

Abstract
OBJECTIVE:
The objective of this study is to investigate in vitro Caco2 permeability, metabolism and in vivo pharmacokinetic (PK) properties of paromomycin to develop an efficient dosage form with improved oral bioavailability.
MATERIALS AND METHODS:
For the purpose, Caco2 permeability assay, mouse microsomal stability assay and in vivo PKs in male BALB/c mice were performed.
RESULTS:
In Caco-2 permeability assay, paromomycin showed negligible permeability in the apical to basolateral (A-to-B) direction and vice versa (B-to-A). Marginal increase in permeability with the use of P-glycoprotein (P-gp) inhibitor, namely, verapamil suggesting paromomycin could be a P-gp substrate. Paromomycin was unstable in liver microsomes of mouse. Paromomycin showed good PK properties after intravenous dose in male BALB/c mice which included low plasma clearance, i.e., <10% of hepatic blood flow in mice, high volume of distribution (Vd), and half-life (T½) of 2.6 h. Following per oral dose, it exhibits low oral bioavailability (0.3%) with carboxymethyl cellulose formulation. Oral plasma exposure increased in mice by 10% and 15% after pretreatment with P-gp inhibitor verapamil and CYP inhibitor 1-Aminobenztriazole, respectively.
CONCLUSION:
Comparatively significant increase in oral plasma exposure of paromomycin was observed with an alternative oral formulation approach, use of P-gp and CYP inhibitors resulting in improved oral bioavailability up to 16%.
Key words: CYP substrate, formulation method, metabolism, paromomycin, permeability, P-glycoprotein substrate, pharmacokinetics
Introduction
Visceral leishmaniasis (VL) is a protozoan disease, which is transmitted through infected female phlebotomized sandflies. VL is also called Kala Azar and widely prevalent in topical countries particularly in East region of Africa and India. VL is widely distributed in various regions of the USA; and to deserts in Asia (West region) with >90% of patients concentrated in Asian countries such as India, Bangladesh, Nepal, Sudan, and Brazil.[1] It affects 0.5 million people per year living in these areas. The key components for control of VL are early diagnosis and treatment.[2,3]
Currently, VL is treated by antimonials, amphotericin B, paromomycin, and miltefosine. Miltefosine is the only drug with oral route of administration while antimonials and amphotericin are given by intravenous (i.v.) route and paromomycin by intramuscular (i.m.) route, which is painful for chronic treatment. Antimonials were withdrawn due to their toxicity although they were used for the treatment for the last 70 years. Amphotericin B was withdrawn due to the development of resistance in >60% of the population in Bihar, India and the other disadvantage of being high treatment cost.[4,5,6] Miltefosine was developed with the objective to treat cancer, and it is the first drug to be orally active against VL that was approved in 2002 in India. Although it has the advantage of being oral treatment, it suffers from major toxicity including gastrointestinal toxicity and teratogenicity.[7]
Paromomycin was approved in Indian clinics in 2007 for VL treatment. Paromomycin administered by i.m. route showed promising results with no nephrotoxicity and reversible ototoxicity.[4] Paromomycin in conjunction with miltefosine can be administered to minimize the resistance with good efficacy.[8,9]
Paromomycin (an aminoglycoside antibiotic) was initially discovered to treat intestinal amebioasis, and later, it was found to be effective against many other infections including VL. Due to its negligible permeability, its use was limited to treatment of local infections. After oral dose administration, paromomycin is negligibly permeable from the intestine and >99% of the drug is excreted in feces.[10,11] Even after multiple dosages and/or after long-term treatment, paromomycin did not show systemic absorption.[12,13,14]
Treatment with paromomycin requires 21-day painful i.m. injection at the dose of 20 mg/kg/day of body weight.[15] About 95% of cure rate was observed after 6 months treatment with paromomycin.[4] No toxicity with minimal adverse effects which can become normal after surging the treatment and cost-effectiveness makes the paromomycin treatment to be promising against VL. The major disadvantage is giving paromomycin as i.m. route for at least 21 days, but the route of administration makes the drug to be less compliant to patients although the cost of medicine is comparatively low.
