

Abstract
Butyrophilin 3A1 (BTN3A1) binds small phosphorous-containing molecules, which initiates transmembrane signaling and activates butyrophilin-responsive cells. We synthesized several phosphinophosphonates and their corresponding tris-pivaloyloxymethyl prodrugs and examined their effects on BTN3A1. An analog of (E)-4-hydroxy-3-methyl-but-2-enyl diphosphate (HMBPP) bound to BTN3A1 with intermediate affinity, which was enthalpy-driven. Docking studies revealed binding to the basic surface pocket and interactions between the allylic hydroxyl group and the BTN3A1 backbone. The phosphinophosphonate stimulated proliferation of Vγ9Vδ2 T cells with moderate activity (EC50 = 26 µM). Cellular potency was enhanced >600-fold in the tris-POM prodrug (EC50 = 0.041 µM). The novel prodrug also induced T cell mediated leukemia cell lysis. Analysis of dose response data reveals HMBPP-induced Hill coefficients of 0.69 for target cell lysis and 0.68 in interferon secretion. Together, tris-POM prodrugs enhance the cellular activity of phosphinophosphonates, reveal structure-activity relationships of butyrophilin ligands, and support a negatively cooperative model of cellular butyrophilin activation.
Graphical Abstract
Introduction
The transmembrane B7 family proteins play critical roles in activation of T cell mediated immune responses.1 The lead members of this family, B7.1 (CD80) and B7.2 (CD86) are ligands of the CD28 receptor that provide requisite co-signaling to allow antigen driven activation of CD4+ and CD8+ cells.2 More recently, the B7 protein PD-L1 has been recognized as inhibitory to T cell activation, and this has led to development of immunotherapies known as “checkpoint inhibitors”, which block the PD-L1 and PD1 interaction and allow for enhanced T cell activation in patients.3, 4 In contrast to these canonical B7 proteins, the butyrophilins are more distantly related and more diverse in their structures.5 Surprisingly, no extracellular butyrophilin ligands have been conclusively identified.
We were among several groups to determine that small molecule activators of gamma delta (γδ) T cells, such as HMBPP (1, Figure 1), are ligands of the internal domain of the transmembrane butyrophilin 3 isoform (BTN3A1).6-11 HMBPP is an essential intermediate of isoprenoid metabolism in organisms that utilize the 2-C-methyl-D-erythritol 4-phosphate pathway, such as microbial pathogens,12 implying that BTN3A1 functions in the human immune response to pathogens. Additionally, BTN3A1 interacts with some intermediates of human isoprenoid metabolism, such as IPP, which may play a role in the human immune response to cancer.13, 14 The precise functions of BTN3A1 in response to these intermediates remains unclear.15
Figure 1.
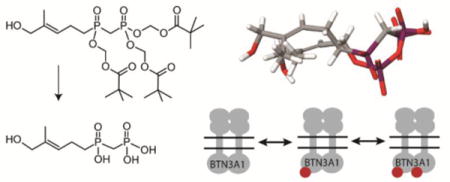
A natural phosphoantigen and some synthetic analogues. HMBPP (1) is the most potent naturally-occurring agonist of BTN3A1. Compound (2) is a bis-POM prodrug which enhances the cellular potency of monophosphonate 3. The novel prodrug 4 (POM3-CC-HMBPP), the focus of the present study, was designed to release a phosphoantigen that more closely mimics HMBPP.
Our previous studies,10 as well as those of Sandstrom9 and Rhodes,16 have demonstrated binding of phosphoantigens to the internal B30.2 domain of BTN3A1. Specifically, Sandstrom’s paper elegantly pinpointed the binding site of the phosphoantigen diphosphate group to a specific basic region of the BTN3A1 B30.2 domain by solving a crystal structure of the complex, while our NMR and isothermal titration calorimetry (ITC) studies support similar interactions in solution. Still, it remains unclear how ligand binding to BTN3A1 leads to T cell activation. Multiple models have been put forward, ranging from a heterotrimeric model17 to a homodimeric model.18 Because there are only two naturally occurring phosphoantigens known at this time, and a minimal number of synthetic analogs,19, 20 further structure activity analysis would likely reveal new information about the biological mechanisms that underlie BTN3A1 activation and its unique inside-out signaling to the T cell receptor.
Our approach in this area has focused on studies of bis-pivaloyloxymethyl (POM) prodrugs of mono-phosphonate HMBPP analogs,10, 21 especially the phosphonate diester 2. Such prodrugs exhibit rapid cellular internalization and subsequent hydrolysis, leading to excellent gains in cellular potency compared to their unprotected and charged analogs (e.g. 3). Here, we set out to test the hypothesis that tris-POM prodrugs of phosphinophosphonate analogs (e.g. 4, which more closely resembles the potent ligand HMBPP) would function as potent phosphoantigens and reveal new insights of the activation of BTN3A1.
Results
Synthesis of phosphinophosphonate analogs of HMBPP and their tris-POM prodrugs
We chose to pursue synthesis of phosphinophosphonates due to their close resemblance to the naturally-occurring and highly potent phosphoantigen HMBPP. Previously, synthesis of compound 8 was reported from a phosphonylphosphonite,22 while compound 10 was once claimed to have moderate phosphoantigen activity.23 Our chemical synthesis of these HMBPP analogs is summarized in Scheme 1. Initially dimethyl homoprenylphosphonate 5 was treated with 1,4-diazabicyclo[2.2.2]octane to yield the monomethyl ester as an ammonium salt, which in turn could be converted to the acid chloride 6 upon reaction with oxalyl chloride. However recent literature24 indicated that dimethyl methylphosphonate could be easily converted to the corresponding acid chloride when allowed to react directly with oxalyl chloride and catalytic dimethylformamide at ambient temperature, and this strategy appeared more straightforward. In fact, treatment of dimethyl homoprenylphosphonate 5 with oxalyl chloride readily afforded the desired phosphonic acid chloride 6 which was used without further purification. Dimethyl methylphosphonate then was allowed to react with n-butyl lithium. After formation of the corresponding anion, addition of the acid chloride 6 afforded the desired phosphinophosphonate 7 in moderate yield. The identity of the phosphinophosphonate 7 was confirmed through analysis of the 13C NMR spectrum, where the central carbon showed splitting by two different phosphorus atoms (J’s of ~136 Hz and 76 Hz).
Scheme 1.
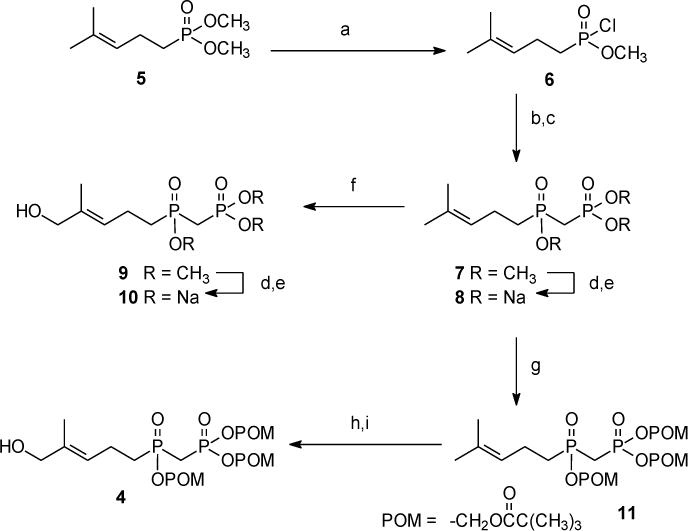
a Reagents and conditions: (a) (COCl)2, DMF (cat.), DCM; (b) n-BuLi, (H3CO)2P(O)CH3; (c) 6, toluene, –78 °C (58% overall); (d) TMSBr; (e) NaOH (91% overall); (f) SeO2, t-BuOOH, 4-HBA, DCM, (10%) (g) POMCl, NaI, ACN, reflux (48%); (h) SeO2, t-BuOOH, DCM; (i) NaBH4, CH3OH (51% overall).
The trimethyl ester 7 served as the central intermediate for further synthesis. The trisodium salt 8 was made available by hydrolysis utilizing conditions reported by McKenna, which involve the use of trimethylsilyl bromide and collidine, followed by treatment with NaOH.25 Traditionally isolation of the sodium salt is achieved through precipitation from aqueous solution by addition of anhydrous acetone.26 However, when this approach was attempted with compound 8, no solid was observed. Similar conditions reported by Boёdec were more rewarding.27 The desired trisodium salt 8 was isolated by dissolving the initial solid in a minimum amount of water. After addition of an anhydrous mixture of isopropyl alcohol and acetonitrile (50/50), a precipitate formed which was easily removed through filtration, and the desired product was recovered from the filtrate.
