which suggests that TGIF might be associated with esophageal tumorigenesis. But, the potential role of TGIF in the proliferation and tumorigenicity of esophageal cancer cells is not clear. In the present study, we knocked down TGIF of EC109 cells with short hairpin RNA (shRNA) lentiviruses and observed the capabilities of proliferation and tumorigenicity of stable TGIF-knocked down EC109 cells in vitro and in vivo. We also observed the effects of TGIF knockdown on cisplatin-induced apoptosis in EC109 cells.
Materials and methods
Cell culture and infection
The esophageal cancer cell line of EC109 was obtained from the Cell Resource Center, Peking Union Medical College (which is the headquarters of National Infrastructure of Cell Line Resource in Beijing, China) and cultured in RPMI-1640 medium with 10% of fetal bovine serum (FBS), 100 µg/ml of streptomycin, 100 U/ml of penicillin, and 2 mM of L-glutamine at 37°C in a 5% CO2 incubator. TGIF shRNA (h) lentiviral particles (sc-36659-V) and control shRNA lentiviral particles-A (sc-108080) were obtained from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). The infection of lentiviral particles was performed in accordance with the manufacturer's instructions. The infected cells were maintained in RPMI-1640 full medium with 10 µg/ml of puromycin (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA). The efficiency of the shRNA lentiviruses targeting TGIF was measured at the level of protein expression. Stable clones were named EC109-shRNA-TGIF and EC109-shRNA-control, respectively.
Measurement of cell proliferation
Cell proliferation was measured according to the methods of our previous study (10). Briefly, 4×104 of cells were seeded into each well of 12-well plate in triplicate. Cells were harvested and counted by using a CASY Cell Counter (Schärfe System GmbH, Reutlingen, Germany) at 24, 48, 72, and 96 h, respectively.
Colony formation assay
Colony formation assay in soft agar was performed as previously described (11). Briefly, 500 cells were suspended in 0.35% low-melting point agarose and plated onto 0.6% low-melting point agarose in 6-well plates. Cells were maintained at 37°C in a 5% CO2 incubator for 18 days. Colonies of ten randomly selected views were counted by an inverted microscope (Leica DM IL LED; Leica Microsystems GmbH, Wetzlar, Germany) at ×100 magnification (11).
Tumor xenograft assay
All of the animal experiments were conducted in the light of the Guide for the Care and Use of Laboratory Animals. The Ethics Committee of Henan Center for Disease Control and Prevention approved this study. Tumor formation assay in nude mice was carried out in accordance with our previous study (11). In brief, male BALB/c nude mice aged 4 weeks were provided by Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). Cells (5×106) in 150 µl of PBS were subcutaneously injected into the back neck of each mouse. Mice were monitored every day and sacrificed at 14 days postinjection (11).
Cell cycle assay
The detailed methods of cell cycle assay were described previously (10). Briefly, cells were plated in 100-mm dishes and harvested when growing to 70–80% confluence. Cells were washed with PBS and fixed in 70% of ethanol at −20°C overnight, following by washing with PBS twice, suspending in 0.5 ml of PBS containing 100 µg/ml of RNase (Invitrogen Life Technologies, Carlsbad, CA, USA) and 50 µg/ml of propidium iodide (PI; Sigma-Aldrich, St. Louis, MO, USA), and incubating at room temperature for 30 min in the dark. The distribution of cell cycle was measured by EPICS XL-MCL ADC flow cytometry (Beckman Coulter, Inc., Brea, CA, USA).
Cisplatin treatment
Cells were plated in 60-mm dishes and treated with 12.5 µg/ml of cisplatin for different time-points indicated. Cells were harvested and washed with PBS, and then were used to analyze protein expression and apoptosis.
Detection of apoptosis
The apoptosis was detected by using the Muse Annexin V and Dead Cell Assay kit (MCH100105; EMD Millipore, Billerica, MA, USA) in Muse™ Cell Analyzer according to the manufacturer's instructions. In details, cells were detached by trypsinization and suspended in at least 1% FBS. The cell samples were incubated with Muse™ Annexin V and Dead Cell Reagent at room temperature for 20 min in the dark. The apoptosis rate was analyzed by using the Muse Cell Analyzer.