The predominant organs for the parasite infection in the human body are liver and spleen. Considering the target organ of parasite infection, the advantage with oral administration for VL treatment led the drug to reach first in liver after intestinal absorption before reaching the systemic circulation unlike other routes of administration.
Paromomycin is relatively a large molecule (molecular weight 615.6 g/mol) with highly hydrophilic in nature making it low bioavailable after oral dose. Due to its absorption limitation, most of the research and clinical trials were performed on paromomycin administered either by i.m. or i.v. route.[4] Extensive literature search on paromomycin reveals that there is scarcity of the research information related to its metabolic stability, CYP inhibition and permeability data, i.e., in general drug metabolism and pharmacokinetics (PKs). With this aim, the authors evaluated in vitro studies to help in designing the formulations to improve oral bioavailability of paromomycin. Till date, PKs data were available after i.v., i.m. or subcutaneous dose administration and only one study was performed following oral administration of paromomycin.[4] Literature review also suggests that the studies were performed with different dosage forms[4] but oral formulations with the excipients to improve the permeability were not explored extensively. No information is available in the reported literature on the permeability of paromomycin in Caco2 assay. The detailed in vitro metabolism and rodent oral PK studies have also not been reported so far in the literature. Therefore, in the present study, we aimed to determined Caco2 permeability, liver metabolism, and oral bioavailability of paromomycin in the presence of P-glycoprotein (P-gp) inhibitor (verapamil) and CYP inhibitor (1-aminobenzotriazole [ABT]) in male BALB/c mice.
Materials and Methods
Chemicals
Paromomycin sulfate (Cat # P9297) was purchased from Sigma, Germany. Loperamide (Cat # L4762) was purchased from Sigma, Germany. Caco-2 cell line was procured from National Centre for Cell Science, Pune, India. Pooled mouse liver microsomes (MLMs) (Cat # 452220) were purchased from BD Gentest, MA, USA (B6C3F1– pool of ~ 100 mice, 9–10 weeks of age; Cat # 452220). Dulbecco's Modified Eagles medium (Cat # D5671), trypsin-EDTA solution (Cat # T4049) and Hank's buffered salt solution (HBSS) Buffer (Cat # H6648) were purchased from Sigma, Germany. Fetal Bovine Serum (Cat # 14-502F) was purchased from Lonza, Walkersville, MD, USA. Glasswares such as T-75 flasks and pipettes were procured from Grenier-Bio-one, Germany. Mill cell-24 well PET membrane 1 μm plates (Cat # PSRP010 R5) were from Millipore Corporation, Billerica MA.
In vitro studies
Caco-2 assay permeability
Caco-2 cell lines (passage 35–50) was used in laboratory and experiments designed to investigate transport/permeability of paromomycin across the monolayers of Caco-2 cell as described previously.[15,16,17] Apical-to-basolateral (PappA-to-B) and vice versa (B-to-A) for the Paromomycin at 25 μM was quantified from the assay samples. The momolayer cells that grown for 21 days in 24 trans-well plate inserts (diameter 6.5 mm) was with a cell count of 0.6 × 105 cells/insert. The monolayer cells integrity was assessed by transepithelial electrical resistance (TEER) value using equipment Millicell-ERS (Costar, Cambridge, MA, USA). Monolayers with TEER value of >230 Ω*cm2, measured before and after each transport experiment, were used for the assay. The paromomycin stock was prepared by dissolving the material in dimethyl sulfoxide (DMSO). The DMSO stock containing paromomycin was added with 10 mL HBSS buffer solution (pH 6.5) with a final concentration of 25 μM of paromomycin and DMSO not >0.25%. Caco2 cells were rinsed thrice with phosphate-buffered saline (37°C), and before assay initiation, the cells were preconditioned with HBSS (pH 7.4). For A-to-B assay, HBSS on the apical side was replaced with 0.5 mL of 25 μM sample solution, and for B-to-A direction, HBSS on basolateral side was replaced with 1.0 mL of 25 μM sample solution. The plate was incubated at 37°C and kept on a plate shaker at 65 rpm. Samples from each well of (50 μL) were removed from both the well sides at following time points 0, 15, 30, 60, and 90 min. A volume of 100 μL HBSS was added to the receiver compartment after sampling of 50 μL at each time point. TEER value was measured at the end of the experiment to check the postexperiment monolayer integrity. A volume of 50 μL sample paromomycin was mixed with 100 μL internal standard followed by thorough mixing using vortexed for 5 min and transferred to 96-well plates for ultra-performance liquid chromatography (UPLC)-MS/MS analysis. The apparent permeability coefficients (Papp) of paromomycin in both apical to basolateral (PappAP-BL) and basolateral to apical (PappBL-AP) directions were measured in triplicate samples.