Allylic oxidation of compound 7 with selenium dioxide gave the expected alcohol 9. Although this compound was isolated in a disappointing yield, sufficient material was obtained to conduct a parallel hydrolysis and obtain the salt 10, again using the modified isolation strategy.27 Finally, treatment of compound 7 with NaI and POMCl afforded the masked phosphinophosphonate 11 as the only isolable product. The POM triester 11 then was treated with SeO2 and tBuOOH to provide the final target compound, the desired E-olefin 4, after a NaBH4 treatment to reduce any aldehyde formed by over-oxidation. Analysis by high performance liquid chromatography and high performance liquid chromatography-mass spectrometry suggests that compound 4 undergoes some minor isomerization in aqueous acetonitrile, which we believe involves transfer of a POM group to the allylic alcohol, but metabolic cleavage of the POM groups from such an isomer would release the same drug moiety.
Phosphinophosphonates and their prodrugs promote expansion of Vγ9Vδ2 T cells
The phosphinophosphonates were evaluated for their ability to trigger proliferation of Vγ9Vδ2 T cells from human peripheral blood (Figure 2). Peripheral blood mononuclear cells were exposed to the test compounds for 72 hours, allowed to grow for 11 days, and the percentage of Vγ9Vδ2 cells at the end of the experiment was assessed by flow cytometry. At a concentration of 0.1 µM, HMBPP, phosphonate 2, and phosphinophosphonate 4 all strongly stimulated proliferation of Vγ9Vδ2 T cells as determined by the appearance of a population that stained positive for both CD3 and the γδ T cell receptor (TCR) (Figure 2A). To determine the potency of the compounds, we performed dose response experiments (Figure 2B, Table 1). Salt 10 stimulated Vγ9Vδ2 T cell proliferation with an EC50 of 26 µM, while no stimulation was observed with compound 8 at concentrations up to 100 µM. As expected, the methyl-protected analog 9 also was inactive up to 100 µM. Importantly, the tris-POM prodrug compound 4, which is expected to release compound 10 upon cellular metabolism, was able to stimulate T cell proliferation with an EC50 of 0.041 µM, providing a 630x increase in potency relative to the unprotected salt 10. Compound 4 exhibited toxicity towards the T cells at concentrations of 10 µM and above (Figure 2B), leading to a selectivity ratio of 85x. No measurable agonist activity was observed in this assay upon treatment with compound 11, which also displayed direct toxicity at similar concentrations to compound 10. Taken together, this data demonstrates that the phosphinate phosphonate analog of HMBPP displays intermediate agonist activity and it tris-POM prodrug 4 dramatically enhances cellular activity.
Figure 2.
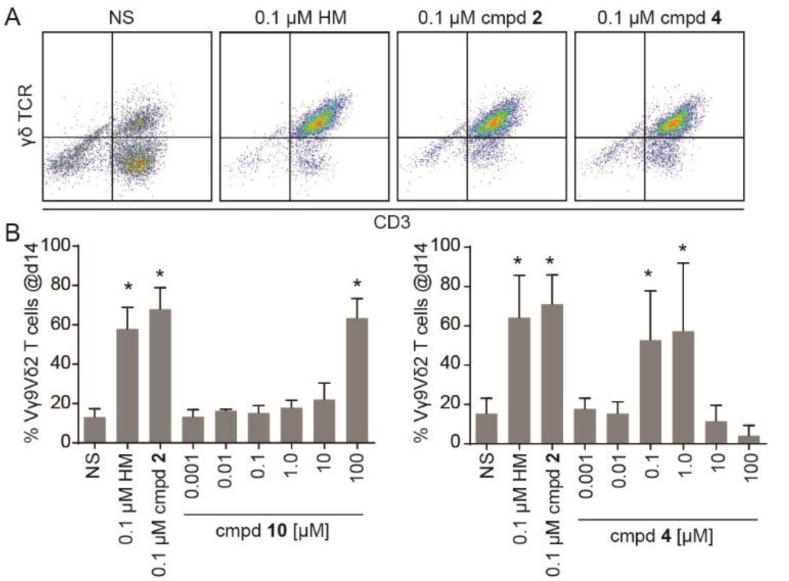
Compound 4 is a potent phosphoantigen. A) Human peripheral blood mononuclear cells were treated with the indicated test compounds, cultured, and analyzed for the presence of CD3+/γδ+ T cells by flow cytometry. Representative data is shown that compares unstimulated cells (NS) that received interleukin 2 versus cells treated with interleukin 2 plus compound 4 for 3 days at 0.1 µM. HMBPP (HM, 1) and compound 2 were used as positive controls. B) Dose response of compound 10. C) Dose response of compound 4. All bars represent mean and standard deviations, n=3. *p < 0.05, ANOVA with Tukey’s post-hoc analysis.
Table 1.
Activity of compounds for expansion of Vγ9Vδ2 T cells from peripheral blood.
| Compound | EC50 (µM) | Fold difference from salt | Fold difference from HMBPP | IC50 (µM) | Selectivity index | Ref |
|---|---|---|---|---|---|---|
| HMBPP (1) | 0.00051 | NA | 1x | 0.5 | 980 | 10 |
| C-HMBP (3) | 4 | NA | 0.00013x | ND | ND | 10 |
| POM2-C-HMBP (2) | 0.0054 | 740x (versus 3) | 0.094x | 0.60 | 110 | 10 |
| 8 | >100 | NA | <0.0000051x | >100 | NA | – |
| 11 | >100 | NA | <0.0000051x | >100 | NA | – |
| CC-HMBPP (10) | 26 | NA | 0.000020x | >100 | >3.8 | – |
| POM3-CC-HMBPP (4) | 0.041 | 630x (versus 10) | 0.012x | 3.5 | 85 | – |
| 9 | >100 | NA | <0.0000051x | >100 | NA | – |
Phosphinophosphonate anions bind to BTN3A1
Phosphoantigens such as HMBPP stimulate Vγ9Vδ2 T cell proliferation by binding to the intracellular domain of BTN3A1. In order to directly assess target binding, we used isothermal titration calorimetry to examine the interaction of compound 10 with the intracellular domain of BTN3A1 (Figure 3A). Similar to HMBPP, the interaction of compound 10 with BTN3A1 was primarily enthalpy driven, with an average ΔH of −74.7 kJ/mol, and a negative entropy contribution (TΔS = −52 kJ/mol) (Table 2). In comparison to HMBPP, the enthalpy of interaction of compound 10 is more favorable. However, changes in the unfavorable entropy term were even larger, leading to a lower affinity binding with the Kd of compound 10 being 111 µM. Therefore, substitution of the central diphosphate oxygen with a carbon atom results in a weaker ligand binding relative to C-HMBPP with known Kd of 1.9 µM, where favorable enthalpy changes are overweighed by detrimental changes in entropy. Still, the binding of compound 10 is stronger than that observed for the natural ligand IPP. Thus, this finding suggests that the presence of the allylic oxygen plays a significant role in the interaction. Additionally, the tris-POM prodrug 4 did not bind to purified BTN3A1 in its prodrug form, further demonstrating that this compound must undergo cellular removal of the phosphonate protecting groups to become active.
Figure 3.
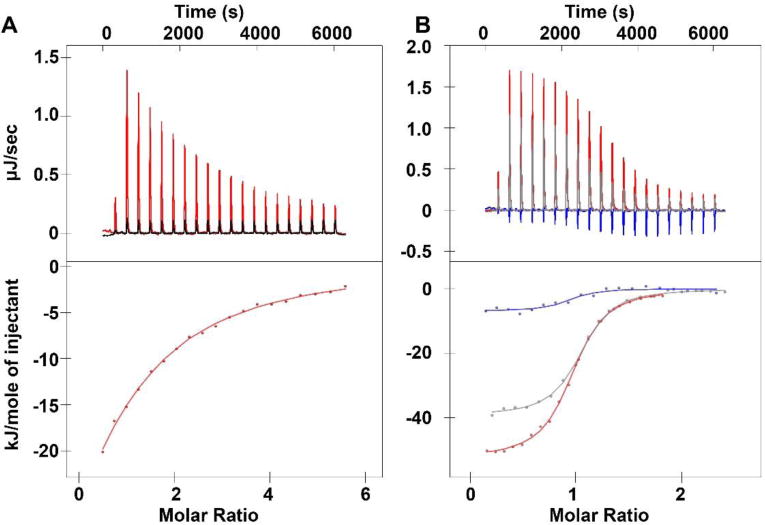
Binding of compounds 8 and 10 to the intracellular domain of BTN3A1. (A) Isothermal titration calorimetry plots for the interaction of compound 10 (red line/dots) or buffer (black line) with the full intracellular domain of BTN3A1. (B) Isothermal titration calorimetry plots for the interaction of the full intracellular domain with HMBPP (1) in the absence of compound 8 (red line/dots), or the presence of 10x compound 8 (gray line/dots), or 100x compound 8 (blue line/dots).