Western blot assay
Western blot analysis was conducted as previously described (10). The primary antibodies used were listed as following: TGIF (sc-9084), Akt (sc-8312), β-catenin (sc-7199), CDK4 (sc-260), cyclin A (sc-751), cyclin B1 (sc-752), cyclin D1 (sc-718), p21 (sc-397), p53 (sc-6243), and β-actin (sc-47778) were obtained from Santa Cruz Biotechnology, Inc., and Rb (#9313S), phospho-Rb (#8516S), c-Myc (#13987S), p65 (#8242S), ERK1/2 (#4695S), Axin1 (#2087S), PARP (#9532S), Bax (#5023S), caspase-3 (#9664S), and caspase-9 (#9508S) were obtained from Cell Signaling Technology, Inc. (Danvers, MA, USA). The secondary antibodies (peroxidase-coupled goat anti-rabbit-IgG and goat anti-mouse IgG) were obtained from ZSGB-BIO (Beijing, China). The membrane was developed by a Bio-Rad Clarity™ Western ECL substrate in the ChemiDoc™ XRS+ Imaging system (both from Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Statistical analysis
Numeric data were presented as mean ± standard deviation (SD) or median. Student's t-test, one-way analysis of variance and Mann-Whitney U test were performed to estimate statistical significance among groups by using the SPSS 13.0 software (SPSS, Inc., Chicago, IL, USA). P<0.05 was considered statistically significant. All the tests were two-sided.
Results
The efficiency of TGIF knockdown in EC109 cells
As shown in Fig. 1A, the expression of TGIF protein was significantly reduced in EC109-shRNA-TGIF cells, compared with EC109-shRNA-control cells, which suggests that a stable TGIF-knocked down EC109 cell line was successfully established.
Figure 1.

(A) The identification of TGIF-knocked down EC109 cells. Our data indicated that shRNA significantly reduced the level of TGIF protein expression in EC109 cells. (B) The effects of TGIF knockdown on EC109 cell proliferation. Our results showed that TGIF knockdown inhibited EC109 cell proliferation from 72 h (*P<0.05 vs. shRNA-control). (C) Effects of TGIF knockdown on colony formation. Our findings indicated that the ability of colony formation was significantly decreased in EC109-shRNA-TGIF cells as compared with EC109-shRNA-control cells. (D) Effects of TGIF knockdown on tumor growth in nude mice. Our results demonstrated that the tumor weight was significantly reduced in EC109-shRNA-TGIF cells as compared with EC109-shRNA-control cells. TGIF, TG-interacting factor; shRNA, short hairpin RNA.
Effects of TGIF knockdown on EC109 cell proliferation
Fig. 1B indicated that EC109-shRNA-TGIF cells grew significantly slowly, compared with EC109-shRNA-control cells from 72 h, which suggests that TGIF knockdown suppressed EC109 cell proliferation.
Effects of TGIF knockdown on colony formation
Results of colony formation were presented in Fig. 1C, which showed that EC109-shRNA-TGIF cells formed significantly less colonies (17±4/10 fields) than EC109-shRNA-control cells did (28±5/10 fields).
Effects of TGIF knockdown on tumor xenograft formation in nude mice
Fig. 1D demonstrated the results of tumor xenograft formation in nude mice. Our data indicated that the significantly decreased tumor weight was observed in EC109-shRNA-TGIF cells, compared with EC109-shRNA-control cells (Fig. 1D), which suggests that TGIF knockdown significantly suppressed tumor formation and tumor growth of EC109 cells in vivo.
Effects of TGIF knockdown on cell cycle distribution
As shown in Fig. 2A, EC109-shRNA-TGIF cells had the significantly increased percentage of G1 phase cells (45.8±3.9%) accompanied with the significantly decreased percentage of S phase cells (47.6±2.7%) as compared with EC109-shRNA-control cells (39.5±1.3 and 53.5±0.7%, respectively). Our data suggested that TGIF knockdown might induce the inhibition of EC109 cell growth by arresting the cell cycle in the G1 phase.
Figure 2.