Metabolic stability
Pooled MLMs procured from BD Gentest (B6C3F1– pool of ~100 mice, 9–10 weeks of age; Cat # 452220) were used for assays. Final pooled MLMs were incubated in triplicates each consisting 0.25 mg/mL protein, NADPH (1 mM) and phosphate buffer (50 mM; pH 7.4) with the final DMSO concentration remained at 0.1%. The liver microsomes along with the paromomycin (25 μM) were placed at 37°C for 5 min. Following incubation, the reaction was initiated by adding NADPH. Samples were withdrawn at 0, 5, 15, 30, and 60 min and quenched with 100 μL of ice-cold 10% perchloric acid in water containing internal standard. Reactions without NADPH and heat inactivated microsomes minus NADPH (at time point of 0 min and 60 min) identify non-NADPH related metabolism or instability associated with incubation buffer. Imipramine is a positive control substrate. Data were subjected to analyzed using GraphPad Prism (version 4.0; GraphPad Software Inc.). Log-linear plots and linear regression of the log-linear curve were plotted. Parameter like half-life (t½) microsomal intrinsic clearance (CL'int) was reported.
In vivo pharmacokinetic studies
Animal husbandry and handling
Pre-approved animal studies by Institutional Animal Ethics Committee were conducted in compliance to guidelines of the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), Government of India. Healthy male animals (BALB/c mice) with 8–12 weeks old, weighing between 25 and 35 g was procured from CPCSEA approved breeder, India. Mice were housed (n = 3) in each cage. Humidity and temperature was maintained at 40%–70% and 22°C ± 3°C, respectively. Illumination was controlled by 12 h light and 12 h dark cycle in a day. Animals were provided with water and standard laboratory rodent diet and ad libitum to the animals.
Formulations
Formulation for i.v. dose was prepared in 100% normal saline. For oral dose paromomycin was prepared in 0.5% carboxymethylcellulose (CMC) in reverse osmosis water and as a solution in 10% Gelucire 44/4, 10% solutol, 30% PG, and 50% normal saline (GSP), and administered as alone. Verapamil and ABT formulations were prepared in normal saline. All formulations were prepared freshly before dose administration.
Pharmacokinetics studies
PKs studies were conducted in male BALB/c mice (25–35 g). Paromomycin was dosed by i. v. route through tail vein at 50 mg/kg and by oral (per oral [p.o.]) route at 500 mg/kg. Mice were pretreated with Verapamil (10 mg/kg/p.o.) and ABT (50 mg/kg/p.o.) to determine whether paromomycin is a P-gp or CYP substrate. Mice were fasted 4 h before dose and food was offered 4 h postdose administration and water ad libitum during the entire period of experiments. A 60 μL of blood sample was withdrawn from retro-orbital plexus at 0.08 (i.v.), 0.25, 0.05, 1, 2, 4, 8 and 24 h (K2 EDTA anticoagulant, 10 μL/mL) by sparse sampling design (n = 3 at each time point). Plasma was immediately harvested from the blood by centrifugation at 4000 rpm for 10 min at 4 ± 2°C and stored below-70 ± 10°C until bioanalysis. The i.v. dosing was done through tail vein and p.o. dose given by oral gavage. The dose volume was 5 mL/kg for i.v. and 10 mL/kg for p.o. dose.
Pharmacokinetic analysis
Noncompartmental analysis module of Phoenix WinNonlin® (Version 6.3) was applied to assess the PK parameters. The trapezoidal areas under the plasma concentration versus time curve (AUC) were calculated by standard linear trapezoidal rule. Terminal at least three plasma concentrations were used to calculate elimination rate constant, ke using regression analysis. The terminal elimination half-life (T½) was estimated using standard formula 0.693/ke; CLi.v.= Dose/AUCinf; Vss = MRT × CLi.v.