Table 2.
Thermodynamic measurements of phosphoantigen interactions with BTN3A1.
| Compound 10 [µM] | kd [µM] | ΔH (kJ/mol) | TΔS (kJ/mol) | ΔG (kJ/mol) | n | |
|---|---|---|---|---|---|---|
| Run 1 | 240 | 145 | −72.1 | −50.1 | −21.9 | 0.978 |
| Run 2 | 480 | 106 | −77.8 | −55.1 | −22.7 | 0.995 |
| Run 3 | 688 | 83.1 | −74.1 | −50.8 | −23.3 | 0.944 |
We also assessed binding of compound 8 to BTN3A1 (Figure 3B). The binding of this compound was weaker than that of compound 10, which was expected because the former compound lacks the allylic hydroxyl group. The heat of the interaction was too low to be measured directly by calorimetry. Therefore, we used higher concentrations of compound 8 to compete with the binding of HMBPP to BTN3A1. As we had observed previously, binding of HMBPP to the BTN3A1 full intracellular domain construct occurred with a ΔH of −52 kJ/mol and TΔS of −19 kJ/mol. In the presence of 10-fold higher compound 8, the ΔH of HMBPP binding was reduced to −39 kJ/mol and while TΔS increased to −4.6 kJ/mol. In the presence of 100-fold molar excess of compound 8, the ΔH of HMBPP binding was reduced to −6.9 kJ/mol and while TΔS increased to +28 kJ/mol. Therefore, while compound 8 is not a strong phosphoantigen, it is capable of disrupting HMBPP binding in a dose-dependent manner. Notably, the binding of both compounds 10 and 8 is stronger than that of the naturally occurring weak phosphoantigen IPP. Therefore, both of the phosphinophosphonates can be categorized as moderate BTN3A1 ligands.
Molecular docking of HMBPP and compound 10
In order to understand the basis for the differences in BTN3A1 binding of compound 10 versus HMBPP, we performed a molecular docking analysis using the publically available crystal structure of the BTN3A1 B30.2 domain in complex with C-HMBPP (PDB:4N7U).9 Our docking of HMBPP revealed similar binding of the beta phosphate to the protein (Figure 4A). The binding model predicts a number of hydrogen bond interactions between HMBPP and BTN3A1 R412, R418, and R469 at the beta phosphate (Figure 4B), in excellent agreement with the binding observed with C-HMBPP in the crystal structure. Additionally, the model predicts an interaction between R418 and the HMBPP oxygen atom that links the alpha and beta phosphates. Compound 10 contains a methylene linker in this position, which is incapable of hydrogen bonding to R418, resulting in changes to the predicted binding affinity and binding orientation relative to HMBPP (Figure 4C). In the context of our calorimetry data, it is likely that even though this particular hydrogen bond is lost, the strength of other bonding interactions (Figure 4D) is increased in compound 10 to contribute to the overall favorable enthalpy gains. At the same time, this different binding orientation of compound 10 is entropically unfavorable. The oxygen that links the alkyl chain to the alpha phosphate in HMBPP was not predicted to form hydrogen bonds with the protein, which implies that the oxygen at this position is non-essential, although it may indirectly affect binding through its influence on the pKa values of the phosphorus acids.
Figure 4.
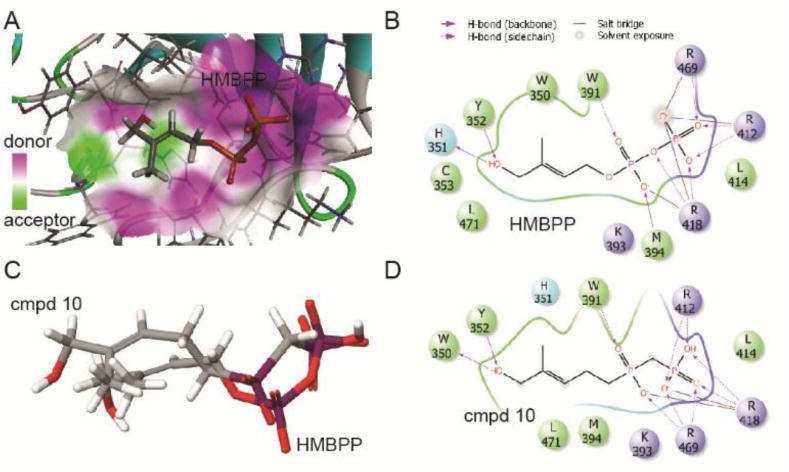
Molecular docking of compound 10 into the B30.2 domain of BTN3A1. A) HMBPP binds to a basic binding pocket on the surface of the BTN3A1 B30.2 domain.9 Surfaces highlighted in purple indicate the presence of hydrogen bond donors, while acceptors are indicated in green. Docking into the published crystal structure (PBD ID: 4N7U) was performed using Schrodinger Glide as described in the methods and rendered using BIOVIA Discovery Studio. B) Best view of HMBPP in binding pocket. HMBPP is predicted to form a variety of molecular interactions with the binding pocket. C) Overlay of HMBPP and compound 10 as oriented in the BTN3A1 intracellular binding pocket. D) Best view of compound 10 in the binding pocket. Rendering was performed using Schrodinger Maestro.
The original crystal structure of the protein ligand complex was unable to determine the orientation of the HMBPP alkyl chain due to poor resolution of the electron density in that part of the ligand.9 However, when we docked HMBPP, we observed that the allylic oxygen was projected to act as a hydrogen bond acceptor from Y352 and a hydrogen bond donor to the backbone of H351 (Figure 4B). The H351 backbone interaction is notable, because this residue is an arginine in the BTN3A3 isoform, which is unable to bind phosphoantigens. Compound 10 maintains the interaction with the backbone near Y352, but because the orientation of compound 10 is altered, its allylic hydroxyl group is predicted to interact with the backbone at W350 rather than H351 (Figure 4D). Taken together, the major difference in binding of compound 10 relative to HMBPP appears to be an entropically unfavorable change in the orientation of the ligand dependent upon formation of a different set of hydrogen bonds brought about by the phosphinophosphonate versus the central oxygen of the diphosphate.
Phosphinophosphonates and their prodrugs promote Vγ9Vδ2 T cell mediated lysis of K562 cells
We next assessed whether the compounds could trigger Vγ9Vδ2 T effector cells to lyse K562 target cells. Here, the K562 cells were exposed to the compounds for two hours, washed, and mixed with T cells for four hours to allow lysis to occur. As expected, the control HMBPP triggered lysis in a dose-dependent manner with an EC50 of 0.0016 µM (Table 3). It should be noted this value is higher than other values reported for HMBPP because the two hour exposure does not allow for maximum uptake of charged phosphoantigens.21, 28 In this assay, compound 10 triggered lysis with an EC50 of 41 µM, while its tris-POM analog 4 triggered lysis with a 0.28 µM EC50, representing a 150-fold increase in cellular potency. Compound 11 also demonstrated activity in this assay, with an EC50 of 7.3 µM.
Table 3.
Activity of compounds for cell mediated lysis of and direct cytotoxicity to K562 cells.
| Lysis of K562 cells by T cells (2 hour treatment/4 hour co-culture) | Direct K562 toxicity (72 hour treatment) | |||||
|---|---|---|---|---|---|---|
| Compound | EC50 (µM) | Fold difference from salt | Fold difference from HMBPP | IC50 (µM) | Fold difference from salt | Fold difference from HMBPP |
| HMBPP (1) | 0.0016 | NA | 1x | >100 | NA | 1x |
| 8 | >100 | NA | <0.000016x | >100 | NA | NA |
| 11 | 7.3 | >14x | 0.00022x | 9.7 | >10x | >10x |
| CC-HMBPP (10) | 41 | NA | 0.000039x | >100 | NA | NA |
| POM3-CC-HMBPP (4) | 0.28 | 146x | 0.0057x | 3.4 | >29x | >29x |
| 9 | >100 | NA | <0.000016x | >100 | NA | NA |
It was surprising that compound 11 was active in the lysis assay but not active in the T cell expansion assay. We hypothesized that this difference was due to direct toxicity of the compound, which at 72 hours of exposure was sufficient to mask its activity as a phosphoantigen, while at the shorter exposure times utilized in the lysis assays, direct toxicity was less likely to be an issue. We tested the ability of the compounds to directly inhibit the proliferation of the target cells (Table 3). We found that compound 4 inhibited K562 cells with a 72 hour IC50 of 3.4 µM, while compound 11 was toxic in a similar range (IC50 = 9.7 µM). Therefore, while compound 11 shows that the allylic hydroxyl functionality is not absolutely required for cellular phosphoantigen activity, the agonist activity of compound 11 is sufficiently weak relative to its direct toxicity to indicate that the allylic hydroxyl group is an important selectivity feature.