(A) Effects of TGIF knockdown on cell cycle distribution. Data showed that TGIF knockdown increased the percentage of EC109 cells in G1 phase and decreased the percentage of EC109 cells in S phase. (B and C) Effects of TGIF knockdown on the expression of interested proteins. Data showed that TGIF knockdown suppressed the expression of phospho-Rb protein. TGIF, TG-interacting factor; shRNA, short hairpin RNA.
TGIF knockdown suppressed the expression of phospho-Rb
Fig. 2B presented the alterations of cell cycle-related protein expression, while TGIF knocking down in EC109 cells. Our findings showed that knockdown of TGIF suppressed the expression of phospho-Rb protein. There was no obvious alterations in the expression of the cyclin A, cyclin B1, cyclin D1, CDK4 and p21 proteins between EC109-shRNA-TGIF cells and EC109-shRNA-control cells. In addition, we did not observe significant difference in the expression of p53, c-Myc, p65, Akt, ERK1/2, Axin1 and β-catenin proteins between EC109-shRNA-TGIF cells and EC109-shRNA-control cells (Fig. 2C).
Cisplatin suppressed the expression of TGIF
Fig. 3 exhibited the effects of cisplatin on the expression of TGIF protein in EC109 cells. Our data showed that 12.5 µg/ml of cisplatin could suppress the expression of TGIF protein from 24 h (Fig. 3).
Figure 3.
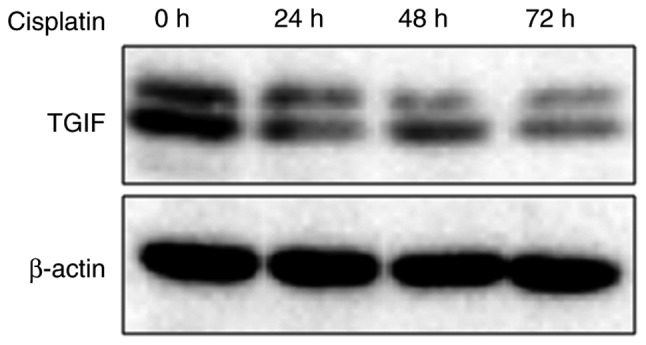
Effects of cisplatin on the expression of TGIF protein. Data showed that cisplatin could reduce the expression of TGIF protein. TGIF, TG-interacting factor.
TGIF knockdown promoted cisplatin-indcued apoptosis
Results from flow cytometry indicated that the percentage of total apoptosis (early apoptosis and late apoptosis) was significantly higher in EC109-shRNA-control cells treated with 12.5 µg/ml of cisplatin than that in negative control (Fig. 4A), which suggests that cisplatin could induce apoptosis in EC109 cells. When TGIF gene was knocked down by shRNA in EC109 cells, we observed that the percentage of total apoptosis was significantly higher in EC109-shRNA-TGIF cells treated with 12.5 µg/ml of cisplatin than that in EC109-shRNA-control cells treated with 12.5 µg/ml of cisplatin (Fig. 4A), which suggests that TGIF knockdown promoted cisplatin-induced apoptosis in EC109 cells.
Figure 4.

(A) Effects of TGIF knockdown on cisplatin-induced EC109 cells apoptosis. Data showed that the percentage of total apoptosis was significantly increased in EC109-shRNA-TGIF cells treated with cisplatin compared with EC109-shRNA-control cells treated with cisplatin. (B) Effects of TGIF knockdown on apoptosis-related markers. TGIF knockdown decreased the expression of full length PARP and increased the expression of cleaved caspase-3 in cisplatin-treated group. TGIF, TG-interacting factor; shRNA, short hairpin RNA.
In addition, we observed the significantly decreased expression of full length PARP in EC109-shRNA-TGIF cells treated with 12.5 µg/ml of cisplatin as compared with EC109-shRNA-control cells treated with 12.5 µg/ml of cisplatin (Fig. 4B). We observed the significantly increased expression of cleaved caspase-3 in EC109-shRNA-TGIF cells treated with 12.5 µg/ml of cisplatin as compared with EC109-shRNA-control cells treated with 12.5 µg/ml of cisplatin (Fig. 4B). Our data suggested that TGIF knockdown had effects on the expression of apoptosis-related markers in EC109 cells treated with cisplatin.