Bioanalysis
Paromomycin and internal standard (antipyrine) were analyzed in both in vitro and in vivo samples using an UPLC (ACQUITY Waters, USA) mass spectrometric (ABSciex, USA). Positive ionization mode was applied with MRM transitions of 455.30/308.60 for paromomycin and 189.4/104.0 for antipyrine. A gradient UPLC method with a short run time of 3 min was used for sample analysis. Mobile phase was acetonitrile (A) and 0.005% Trifluoroacetic acid in water (B) with 0.2 mL/min flow rate. The mobile phase run started with 80:20 (A:B) maintained up to 0.4 min with gradual change to 20:80 by 0.6 min. The mobile phase was maintained at 20:80 till 1.8 min with a gradual change to 80:20 from 2 min. The mobile phase was maintained at 80:20 for up to 3 min. Chromatographic separation was obtained using ACQUITY, BEH, HILIC column (50 × 2.1 mm, 1.7 μm) maintained at 45°C with an injection volume of 5 μL for all the study samples.
In vitro and in vivo samples
In vitro assay samples were extracted using protein precipitation technique. A volume of 25 μL of sample was added to 100 μL of perchloric acid (10% prepared in water) into it and vortexed for 5 min. Samples were subjected to centrifugation for 10 min at 4000 rpm at 4°C. A clear supernatant after centrifugation of the samples were transferred into 96 well plates and quantified using LC-MS/MS. The peak area ratios were analyzed.
In vivo plasma samples were extracted by using protein precipitation technique. A volume of 25 μL of plasma sample was added to 25 μL of internal standard prepared in water (Antipyrine, 500 ng/mL) and vortexed. A 100 μL of perchloric acid (10% prepared in water) was spiked and vortexed for 5 min. Samples were subjected to centrifugation for 10 min at a speed of 4000 rpm at 4°C. A clear supernatant of 100 μL volume was transferred to 96 well plates for quantification using LC-MS/MS.
Lower limit of quantification was 50.6 ng/mL, and the calibration curve was linear over a 1000-fold concentration range.
Statistical analysis
Mean values reported with standard error mean (mean ± standard error of the mean). Data were subjected statistically analysis of variance followed by the two-way unpaired test.
Results
In vitro Caco2 permeability study
Bi-directional permeability assay using Caco-2 monolayer cell was examined to assess the permeability of paromomycin. Caco-2 assay is necessary to know how the paromomycin permeability in Caco-2 cells in both directions. Bidirectional movement of the paromomycin in Caco-2 cells from A-to-B and B-to-A are summarized in Table 1. Paromomycin showed low permeability from A-to-B and B-to-A, i.e., in both the directions [Table 1] and the efflux ratio (B-to-A/A-to-B) was found to be 2.9. The efflux ratio >2 reveals that paromomycin could be a P-gp substrate.
Table 1.
Permeability of paromomycin across the Caco-2 cell monolayers

In vitro mouse liver microsomes stability assay
Metabolism of paromomycin was investigated using pooled MLM and assay was performed with and without the addition of NADPH cofactor. Microsomal t½ value was measured by monitoring substrate disappearance over time [Table 2]. In MLM, paromomycin microsomal t½ values appeared to be concentration dependent, i.e., longer half-lives at higher substrate concentrations, indicating a saturable metabolism in the tested concentration range of 0.1–25 μM. Metabolic stability results are shown in Table 1. The data suggest that paromomycin was unstable in MLM (CL'intr; 150 mL/min/kg).
Table 2.