Cellular phosphoantigen-induced BTN3A1 activation is negatively cooperative
During these studies, we performed a number of phosphoantigen dose response curves. Because it was surprising that a small difference in phosphoantigen Kd led to a large difference in cellular activity, we analyzed our dose response data using a four parameter non-linear regression which allows for a variable slope, corresponding to the Hill coefficient, in contrast to the usual three parameter analysis in which the Hill coefficient n defaults to 1 (Figure 5). Here, we found that dose response curves of K562 lysis in response to HMBPP were more accurately modeled using a four parameter equation, as the three parameter model gives an R2 value of 0.98 while the four parameter model gives an R2 value of 0.99 and a calculated Hill slope of 0.69 (Figure 5A). Likewise, production of interferon γ in response to HMBPP stimulation also fits the four parameter model (R2 = 0.99) better than the three parameter model (R2 = 0.98). Although there is an order of magnitude difference in cytokine production versus lysis activity, the Hill slope in this assay was 0.68. Taken together, Hill slopes well below 1 were observed in two independent assays of HMBPP induced T cell activation, demonstrating that phosphoantigen induced γδ T cell activation via BTN3A1 is a negatively cooperative event (Figure 6).
Figure 5.
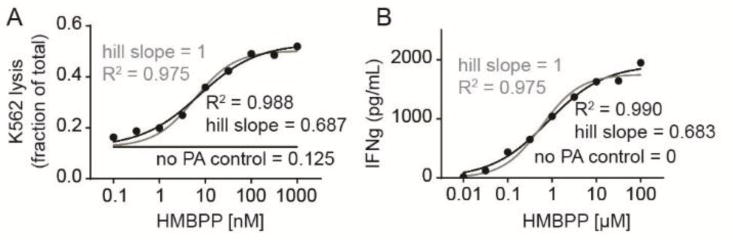
HMBPP-induced K562 cell lysis and interferon production display Hill slopes of less than 1, indicative of negative cooperativity. A) Dose response curve of K562 lysis by γδ T cells in response to various concentrations of HMBPP. Gray line represents a three parameter non-linear regression in which the Hill slope (n) is equal to 1. Black line represents a four parameter non-linear regression in which the Hill slope is calculated. B) Dose response curve of interferon production by γδ T cells in response to K562 cells loaded for 2 hours with HMBPP. Gray line represents a three parameter non-linear regression in which the Hill slope (n) is equal to 1. Black line represents a four parameter non-linear regression in which the Hill slope is calculated. Underlying data in panel B is used with permission.28
Figure 6.
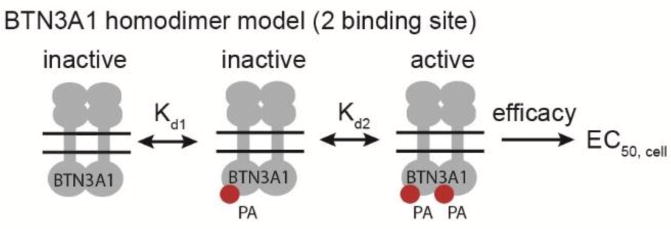
Hypothetical homodimer model of BTN3A1 activation. If BTN3A1 exists as a homodimer, it would contain two ligand binding sites. Cellular activity may be a function of two ligand binding events and their efficacy for activating the γδ TCR. The presence of multiple binding sites would afford the opportunity for cooperative ligand binding. As described in the text, our data is consistent with a model of negative cooperativity.
Discussion and Conclusions
Vγ9Vδ2 T cells have untapped clinical potential as cytotoxic T cells that can kill malignant cells in a way that is inducible by small molecule butyrophilin agonists. However, the mechanisms controlling butyrophilin activation remain largely undefined. Additionally, the most potent naturally occurring BTN3A1 agonist, HMBPP, contains a diphosphate that limits both its stability and its access to the intracellular BTN3A1 binding site.20 We hypothesized herein that tris-POM phosphinophosphonate prodrugs (e.g. 4), which closely mimic the structure of HMBPP, would produce a strong cellular response due to the increased stability of the -C-P-C-P linkage versus the -O-P-O-P substructure found in HMBPP, and the improved cell permeability of the prodrug form. Indeed, we observed that the phosphinophosphonate analog of HMBPP (compound 10) can trigger activation of Vγ9Vδ2 T cells. The tris-POM delivery strategy (compound 4) was even more effective in that it increased cellular proliferation by 630 fold following a 3 day stimulation and it increased cell lysis by 150 fold following a 2 hour exposure. Based on these findings, we conclude that phosphinophosphonates are viable diphosphate analogs in this system, and that tris-POM prodrugs are an effective route to enhance their cellular activity.
The data we presented shows that phosphinophosphonate analogs of HMBPP trigger activation of Vγ9Vδ2 T cells. Chemical synthesis of two novel phosphinophosphonate prodrugs was achieved with acceptable yield and good purity. Agonist activity was observed in two complementary cellular assays (expansion and lysis) with good potencies that fall between those of the natural ligands HMBPP and IPP. Both compound 10 and 8, which are analogs of HMBPP and DMAPP, respectively, bind to the molecular target, BTN3A1 with intermediate affinity. Our negative control, the trimethyl ester 9, neither binds to the protein nor is biologically active, indicating that ligand binding requires an anionic oxygen or is sterically blocked by the methyl groups. These findings are consistent with those of Gossman29 and Reichenberg.30 The novel tris-POM prodrugs of both compounds (4 and 11) effectively deliver both of these compounds into cells, enhancing cellular activity. Importantly, we have noted a 26-fold difference in cellular activity between compound 4 and 11, which only differ by the presence or absence of the allylic hydroxyl group. This implies a critical role for the allylic hydroxyl group in binding to BTN3A1.
Our modeling data suggest that a direct interaction is possible between the allylic hydroxyl group of the phosphoantigen and the backbone near BTN3A1 H351. This interaction was not observed in the crystal structure because of undefined electron density within the phosphoantigen alkyl chain, which in part may have been impacted by a crosslinking step that was necessary to prevent dissolution of the crystals. Thus, while we cannot say with certainty that this interaction occurs, both the H351 residue and the phosphoantigen allylic hydroxyl group are essential for strong phosphoantigen activity, and these two components are close enough in space in the ligand bound complex for an interaction to occur.
We have previously observed that bis-POM prodrugs of mono-phosphonate HMBPP analogs (e.g. compound 2) potently trigger cellular activation of Vγ9Vδ2 T cells.10, 21 However, when drugs are designed to interact with phosphate binding sites, it is often desirable to deliver a diphosphate analog rather than a monophosphate analog to avoid the need for cellular phosphorylation (i.e. kinase bypass).31 Our initial hypothesis in the current studies was that prodrug 4 would exhibit even stronger potency due to its ability to deliver a molecule that more closely mimics the natural ligand, which also contains two phosphorus groups rather than one. Indeed, prodrug 4 also imparts a strong fold increase in activity relative to the unprotected sodium salt 10. However, while stronger BTN3A1 binding was observed for compound 10 relative to compound 3, and the prodrugs gave a desired increase in cellular activity, the activity of both salt 10 and prodrug 4 was still weaker than that of salt 3 and prodrug 2, respectively. Based on this finding, it is possible that C-HMBP (3) may undergo phosphorylation to the phosphonate phosphate (C-HMBPP) in order to achieve its full biological activity. This metabolite would retain its central oxygen atom, allowing it to interact with R418 in a mode more similar to HMBPP.
Interestingly, both prodrug 4 and 11 demonstrated some measurable direct toxicity to the K562 cells, with 72 hour IC50 values of 3.4 and 9.7 μM, respectively. This is in contrast to HMBPP, IPP and prodrug 2, which are not directly toxic, but similar to the nitrogenous bisphosphonates such as zoledronate and risedronate which do exhibit direct toxicity. It remains to be seen whether direct toxicity is a desirable feature in this class of compounds. In the context of cancer chemotherapy, the direct toxicity may be beneficial because it potentially adds a second anticancer mechanism. However, in other applications such as ex vivo expansion for use in cell-based therapies, toxicity of the stimulant may need to be avoided.