Discussion
In our study, the functional role of TGIF in the proliferation and tumorigenicity of esophageal cancer cell line of EC109 was investigated. The abilities of EC109 cell growth and tumor formation in vitro and in vivo were inhibited when the expression of TGIF was knocked down by shRNA specifically targeting TGIF, which suggests that TGIF may act as an oncogene in the development of esophageal cancer. Knockdown of TGIF arrested the cell cycle of EC109 cells in the G1 phase by downregulating phospho-Rb. In addition, knockdown of TGIF promoted cisplatin-induced apoptosis of EC109 cells.
Cell cycle arrest is one of the major causes of cancer cell proliferation inhibition (23,24). Dysregualtion of several key factors, including CDK4, cyclin D1, p21 and phospho-Rb could result in G1 phase arrest (25,26). In this study, we observed that knockdown of TGIF induced cell cycle arrest in the G1 phase accompanied with significantly decreased expression of phospho-Rb protein, while other proteins such as CDK4, cyclin D1 and p21 did not significantly change. Studies have shown that activation of cyclin D1-CDK4 complex can phosphorylate Rb and keep Rb inactivation, thus promote G1/S phase transition (27,28). Our previous data showed that silencing of TGIF induced G1 phase cell cycle arrest along with the decreased expression of phospho-Rb, cyclin D1 and CDK4 in lung cancer cells (10). Together, the current observations suggests that knockdown of TGIF led to the decreased expression of phospho-Rb not through regulating CDK4 and cyclin D1 expression in esophageal cancer cells. Further studies should focus on the mechanisms linking TGIF and phospho-Rb in esophageal cancer.
Previous studies have shown that wnt/β-catenin pathway is involved in the development of esophageal cancer (29,30) and β-catenin is the key regulator in the wnt/β-catenin signaling pathway. Deng et al reported that aberrant expression of β-catenin was identified in 54.3% (114 of 265) of ESCC (31). The level of β-catenin expression in ESCC was significantly higher than that in the adjacent non-cancerous tissues (32,33). The overexpression of β-catenin was aggressively associated with lymph node metastasis, advanced pathological stage and prognosis of the patients with ESCC (32). In addition, Xu and Lu reported that β-catenin was involved in miR-214 inhibiting esophageal cancer cell growth and invasion (33). Jia et al found that RAP1B activated wnt/β-catenin signaling in ESCC (34). However, in this present study, we found that knockdown of TGIF had no obvious effects on the expression of β-catenin and Axin1 proteins in esophageal cancer cells, which suggests that the wnt/β-catenin signaling pathway might not be involved in knockdown of TGIF inhibiting the tumorigenicity of esophageal cancer cells. Previous studies showed that TGIF could regulate the expression of β-catenin protein in breast cancer (9) and lung cancer (10,12). Taken together, the regulation of β-catenin by TGIF might be dependent on tumor types.
In this study, we observed that knockdown of TGIF suppressed the tumorigenicity of esophageal cancer cell of EC109 and cisplatin could repress the expression of TGIF protein. We further investigated the potential role of TGIF in cisplatin-induced apoptosis of EC109 cells. Our data showed that knockdown of TGIF promoted cisplatin-induced apoptosis of EC109 cells, along with the alterations of apoptosis-related markers, such as the decreased level of full length PARP protein expression and the increased level of cleaved caspase-3 protein expression. Studies suggested the cleavage of caspase-3 was an early event in apoptosis induced by chemotherapeutic agents (35). Activation of caspase-3 was partially or totally responsible for proteolytic cleavage of many key proteins such as PARP (36,37). Liu et al reported that knockdown of TGIF enhanced arsenic trioxide-induced apoptosis in HepG2 cells (38). Together, our findings indicated that TGIF was likely to be a potential therapeutic target for the treatment of esophageal cancer.
To the best of our knowledge, only one published study reported the association of TGIF amplifications with esophageal cancer (22). Although, in this study, we primarily obtained exciting data on the potential role of TGIF in the proliferation and tumorigenicity of esophageal cancer cells, some limitations should be acknowledged. First, our data are only based on one esophageal cancer cell line, other cell lines should be applied to verify our findings. Second, the pattern of TGIF expression in esophageal cancer tissue should be investigated in future studies. Third, the effects of TGIF overexpression on the proliferation and tumorigenicity in esophageal cancer cells should be addressed further. In addition, transgenic animal model could be applied to assess the functional role of TGIF in esophageal tumorigenesis.