Metabolic stability of paromomycin in mouse liver microsomes

In vivo pharmacokinetics
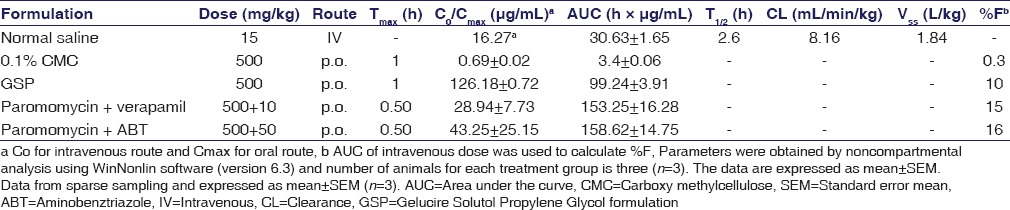
Following i.v. administration through tail vein in mice, paromomycin showed a biphasic disposition [Figure 1] and low systemic clearance (CL <10% of liver blood flow[18,19,20]) and high volume of distribution (Vss >0.7 L/kg[11]), with a t½ of 2.6 h. Following oral administration, paromomycin showed very poor oral bioavailability (F) of 0.3%.
Figure 1.
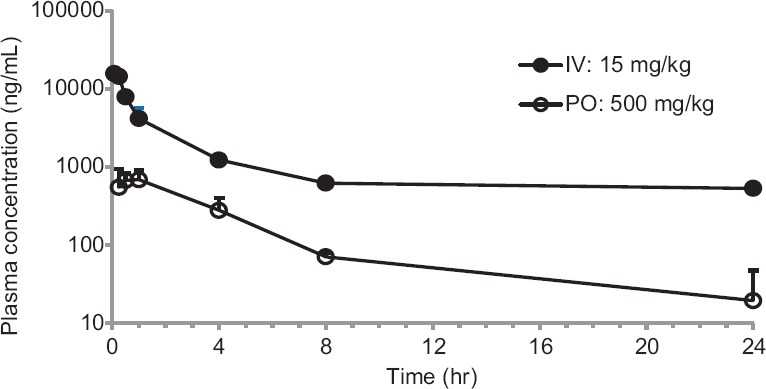
Concentration-time profiles of Paromomycin after intravenous and peroral dose administration to mice. Bars represent the standard deviation (n = 3). The data are expressed as mean ± standard error of the mean
Paromomycin showed low plasma clearance (8 mL/min/kg). The Vd was 2-fold greater compared to the total body water (0.7 L/kg) suggesting that paromomycin is well distributed into tissues. Oral plasma exposure with CMC formulation was very low (%F = 0.3) while the oral exposure was increased 29 folds with GSP based formulation. The concentration-time profiles for Paromomycin are provided in Figure 1. Paromomycin oral exposure increased by 1.5 times when pretreated with P-gp inhibitor, verapamil. Oral exposure increased by 1.6 times when paromomycin was pretreated with CYP inhibitor, i.e., ABT. The concentration-time profiles for paromomycin pretreated with verapamil and ABT are shown in Figure 2. The oral exposure of paromomycin with GSP formulation showed 2-folds higher than CMC formulation and 1.6-fold higher after pretreatment with verapamil and ABT [Table 3].
Figure 2.
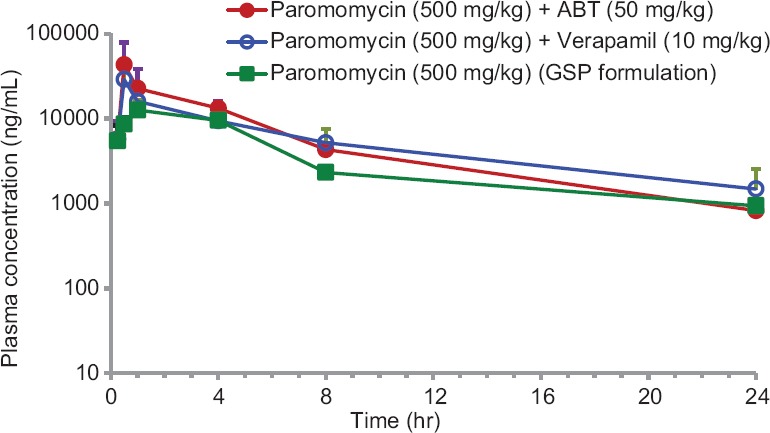
Mean plasma concentration-time profiles of Paromomycin after per oral dose (15 mg/kg) administration to mice in the absence or presence of ABT (50 mg/kg), or verapamil (10 mg/kg) or GSP formulation. Bars represent the standard deviation (n = 3). The data are expressed as mean ± standard error of the mean
Table 3.