Our data clearly shows that tris-POM prodrugs effectively increase cellular activity of phosphinophosphonates in both primary blood cells and a leukemia cell line. This effect is presumed to be a result of metabolism of the prodrug by cellular esterases, which promotes the intracellular accumulation of the charged drug in its free acid form. Because esterases frequently lack substrate specificity, multiple enzymes may be capable of this metabolism. A prior study which examined the disoproxil prodrug of tenofovir found that carboxyesterase rapidly generated the mono-acid form of the drug, while phosphodiesterase was able to convert the mono-acid to the di-acid form.32 More recent studies have specifically pointed to the carboxyesterase isoform hCE2/CES2 in this metabolism,33, 34 though roles of other cellular esterases cannot be ruled out.
While the phosphoantigen activity of compound 10 was weaker than expected, this finding directly leads to a fascinating biological interpretation- that multiple ligand binding events are required for cellular activation of BTN3A1. We believe that our data supports this mechanism because while HMBPP (1) binds to BTN3A1 with 74-fold greater potency than compound 10, the cellular activity of HMBPP is 26000- to 51000-fold greater than compound 10, depending on the assay. In other words, the cellular activity is not directly proportional to ligand affinity as has been proposed.9 Rather, cellular activity is strongly enhanced by small increases to binding affinity, which better fits an exponential relationship (Figure 6). This conclusion is based on the assumption that HMBPP and compound 10 display similar efficacies, which is likely given that the compounds produce the same maximum response (Figure 2). We also assume that HMBPP and compound 10 display similar rates of cellular internalization, which is likely given their similar size and their expected charge in endocytic vesicles.28
As our data suggests (Figure 5), HMBPP induced T cell activation fits more accurately to a four parameter model in which the Hill slope, or cooperatively factor, is allowed to vary.35 The Hill coefficient is less than 1, which provides a second line of evidence that is consistent with a model of negative cooperativity at the cellular level.36 The Hill equation states Y = [L]n/(Kd + [L]n), where Y is the fraction of occupied receptors, [L] is the ligand concentration, Kd is the dissociation constant. Therefore, when n is less than 1, small differences in Kd values would lead to much larger differences in cellular activity. This phenomena is consistent with our observations (e.g. 74x versus 51000x). We propose that this finding is more indicative of a homo-dimerization model of BTN3A1 as described by Palakodeti and Adams37 rather than the heterotrimerization model put forth by Rhodes and Trowsdale17 because the hetero-trimer would only contain one ligand binding site while the homo-dimer would contain two. Further studies in this area are necessary because we cannot say with certainty that the negative cooperativity results from receptor ligand interactions rather than other cellular phenomena including compound metabolism.
It also should be noted that because the cellular potency of HMBPP greatly exceeds the affinity of the receptor for HMBPP binding, cells likely contain a large percentage of “spare” BTN3A1 receptors. Only a small frequency of the BTN3A1 receptor would need to be in the active ligand bound state at any time to trigger a maximal response from the T cell. When combined with the potential for tight control of active BTN3A1 through multi-ligand binding and negative cooperativity, this model is one way to account for both the exquisite sensitivity of Vγ9Vδ2 T cells to HMBPP and the tremendous range of phosphoantigen cellular potencies that covers over 7 orders of magnitude.20
Experimental Section
General Experimental Procedures
Methylene chloride and acetonitrile were distilled from calcium hydride prior to use, while toluene and DMF were dried over molecular sieves. The NaI was oven dried overnight and solutions of n–BuLi were purchased from a commercial source and titrated with diphenylacetic acid prior to use. All other reagents and solvents were purchased from commercial sources and used without further purification. All reactions in non-aqueous solvents were conducted in flame-dried glassware under a positive pressure of argon and with a magnetic stir bar. The NMR spectra were obtained at 300, 400 or 500 MHz for 1H, 75, 100 or 125 MHz for 13C NMR, and 121 or 202 MHz for 31P, in CDCl3 with (CH3)4Si (1H, 0.00 ppm) or CDCl3 (1H, 7.26 ppm; 13C NMR; 77.0 ppm), as the internal standards. The 31P NMR chemical shifts are recorded in ppm relative to 85% H3PO4 (external standard). High-resolution mass spectra were obtained by GC-TOF. Silica gel (60 Å, 0.040 – 0.063 mm) was used for flash column chromatography. Compounds were assessed for purity by HPLC or LC-MS. All compounds that were tested in biological assays met or exceeded 95% purity.
Chemical Synthesis
(E)-(((((5-Hydroxy-4-methylpent-3-en-1-yl)((pivaloyloxy)methoxy)phosphoryl)methyl)phosphoryl)bis(oxy))bis(methylene) bis(2,2-dimethylpropanoate) (4)
Phosphonate 11 (0.35 g, 0.60 mmol), selenium dioxide (50 mg, 0.45 mmol), and a 70% solution of tert-butylhydroperoxide (0.25 mL, 1.80 mmol) were dissolved in dichloromethane (5 mL) and the solution was allowed to react at room temperature for 3 days. The reaction was quenched by addition of brine and extracted with dichloromethane. The combined organic portions were washed with Na2SO3, dried (Na2SO4), and filtered, and the filtrate was concentrated in vacuo. The resulting material was re-dissolved in methanol (5 mL) and allowed to react with sodium borohydride (0.02 g, 0.60 mmol) for 60 minutes at room temperature. This solution was then quenched by addition of aqueous ammonium chloride and extracted with dichloromethane. The organic layer was dried with Na2SO4, filtered over celite, and concentrated in vacuo. The residue was purified by column chromatography (5% MeOH in ether) and the product 4 was isolated as a clear oil in 51% yield (182 mg): 1H NMR (500 MHz, CDCl3) δ 5.77–5.63 (m, 6H), 5.45–5.40 (m, 1H), 3.98 (s, 2H), 3.58 (br s, 1H), 2.69–2.50 (m, 2H), 2.46–2.31 (m, 2H), 2.12–2.02 (m, 2H), 1.67 (s, 3H), 1.23 (s, 27H); 13C NMR (125 MHz, CDCl3) δ 177.0, 176.9 (d, JPC = 4.1 Hz, 2C), 136.8, 123.1 (d, JPC = 14.3 Hz), 81.8 (d, JPC = 5.3 Hz, 2C), 80.9 (d, JPC = 6.3 Hz), 68.2, 38.7–38.6 (m, 3C), 30.0 (d, JPC = 96.7 Hz), 29.1 (dd, JPC = 135.2 Hz, 74.9 Hz), 26.8 (3C), 26.8 (6C), 19.6 (d, JPC = 4.4 Hz), 13.6; 31P NMR (202 MHz, CDCl3) δ +48.4 (d, JPP = 4.0 Hz), +19.3 (d, JPP = 4.0 Hz); HRMS (ES+) calculated for C25H46O12P2 [ M+ + Na] 623.2362; found 623.2372.
Dimethyl ((methoxy(4-methylpent-3-en-1-yl)phosphoryl)methyl)phosphonate (7)
Dimethyl (4-methylpent-3-en-1-yl) phosphonate (5, 0.59 g, 3.11 mmol) and oxalyl chloride (0.80 mL, 9.35 mmol) were dissolved in anhydrous dichloromethane (20 mL) and dimethylformamide (0.02 mL) and the solution was cooled to 0 °C. After the reaction mixture was stirred overnight, the solution was concentrated in vacuo to produce compound 6 and then was used without further purification.24 To a stirred solution of n–BuLi (6.11 mL, 15.2 mmol) in toluene at –78 °C, dimethyl methylphosphonate (1.69 mL, 15.2 mmol) was added dropwise. The resulting solution was allowed to stir for 30 minutes and then acid chloride 6 (3.11 mmol) was added dropwise. The reaction temperature was held at –78 °C for 1 hour and then allowed to warm unassisted while it stirred overnight. The reaction was quenched by addition of aqueous NH4Cl and extracted with dichloromethane. The combined organic portions were dried (MgSO4) and filtered, and the filtrate was concentrated in vacuo. The residue was purified by column chromatography (10% EtOH in hexanes) and the desired product 7 was isolated as a clear oil in 58% yield (0.52 g): 1H NMR (500 MHz, CDCl3) δ 5.12–5.06 (m, 1H), 3.78 (d, JPH = 10.1 Hz, 6H), 3.74 (d, JPH = 11.3 Hz, 3H), 2.44–2.34 (m, 2H), 2.33–2.22 (m, 2H), 1.99–1.90 (m, 2H), 1.66 (s, 3H), 1.61 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 133.1 (d, JPC = 1.4 Hz), 122.6 (d, JPC = 15.7 Hz), 52.9 (t, JPC = 6.2 Hz, 2C), 51.4 (d, JPC = 6.8 Hz), 29.2 (d, JPC = 97.9 Hz), 26.2 (dd, JPC = 135.6, 76.5 Hz), 25.5, 20.1 (d, JPC = 4.2 Hz), 17.6; 31P NMR (202 MHz, CDCl3) δ +48.2 (d, JPP = 5.0 Hz), +22.8 (d, JPP = 5.0 Hz); HRMS (ES+) calculated for C10H23O5P2 [M+ +H] 285.1021; found 285.1021.