In conclusion, the present study indicates that knockdown of TGIF induces growth inhibition of EC109 cells via arresting cell cycle in the G1 phase by downregulating phospho-Rb. This study also indicates that TGIF plays an important role in modulating the tumorigenicity of EC109 cells and cisplatin-induced apoptosis. Therefore, this study enriches our understanding of the oncogenesis of esophageal cancer and suggests that TGIF is likely to be a new therapeutic target for esophageal cancer treatment.
Acknowledgements
This study was supported by a grant from National Natural Science Foundation of China (no. U1404815). The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Xie SH, Wahlin K, Lagergren J. Cause of death in patients diagnosed with esophageal cancer in Sweden: A population-based study. Oncotarget. 2017;8:51800–51809. doi: 10.18632/oncotarget.15270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 3.Wang M, Hao C, Ma Q, Song G, Ma S, Zhao D, Zhao L, Li X, Wei W. DNA image cytometry test for primary screening of esophageal cancer: A population-based multi-center study in high-risk areas in China. Chin J Cancer Res. 2016;28:404–412. doi: 10.21147/j.issn.1000-9604.2016.04.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sohda M, Kuwano H. Current status and future prospects for esophageal cancer treatment. Ann Thorac Cardiovasc Surg. 2017;23:1–11. doi: 10.5761/atcs.ra.16-00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Song G, Liu K, Yang X, Mu B, Yang J, He L, Hu X, Li Q, Zhao Y, Cai X, Feng G. SATB1 plays an oncogenic role in esophageal cancer by up-regulation of FN1 and PDGFRB. Oncotarget. 2017;8:17771–17784. doi: 10.18632/oncotarget.14849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bertolino E, Reimund B, Wildt-Perinic D, Clerc RG. A novel homeobox protein which recognizes a TGT core and functionally interferes with a retinoid-responsive motif. J Biol Chem. 1995;270:31178–31188. doi: 10.1074/jbc.270.52.31178. [DOI] [PubMed] [Google Scholar]
- 7.Gripp KW, Wotton D, Edwards MC, Roessler E, Ades L, Meinecke P, Richieri-Costa A, Zackai EH, Massagué J, Muenke M, Elledge SJ. Mutations in TGIF cause holoprosencephaly and link NODAL signalling to human neural axis determination. Nat Genet. 2000;25:205–208. doi: 10.1038/76074. [DOI] [PubMed] [Google Scholar]
- 8.Wotton D, Lo RS, Lee S, Massagué J. A Smad transcriptional corepressor. Cell. 1999;97:29–39. doi: 10.1016/S0092-8674(00)80712-6. [DOI] [PubMed] [Google Scholar]
- 9.Zhang MZ, Ferrigno O, Wang Z, Ohnishi M, Prunier C, Levy L, Razzaque M, Horne WC, Romero D, Tzivion G, et al. TGIF governs a feed-forward network that empowers Wnt signaling to drive mammary tumorigenesis. Cancer Cell. 2015;27:547–560. doi: 10.1016/j.ccell.2015.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Y, Pan T, Wang H, Li L, Li J, Zhang C, Yang H. Silencing of TGIF attenuates the tumorigenicity of A549 cells in vitro and in vivo. Tumour Biol. 2016;37:12725–12730. doi: 10.1007/s13277-016-5222-8. [DOI] [PubMed] [Google Scholar]
- 11.Wang Y, Wang H, Gao H, Xu B, Zhai W, Li J, Zhang C. Elevated expression of TGIF is involved in lung carcinogenesis. Tumour Biol. 2015;36:9223–9231. doi: 10.1007/s13277-015-3615-8. [DOI] [PubMed] [Google Scholar]