Pharmacokinetic parameters of paromomycin after intravenous and p.o dose administration in male Balb/c mice
Discussion
In vitro Caco-2 permeability results showed paromomycin is low permeable drug suggesting that the bioavailability would be permeability limited in vivo. Paromomycin permeability was low in both the directions from suggested that the efflux ratio was 2.9. The permeability of paromomycin in the presence of P-gp inhibitor verapamil increased by 2 times from A-to-B direction and Papp value was similar with B-to-A, it reveals that paromomycin could be a P-gp substrate.
In vitro intrinsic clearance in MLM was high (150 mL/min/kg), suggesting paromomycin is a CYP substrate. Till now, there was no experimental evidence of paromomycin being a CYP substrate. Our research project was the first to report these findings that due to the combined effect of negligible permeability and CYP mediated metabolism (instability) of paromomycin, led to make it difficult for oral treatment.
Paromomycin showed low plasma clearance (8 mL/min/kg) in mice PKs model. We selected high dose due to less sensitive bioanalytical method for quantification of paromomycin. Paromomycin showed 2.5-fold high volume of distribution as than total body water (0.7 L/kg) suggesting paromomycin being extensively distributed into tissues. Tissue distribution is critical for paromomycin treatment where parasite resides in tissues such as liver, spleen, and pancreas. Oral bioavailability of paromomycin was very low (0.3%) when administered in 0.1% CMC, suggesting permeability limited absorption. This is in accordance with the in vitro Caco-2 permeability results. Furthermore, these results are in accordance with the data published by Bissuel where he studied the oral absorption of paromomycin in chronic treatment of AIDS patients.[9] The compound that has entered into the systemic circulation might have undergone metabolism by CYP enzymes which is supported by the in vitro metabolism data and or excreted into feces. Therefore, the low oral exposure could be due to the synergistic effect of limited permeability and first pass metabolism mediated by CYP.
To evaluate the impact of CYP on the oral bioavailability, ABT pretreatment was given before administration of paromomycin in mice. ABT a nonspecific suicidal CYP inhibitor and can be used to elucidate oxidative metabolism in vitro and in vivo models.[20] ABT inhibits CYP enzymes in intestinal lumen and liver. Paromomycin oral exposure increased 1.6 times after pretreatment with ABT compared to paromomycin alone treatment. Thus the significant increase in mice plasma exposure may be due to enzymatic inhibition of metabolism in the intestinal lumen and liver. This suggests that paromomycin is a CYP substrate and metabolism could be the possible reasons for lower exposure following oral administration which was revealed by low % F.
We also performed in vivo study to evaluate whether paromomycin is a P-gp substrate or not. To confirm it, verapamil was administered orally before paromomycin dose. Overall, 50% increase in oral exposure with verapamil pre-treated animals compared to paromomycin alone [Table 3] confirms that paromomycin is a P-gp substrate and its oral exposure could be improved by the concurrent administration of P-gp inhibitors.[21] Paromomycin being a substrate for both CYP enzymes and P-gp efflux transporter, even at a high dose of 500 mg/kg compound exhibited low oral bioavailability. Due to permeability limitation, this compound showed low bioavailability and whatever the amount being permeable at high dose is being affected by both efflux and CYP metabolism mechanisms.[20] The synergistic effect of P-gp efflux and CYP metabolism might lead to poor oral bioavailability of the compound.
To improve the oral bioavailability, a formulation approach encompassing permeability enhancers and P-gp inhibitors was used since Paromomycin was found to have permeability limitation and was also a P-gp substrate in vivo. The employed excipients were either a P-gp inhibitor (Gelucire 44/14) or absorption enhancers (Solutol HS-15 and propylene glycol)[22,23,24] Gelucire 44/14 is a combination of PEG-esters and glycerides which downregulates P-gp protein expression and MDR1 gene expression in vitro.[21] Solutol HS-15 is a Macrogol 15 hydroxy stearate that improves permeability by disrupting the membrane structure/fluidity change.[22] Polyethylene glycol enhances the permeability by disrupting the lipid bilayer of the intestinal lumen.[23] The oral plasma exposure increased by 2.5 times with the GSP-based formulation as compared to CMC formulation. The incorporation of Gelucire 44/14 a P-gp inhibitor and absorption enhancer's solutol HS-15 and PG helped the compound to show an increase in the plasma exposure. Since paromomycin bioavailability was constrained by absorption limitation and P-gp efflux transporters, formulation with excipients that inhibit P-gp and improve the absorption by changing the membrane fluidity helped to increase the oral exposure significantly.