Sodium (((4-methylpent-3-en-1-yl)oxidophosphoryl)methyl)phosphonate (8)
To a solution of 2, 4, 6-collidine (0.17 mL, 1.31 mmol) in dichloromethane at 0° C was added trimethylsilyl bromide (0.26 mL, 1.96 mmol) and phosphonate 7 (92 mg, 0.32 mmol) and the solution was allowed to stir overnight. The volatiles were removed and toluene was added and then removed in vacuo. The resulting residue was treated with NaOH (3 M, 0.33 mL, 0.98 mmol) and the solution was stirred overnight. The reaction mixture was dried on a lyophilizer to obtain a residue which was dissolved in a small amount of water and precipitated by addition of a 2-propanol/acetonitrile mixture (1:1). The mother liquor was concentrated in vacuo to give the desired product 8 as a white solid in 91% yield (91 mg): 1H NMR (400 MHz, D2O) δ 5.31–5.24 (m, 1H), 2.20 (br s, 2H), 2.00– 1.82(m, 2H), 1.80–1.67 (m, 5H), 1.65 (s, 3H); 13C NMR (100 MHz, D2O) δ 133.2, 125.1 (d, JPC = 14.3 Hz), 32.2–30.0(m), 24.8, 24.2 (d, JPC = 111.0 Hz), 20.6, 16.9; 31P NMR (161 MHz, D2O) δ +40.4 (s) +12.6 (s); HRMS (ES-) calculated for C7H15O5P2 [M- - H] 241.0395; found 241.0404.
Dimethyl (E)-(((5-hydroxy-4-methylpent-3-en-1-yl)(methoxy)phosphoryl)methyl)phosphonate (9)
Phosphonate 7 (0.47 g, 1.66 mmol), selenium dioxide (0.14 g, 1.24 mmol), and tert-butylhydroperoxide (1.07 mL, 6.64 mmol) were dissolved in dichloromethane (10 mL) and the solution was allowed to stir overnight. The reaction was quenched by addition of brine and extracted with diethyl ether. The combined organic portions were washed with Na2S2O3, dried (Na2SO4), and filtered, and the filtrate was concentrated in vacuo. The resulting oil was added to a solution of NaBH4 (0.13 g, 3.31 mmol) in methanol (5 mL) at room temperature. After 2 hours, the reaction was concentrated in vacuo and then quenched by addition of NH4Cl and extracted with ether. The combined organic portions were dried (MgSO4) and filtered, and the filtrate was concentrated in vacuo. The residue was purified by column chromatography (20% EtOH in hexanes) and the product 9 was isolated as a clear oil in 10% yield (42 mg). 1H NMR (400 MHz, CDCl3) δ 5.48–5.42 (m, 1H), 3.99 (s, 2H), 3.82 (d, JPH = 11.4 Hz, 3H), 3.81 (d, JPH = 11.4 Hz, 3H), 3.76 (d, JPH = 11.0 Hz, 3H), 2.48–2.34 (m, 2H), 2.09–1.98 (m, 2H), 1.69 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 136.5 (d, JPC = 1.1 Hz), 123.5 (d, JPC = 14.1 Hz), 68.1, 53.0 (d, JPC = 6.6 Hz, 2C), 51.4 (d, JPC = 6.9 Hz), 28.9 (d, JPC = 98.3 Hz), 26.3 (dd, JPC = 135.5, 75.9 Hz), 19.8 (d, JPC = 4.2 Hz), 13.6; 31P NMR (161 MHz, CDCl3) δ +48.1 (d, JPP = 4.4 Hz, 1P), +22.7 (d, JPP = 4.4 Hz, 1P); HRMS (ES+) calculated for C10H22O6P2 [ M+ + Na] 323.0789; found 323.0787.
Sodium (E)-(((5-hydroxy-4-methylpent-3-en-1-yl)oxidophosphoryl)methyl)phosphonate (10)
To a solution of 2, 4, 6-collidine (36 μL, 0.27 mmol) in dichloromethane at 0° C was added trimethylsilyl bromide (54 μL, 0.42 mmol) and phosphonate 9 (21 mg, 0.07 mmol). The solution was allowed to stir overnight at ambient temperature. After the volatiles were removed in vacuo, toluene was added and then removed in vacuo. The resulting residue was treated with NaOH (1M, 36 μL, 0.27 mmol) and the solution was stirred overnight. The reaction mixture was dried on a lyophilizer to obtain a residue which was dissolved in a small amount of water and precipitated by addition of a 2-propanol/acetonitrile mixture (1:1). The mother liquor was concentrated in vacuo to give the desired product 10 as a white solid in 40% yield (9 mg): 1H NMR (500 MHz, D2O) δ 5.56–5.51 (m, 1H), 3.99 (s, 2H), 2.35–2.25 (m, 2H), 2.05–1.95 (m, 2H), 1.85–1.75 (m, 2H), 1.70 (s, 3H); 13C NMR (125 MHz, D2O) δ 133.7, 126.7 (d, JPC = 16.3 Hz), 67.0, 32.3–27.3 (m, 2C), 19.8–19.7 (m), 12.4; 31P NMR (202 MHz, D2O) δ +39.2, +13.6; HRMS (ES+) calculated for C7H15O6P2 [ M-] 257.0344; found 257.0357.
(((((4-Methylpent-3-en-1-yl)((pivaloyloxy)methoxy)phosphoryl)methyl)phosphoryl)bis(oxy))bis(methylene) bis(2,2-dimethylpropanoate) (11)
The trimethyl ester 7 (0.52 g, 1.82 mmol), sodium iodide (1.09 g, 7.30 mmol), and chloromethyl pivalate (1.06 mL, 7.30 mmol) were dissolved in acetonitrile (2 mL) and the solution was heated at reflux overnight. The reaction was quenched by addition of water and extracted with diethyl ether. The combined organic portions were washed with Na2S2O3, dried (Na2SO4), and filtered, and the filtrate was concentrated in vacuo. The residue was purified by column chromatography (40% EtOAc in hexanes) and the product 11 was isolated as a clear oil in 48% yield (0.51 g): 1H NMR (500 MHz, CDCl3) δ 5.76–5.66 (m, 6H), 5.12–−5.09 (m, 1H), 2.69–−2.52 (m, 2H), 2.40–2.25 (m, 2H), 2.10–1.95 (m, 2H), 1.68 (s, 3H), 1.63 (s, 3H), 1.24 (s, 27H); 13C NMR (125 MHz, CDCl3) δ 177.0, 176.8 (2C), 133.6, 122.3 (d, JPC = 16.1 Hz), 81.9 (d, JPC =5.6 Hz, 2C), 81.0 (d, JPC = 5.6 Hz), 38.7 (2C), 38.7, 30.3 (d, JPC = 147.5 Hz), 29.8 (dd, JPC = 84.5, 41.0 Hz), 26.9 (3C), 26.8 (6C), 25.6, 19.9 (d, JPC = 3.5 Hz), 17.7; 31P NMR (202 MHz, CDCl3) δ +48.9-49.1 (br s), +19.5 (d, JPP = 3.4 Hz); HRMS (ES+) calculated for C25H47O11P2 [M+ + H] 585.2594; found 585.2599.
Biological assays
Materials and supplies
Human peripheral blood mononuclear cell (PMBCs) were isolated from blood obtained from Research Blood Components (Boston, MA). K562 cells were from the American Type Culture Collection. HMBPP (Echelon) was purchased from Fisher. C-HMBP and POM2-C-HMBP were synthesized as described previously.10
K562 proliferation
Proliferation assays were performed using K562 cells in the presence or absence of test compounds. Cells (1 × 104 cells/100 µL) were distributed into each well of a 96-well plate. Test compounds were added for 72 hours, during the last 2 hours the cell quantiblue reagent was added, and signals quantified by fluorescence spectroscopy. Viable cells were expressed as a percentage of untreated control cells following subtraction of a media-only blank.