- 12.Xiang G, Yi Y, Weiwei H, Weiming W. TGIF1 promoted the growth and migration of cancer cells in nonsmall cell lung cancer. Tumour Biol. 2015;36:9303–9310. doi: 10.1007/s13277-015-3676-8. [DOI] [PubMed] [Google Scholar]
- 13.Borlak J, Meier T, Halter R, Spanel R, Spanel-Borowski K. Epidermal growth factor-induced hepatocellular carcinoma: Gene expression profiles in precursor lesions, early stage and solitary tumours. Oncogene. 2005;24:1809–1819. doi: 10.1038/sj.onc.1208196. [DOI] [PubMed] [Google Scholar]
- 14.Liu ZM, Tseng HY, Tsai HW, Su FC, Huang HS. Transforming growth factor β-interacting factor-induced malignant progression of hepatocellular carcinoma cells depends on superoxide production from Nox4. Free Radic Biol Med. 2015;84:54–64. doi: 10.1016/j.freeradbiomed.2015.03.028. [DOI] [PubMed] [Google Scholar]
- 15.Huang HS, Liu ZM, Chen PC, Tseng HY, Yeh BW. TG-interacting factor-induced superoxide production from NADPH oxidase contributes to the migration/invasion of urothelial carcinoma. Free Radic Biol Med. 2012;53:769–778. doi: 10.1016/j.freeradbiomed.2012.06.014. [DOI] [PubMed] [Google Scholar]
- 16.Yeh BW, Wu WJ, Li WM, Li CC, Huang CN, Kang WY, Liu ZM, Huang HS. Overexpression of TG-interacting factor is associated with worse prognosis in upper urinary tract urothelial carcinoma. Am J Pathol. 2012;181:1044–1055. doi: 10.1016/j.ajpath.2012.05.024. [DOI] [PubMed] [Google Scholar]
- 17.Hamid R, Brandt SJ. Transforming growth-interacting factor (TGIF) regulates proliferation and differentiation of human myeloid leukemia cells. Mol Oncol. 2009;3:451–463. doi: 10.1016/j.molonc.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hamid R, Patterson J, Brandt SJ. Genomic structure, alternative splicing and expression of TG-interacting factor, in human myeloid leukemia blasts and cell lines. Biochim Biophys Acta. 2008;1779:347–355. doi: 10.1016/j.bbagrm.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 19.Willer A, Jakobsen JS, Ohlsson E, Rapin N, Waage J, Billing M, Bullinger L, Karlsson S, Porse BT. TGIF1 is a negative regulator of MLL-rearranged acute myeloid leukemia. Leukemia. 2015;29:1018–1031. doi: 10.1038/leu.2014.307. [DOI] [PubMed] [Google Scholar]
- 20.Libório TN, Ferreira EN, Xavier Aquino FC, Carraro DM, Kowalski LP, Soares FA, Nunes FD. TGIF1 splicing variant 8 is overexpressed in oral squamous cell carcinoma and is related to pathologic and clinical behavior. Oral Surg Oral Med Oral Pathol Oral Radiol. 2013;116:614–625. doi: 10.1016/j.oooo.2013.07.014. [DOI] [PubMed] [Google Scholar]
- 21.Matizonkas-Antonio LF, Libório TN, Xavier Aquino FC, Silva-Valenzuela Md, Michaluarte-Júnior P, Nunes FD. Detection of TGIF1 homeobox gene in oral squamous cell carcinoma according to histologic grading. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2011;111:218–224. doi: 10.1016/j.tripleo.2010.10.003. [DOI] [PubMed] [Google Scholar]
- 22.Nakakuki K, Imoto I, Pimkhaokham A, Fukuda Y, Shimada Y, Imamura M, Amagasa T, Inazawa J. Novel targets for the 18p11.3 amplification frequently observed in esophageal squamous cell carcinomas. Carcinogenesis. 2002;23:19–24. doi: 10.1093/carcin/23.1.19. [DOI] [PubMed] [Google Scholar]
- 23.Chan AS, Mowla SN, Arora P, Jat PS. Tumour suppressors and cellular senescence. IUBMB Life. 2012;66:812–822. doi: 10.1002/iub.1335. [DOI] [PubMed] [Google Scholar]