Expansion of Vγ9Vδ2 T cells from peripheral blood
The compounds were tested for their ability to trigger expansion of human Vγ9Vδ2 T cells from peripheral blood as described previously.10 In each experiment, 100 nM of HMBPP and 100 nM of POM2-C-HMBP were used as positive controls. Negative controls contained cells without or with interleukin 2 in the absence of test compounds. EC50 values were determined as the concentration that induced 50% of the maximum increase observed after interleukin 2 controls were subtracted. All experiments were performed at least three independent times using cells from at least 2 different donors.
Lysis assays
Lysis assays were performed using the Cytotox 96 assay kit (Promega) according to manufacturer’s protocol with some modifications. Vγ9Vδ2 T cells were expanded using a 3 day stimulation with 100 nM HMBPP followed by 4-11 additional days of culture in the presence of interleukin 2 (5 ng/mL). T cells were purified by negative selection (Miltenyi). K562 cells were exposed to test compounds for 2 hours, washed twice in media, and mixed with the purified effector T cells for 4 hours in a 96-well plate. Each well contained a 3:1 E:T ratio in 100 µL. The cell mixture was exposed to the substrate solution for 1 hour, following which the reaction was stopped and quantified by absorbance at 550 nM. After subtracting a media blank, the percentage of T cell mediated lysis was expressed relative to a positive control in which all cells were lysed using detergent.
Interferon γ
Interferon γ was measured by enzyme-linked immunosorbent assay as previously described.28 Existing data was re-analyzed with permission.
Isothermal titration calorimetry
The full intracellular domain of BTN3A1 was purified and used for calorimetry experiments as described previously.10 For each compound, three independent titrations were performed, using a protein concentration of 43 µM and compound concentrations between 300 and 860 µM.
Modeling
Molecular docking was carried out by Glide in the Schrodinger suite. The crystal structure of the intracellular domain of BTN3A1 in complex with C-HMBPP was obtained from the Protein Database (PBD ID: 4N7U). Water and glycerol molecules were removed from the raw protein structure which was then prepared using Protein-Preparation Wizard in the Schrodinger suite and OPLS2005 force fields. 3D sketches of HMBPP and compound 10 were built with Marvin Sketch and were prepared with Gaussian according to density function theory calculations. The grid box for docking was determined using the position of C-HMBPP which co-crystalized with the B30.2 domain of BTN3A1.
Supplementary Material
Acknowledgments
We appreciate the assistance of. Dr. Radha Charan Dash and Prof. M. Kyle Hadden with the molecular docking studies. Research reported in this publication was supported by the National Cancer Institute of the United States National Institutes of Health under Award Number R01CA186935 (A.J.W., P.I.), and by a Research Program of Excellence Award from the Roy J. Carver Charitable Trust (D. F. W., P. I.).
Abbreviations
- BTN
butyrophilin
- C-HMBPP
(E)-4-hydroxy-3-methyl-but-2-enyl phosphonophosphate
- CC-HMBPP
(E)-4-hydroxy-3-methyl-but-2-enyl phosphinophosphonate
- DMAPP
dimethylallyl diphosphate
- HMBPP
(E)-4-hydroxy-3-methyl-but-2-enyl diphosphate
- IPP
isopentenyl diphosphate
- ITC
isothermal titration calorimetry
- PD-1
programmed cell death protein 1
- PD-L1
programmed death-ligand 1
- POM
pivaloyloxymethyl
Footnotes
Supporting Information
Molecular Formula Strings and NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
A.J.W. and D. F. W. own shares in Terpenoid Therapeutics, Inc. The current work did not involve the company.
The other authors have no financial conflicts of interest.
References
- 1.Schwartz RH. Costimulation of T Lymphocytes: The Role of Cd28, Ctla-4, and B7/Bb1 in Interleukin-2 Production and Immunotherapy. Cell. 1992;71:1065–1068. doi: 10.1016/s0092-8674(05)80055-8. [DOI] [PubMed] [Google Scholar]
- 2.Krummel MF, Allison JP. Cd28 and Ctla-4 Have Opposing Effects on the Response of T Cells to Stimulation. J Exp Med. 1995;182:459–465. doi: 10.1084/jem.182.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sharma P, Allison JP. Immune Checkpoint Targeting in Cancer Therapy: Toward Combination Strategies with Curative Potential. Cell. 2015;161:205–214. doi: 10.1016/j.cell.2015.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Joyce JA, Fearon DT. T Cell Exclusion, Immune Privilege, and the Tumor Microenvironment. Science. 2015;348:74–80. doi: 10.1126/science.aaa6204. [DOI] [PubMed] [Google Scholar]
- 5.Abeler-Dorner L, Swamy M, Williams G, Hayday AC, Bas A. Butyrophilins: An Emerging Family of Immune Regulators. Trends Immunol. 2012;33:34–41. doi: 10.1016/j.it.2011.09.007. [DOI] [PubMed] [Google Scholar]
- 6.Harly C, Guillaume Y, Nedellec S, Peigne CM, Monkkonen H, Monkkonen J, Li J, Kuball J, Adams EJ, Netzer S, Dechanet-Merville J, Leger A, Herrmann T, Breathnach R, Olive D, Bonneville M, Scotet E. Key Implication of Cd277/Butyrophilin-3 (Btn3a) in Cellular Stress Sensing by a Major Human Gammadelta T-Cell Subset. Blood. 2012;120:2269–2279. doi: 10.1182/blood-2012-05-430470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang H, Henry O, Distefano MD, Wang YC, Raikkonen J, Monkkonen J, Tanaka Y, Morita CT. Butyrophilin 3a1 Plays an Essential Role in Prenyl Pyrophosphate Stimulation of Human Vgamma2vdelta2 T Cells. J Immunol. 2013;191:1029–1042. doi: 10.4049/jimmunol.1300658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vavassori S, Kumar A, Wan GS, Ramanjaneyulu GS, Cavallari M, El Daker S, Beddoe T, Theodossis A, Williams NK, Gostick E, Price DA, Soudamini DU, Voon KK, Olivo M, Rossjohn J, Mori L, De Libero G. Butyrophilin 3a1 Binds Phosphorylated Antigens and Stimulates Human Gammadelta T Cells. Nat Immunol. 2013;14:908–916. doi: 10.1038/ni.2665. [DOI] [PubMed] [Google Scholar]
- 9.Sandstrom A, Peigne CM, Leger A, Crooks JE, Konczak F, Gesnel MC, Breathnach R, Bonneville M, Scotet E, Adams EJ. The Intracellular B30.2 Domain of Butyrophilin 3a1 Binds Phosphoantigens to Mediate Activation of Human Vgamma9vdelta2 T Cells. Immunity. 2014;40:490–500. doi: 10.1016/j.immuni.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hsiao CH, Lin X, Barney RJ, Shippy RR, Li J, Vinogradova O, Wiemer DF, Wiemer AJ. Synthesis of a Phosphoantigen Prodrug That Potently Activates Vgamma9vdelta2 T-Lymphocytes. Chem Biol. 2014;21:945–954. doi: 10.1016/j.chembiol.2014.06.006. [DOI] [PubMed] [Google Scholar]
- 11.Bonneville M, Chen ZW, Dechanet-Merville J, Eberl M, Fournie JJ, Jameson JM, Lopez RD, Massaia M, Silva-Santos B. Chicago 2014–30 Years of Gammadelta T Cells. Cell Immunol. 2015;296:3–9. doi: 10.1016/j.cellimm.2014.11.001. [DOI] [PubMed] [Google Scholar]
- 12.Wiemer AJ, Hsiao C-HC, Wiemer DF. Isoprenoid Metabolism as a Therapeutic Target in Gram-Negative Pathogens. Curr Top Med Chem. 2010;10:1858–1871. doi: 10.2174/156802610793176602. [DOI] [PubMed] [Google Scholar]
- 13.Parente-Pereira AC, Shmeeda H, Whilding LM, Zambirinis CP, Foster J, van der Stegen SJ, Beatson R, Zabinski T, Brewig N, Sosabowski JK, Mather S, Ghaem-Maghami S, Gabizon A, Maher J. Adoptive Immunotherapy of Epithelial Ovarian Cancer with Vgamma9vdelta2 T Cells, Potentiated by Liposomal Alendronic Acid. J Immunol. 2014;193:5557–5566. doi: 10.4049/jimmunol.1402200. [DOI] [PubMed] [Google Scholar]
- 14.Sebestyen Z, Scheper W, Vyborova A, Gu S, Rychnavska Z, Schiffler M, Cleven A, Cheneau C, van Noorden M, Peigne CM, Olive D, Lebbink RJ, Oostvogels R, Mutis T, Schuurhuis GJ, Adams EJ, Scotet E, Kuball J. Rhob Mediates Phosphoantigen Recognition by Vgamma9vdelta2 T Cell Receptor. Cell Rep. 2016;15:1973–1985. doi: 10.1016/j.celrep.2016.04.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Riganti C, Massaia M, Davey MS, Eberl M. Human Gammadelta T-Cell Responses in Infection and Immunotherapy: Common Mechanisms, Common Mediators. Eur J Immunol. 2012;42:1668–1676. doi: 10.1002/eji.201242492. [DOI] [PubMed] [Google Scholar]
- 16.Rhodes DA, Chen HC, Price AJ, Keeble AH, Davey MS, James LC, Eberl M, Trowsdale J. Activation of Human Gammadelta T Cells by Cytosolic Interactions of Btn3a1 with Soluble Phosphoantigens and the Cytoskeletal Adaptor Periplakin. J Immunol. 2015;194:2390–2398. doi: 10.4049/jimmunol.1401064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rhodes DA, Reith W, Trowsdale J. Regulation of Immunity by Butyrophilins. Annu Rev Immunol. 2016;34:151–172. doi: 10.1146/annurev-immunol-041015-055435. [DOI] [PubMed] [Google Scholar]
- 18.Wang H, Morita CT. Sensor Function for Butyrophilin 3a1 in Prenyl Pyrophosphate Stimulation of Human Vgamma2vdelta2 T Cells. J Immunol. 2015;195:4583–4594. doi: 10.4049/jimmunol.1500314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boedec A, Sicard H, Dessolin J, Herbette G, Ingoure S, Raymond C, Belmant C, Kraus JL. Synthesis and Biological Activity of Phosphonate Analogues and Geometric Isomers of the Highly Potent Phosphoantigen (E)-1-Hydroxy-2-Methylbut-2-Enyl 4-Diphosphate. J Med Chem. 2008;51:1747–1754. doi: 10.1021/jm701101g. [DOI] [PubMed] [Google Scholar]
- 20.Wiemer DF, Wiemer AJ. Opportunities and Challenges in Development of Phosphoantigens as Vgamma9vdelta2 T Cell Agonists. Biochem Pharmacol. 2014;89:301–312. doi: 10.1016/j.bcp.2014.03.009. [DOI] [PubMed] [Google Scholar]
- 21.Wiemer AJ, Shippy RR0, Kilcollins AM, Li J, Hsiao CH, Barney RJ, Geng ML, Wiemer DF. Evaluation of a 7-Methoxycoumarin-3-Carboxylic Acid Ester Derivative as a Fluorescent, Cell-Cleavable, Phosphonate Protecting Group. Chem BioChem. 2016;17:52–55. doi: 10.1002/cbic.201500484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McClard RW, Fujita TS, Stremler KE, Poulter CD. Novel Phosphonylphosphinyl (P-C-P-C-) Analogs of Biochemically Interesting Diphosphates. Syntheses and Properties of P-C-P-C-, Analogs of Isopentenyl Diphosphate and Dimethylallyl Diphosphate. J Am Chem Soc. 1987;109:5544–5545. [Google Scholar]
- 23.Hassan J, Eberl M, Altincicek B, Hintz M, Wolf O, Kollas AK, Reichenberg A, Wiesner J. PCT Int Appl WO2003009855. 2003 [Google Scholar]
- 24.Nemeth G, Greff Z, Sipos A, Varga Z, Szekely R, Sebestyen M, Jaszay Z, Beni S, Nemes Z, Pirat JL, Volle JN, Virieux D, Gyuris A, Kelemenics K, Ay E, Minarovits J, Szathmary S, Keri G, Orfi L. Synthesis and Evaluation of Phosphorus Containing, Specific Cdk9/Cyct1 Inhibitors. J Med Chem. 2014;57:3939–3965. doi: 10.1021/jm401742r. [DOI] [PubMed] [Google Scholar]
- 25.Mckenna CE, Higa MT, Cheung NH, Mckenna MC. Facile Dealkylation of Phosphonic Acid Dialkyl Esters by Bromotrimethylsilane. Tetrahedron Lett. 1977:155–158. [Google Scholar]
- 26.Zhou X, Ferree SD, Wills VS, Born EJ, Tong H, Wiemer DF, Holstein SA. Geranyl and Neryl Triazole Bisphosphonates as Inhibitors of Geranylgeranyl Diphosphate Synthase. Bioorg Med Chem. 2014;22:2791–2798. doi: 10.1016/j.bmc.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boedec A, Sicard H, Dessolin J, Herbette G, Ingoure S, Raymond C, Belmant C, Kraus JL. Synthesis and Biological Activity of Phosphonate Analogues and Geometric Isomers of the Highly Potent Phosphoantigen (E)-1-Hydroxy-2-Methylbut-2-Enyl 4-Diphosphate. J Med Chem. 2008;51:1747–1754. doi: 10.1021/jm701101g. [DOI] [PubMed] [Google Scholar]
- 28.Kilcollins AM, Li J, Hsiao CH, Wiemer AJ. Hmbpp Analog Prodrugs Bypass Energy-Dependent Uptake to Promote Efficient Btn3a1-Mediated Malignant Cell Lysis by Vgamma9vdelta2 T Lymphocyte Effectors. J Immunol. 2016;197:419–428. doi: 10.4049/jimmunol.1501833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gossman W, Oldfield E. Quantitative Structure–Activity Relations for Gammadelta T Cell Activation by Phosphoantigens. J Med Chem. 2002;45:4868–4874. doi: 10.1021/jm020224n. [DOI] [PubMed] [Google Scholar]
- 30.Reichenberg A, Hintz M, Kletschek Y, Kuhl T, Haug C, Engel R, Moll J, Ostrovsky DN, Jomaa H, Eberl M. Replacing the Pyrophosphate Group of Hmb-Pp by a Diphosphonate Function Abrogates Its Potential to Activate Human Gammadelta T Cells but Does Not Lead to Competitive Antagonism. Bioorg Med Chem Lett. 2003;13:1257–1260. doi: 10.1016/s0960-894x(03)00138-0. [DOI] [PubMed] [Google Scholar]
- 31.Wiemer AJ, Wiemer DF. Prodrugs of Phosphonates and Phosphates: Crossing the Membrane Barrier. Top Curr Chem. 2015;360:115–160. doi: 10.1007/128_2014_561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Naesens L, Bischofberger N, Augustijns P, Annaert P, Van den Mooter G, Arimilli MN, Kim CU, De Clercq E. Antiretroviral Efficacy and Pharmacokinetics of Oral Bis(Isopropyloxycarbonyloxymethyl)-9-(2-Phosphonylmethoxypropyl)Adenine in Mice. Antimicrob Agents Chemother. 1998;42:1568–1573. doi: 10.1128/aac.42.7.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Laizure SC, Herring V, Hu Z, Witbrodt K, Parker RB. The Role of Human Carboxylesterases in Drug Metabolism: Have We Overlooked Their Importance? Pharmacotherapy. 2013;33:210–222. doi: 10.1002/phar.1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shen Y, Yan B. Covalent Inhibition of Carboxylesterase-2 by Sofosbuvir and Its Effect on the Hydrolytic Activation of Tenofovir Disoproxil. J Hepatol. 2016;66:660–661. doi: 10.1016/j.jhep.2016.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hill AV. The Possible Effects of the Aggregation of the Molecules of Haemoglobin on Its Dissociation Curves. J Physiol. 1910;40:iv–vii. [Google Scholar]
- 36.Conway A, Koshland DE., Jr Negative Cooperativity in Enzyme Action. The Binding of Diphosphopyridine Nucleotide to Glyceraldehyde 3-Phosphate Dehydrogenase. Biochemistry. 1968;7:4011–4023. doi: 10.1021/bi00851a031. [DOI] [PubMed] [Google Scholar]
- 37.Palakodeti A, Sandstrom A, Sundaresan L, Harly C, Nedellec S, Olive D, Scotet E, Bonneville M, Adams EJ. The Molecular Basis for Modulation of Human Vgamma9vdelta2 T Cell Responses by Cd277/Butyrophilin-3 (Btn3a)-Specific Antibodies. J Biol Chem. 2012;287:32780–32790. doi: 10.1074/jbc.M112.384354. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.