- 24.López-Sáez JF, de la Torre C, Pincheira J, Giménez-Martin G. Cell proliferation and cancer. Histol Histopathol. 1998;13:1197–1214. doi: 10.14670/HH-13.1197. [DOI] [PubMed] [Google Scholar]
- 25.Reed SI, Bailly E, Dulic V, Hengst L, Resnitzky D, Slingerland J. G1 control in mammalian cells. J Cell Sci Suppl. 1994;18:69–73. doi: 10.1242/jcs.1994.Supplement_18.10. [DOI] [PubMed] [Google Scholar]
- 26.Reed SI. Control of the G1/S transition. Cancer Surv. 1997;29:7–23. [PubMed] [Google Scholar]
- 27.Sherr CJ. Mammalian G1 cyclins. Cell. 1993;73:1059–1065. doi: 10.1016/0092-8674(93)90636-5. [DOI] [PubMed] [Google Scholar]
- 28.Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- 29.Zhang M, Linghu E, Zhan Q, He T, Cao B, Brock MV, Herman JG, Xiang R, Guo M. Methylation of DACT2 accelerates esophageal cancer development by activating Wnt signaling. Oncotarget. 2016;7:17957–17969. doi: 10.18632/oncotarget.7647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ge C, Wu S, Wang W, Liu Z, Zhang J, Wang Z, Li R, Zhang Z, Li Z, Dong S, et al. miR-942 promotes cancer stem cell-like traits in esophageal squamous cell carcinoma through activation of Wnt/β-catenin signalling pathway. Oncotarget. 2015;6:10964–10977. doi: 10.18632/oncotarget.3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deng F, Zhou K, Cui W, Liu D, Ma Y. Clinicopathological significance of wnt/β-catenin signaling pathway in esophageal squamous cell carcinoma. Int J Clin Exp Pathol. 2015;8:3045–3053. [PMC free article] [PubMed] [Google Scholar]
- 32.Lv J, Cao XF, Ji L, Zhu B, Wang DD, Tao L, Li SQ. Association of β-catenin, Wnt1, Smad4, Hoxa9, and Bmi-1 with the prognosis of esophageal squamous cell carcinoma. Med Oncol. 2012;29:151–160. doi: 10.1007/s12032-010-9816-5. [DOI] [PubMed] [Google Scholar]
- 33.Xu Y, Lu S. Regulation of β-catenin-mediated esophageal cancer growth and invasion by miR-214. Am J Transl Res. 2015;7:2316–2325. [PMC free article] [PubMed] [Google Scholar]
- 34.Jia Z, Yang Y, Dengyan Z, Chunyang Z, Donglei L, Kai W, Song Z. RAP1B, a DVL2 binding protein, activates Wnt/beta-catenin signaling in esophageal squamous cell carcinoma. Gene. 2017;611:15–20. doi: 10.1016/j.gene.2017.01.021. [DOI] [PubMed] [Google Scholar]
- 35.Henkels KM, Turchi JJ. Cisplatin-induced apoptosis proceeds by caspase-3-dependent and -independent pathways in cisplatin-resistant and -sensitive human ovarian cancer cell lines. Cancer Res. 1999;59:3077–3083. [PubMed] [Google Scholar]
- 36.Horky M, Wurzer G, Kotala V, Anton M, Vojtĕsek B, Vácha J, Wesierska-Gadek J. Segregation of nucleolar components coincides with caspase-3 activation in cisplatin-treated HeLa cells. J Cell Sci. 2001;114:663–670. doi: 10.1242/jcs.114.4.663. [DOI] [PubMed] [Google Scholar]
- 37.Rheaume E, Cohen LY, Uhlmann F, Lazure C, Alam A, Hurwitz J, Sekaly RP, Denis F. The large subunit of replication factor C is a substrate for caspase-3 in vitro and is cleaved by a caspase-3-like protease during Fas-mediated apoptosis. EMBO J. 1997;16:6346–6354. doi: 10.1093/emboj/16.21.6346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu ZM, Tseng JT, Hong DY, Huang HS. Suppression of TG-interacting factor sensitizes arsenic trioxide-induced apoptosis in human hepatocellular carcinoma cells. Biochem J. 2011;438:349–358. doi: 10.1042/BJ20101653. [DOI] [PubMed] [Google Scholar]