Conclusion
This is for the first time authors report that paromomycin is a substrate of CYP and P-gp. Thus, our research work clearly suggests that paromomycin oral dose is possible and its poor oral exposure could be improved by the concurrent administration of P-gp inhibitors and or by adopting different formulation approaches encompassed with the excipients that can improve the oral bioavailability.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Maltezou HC. Drug resistance in Visceral leishmaniasis. J Biomed Biotechnol. 2010;2010:617521. doi: 10.1155/2010/617521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Croft SL, Sundar S, Fairlamb AH. Drug resistance in leishmaniasis. Clin Microbiol Rev. 2006;19:111–26. doi: 10.1128/CMR.19.1.111-126.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sundar S, More DK, Singh MK, Singh VP, Sharma S, Makharia A, et al. Failure of pentavalent antimony in Visceral leishmaniasis in India: Report from the center of the Indian epidemic. Clin Infect Dis. 2000;31:1104–7. doi: 10.1086/318121. [DOI] [PubMed] [Google Scholar]
- 4.Sundar S, Jha TK, Thakur CP, Sinha PK, Bhattacharya SK. Injectable paromomycin for Visceral leishmaniasis in India. N Engl J Med. 2007;356:2571–81. doi: 10.1056/NEJMoa066536. [DOI] [PubMed] [Google Scholar]
- 5.Wiwanitkit V. Interest in paromomycin for the treatment of Visceral leishmaniasis (kala-azar) Ther Clin Risk Manag. 2012;8:323–8. doi: 10.2147/TCRM.S30139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chappuis F, Sundar S, Hailu A, Ghalib H, Rijal S, Peeling RW, et al. Visceral leishmaniasis: What are the needs for diagnosis, treatment and control? Nat Rev Microbiol. 2007;5:873–82. doi: 10.1038/nrmicro1748. [DOI] [PubMed] [Google Scholar]
- 7.Dorlo TP, Balasegaram M, Beijnen JH, de Vries PJ. Miltefosine: A review of its pharmacology and therapeutic efficacy in the treatment of leishmaniasis. J Antimicrob Chemother. 2012;67:2576–97. doi: 10.1093/jac/dks275. [DOI] [PubMed] [Google Scholar]
- 8.Seifert K, Croft SL. In vitro and in vivo interactions between miltefosine and other antileishmanial drugs. Antimicrob Agents Chemother. 2006;50:73–9. doi: 10.1128/AAC.50.1.73-79.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bissuel F, Cotte L, de Montclos M, Rabodonirina M, Trepo C. Absence of systemic absorption of oral paromomycin during long-term, high-dose treatment for cryptosporidiosis in AIDS. J Infect Dis. 1994;170:749–50. doi: 10.1093/infdis/170.3.749. [DOI] [PubMed] [Google Scholar]
- 10.Iwaki S, Honke K, Nishida N, Taniguchi N. The absorption, excretion and influence on bowel flora of oral paromomycin sulfate (author's transl) Jpn J Antibiot. 1981;34:1078–81. [PubMed] [Google Scholar]
- 11.Takahashi K, Tamagawa S, Katagi T, Yoshitomi H, Kamada A, Rytting JH, et al. In vitro transport of sodium diclofenac across rat abdominal skin: Effect of selection of oleaginous component and the addition of alcohols to the vehicle. Chem Pharm Bull (Tokyo) 1991;39:154–8. doi: 10.1248/cpb.39.154. [DOI] [PubMed] [Google Scholar]
- 12.Musa AM, Younis B, Fadlalla A, Royce C, Balasegaram M, Wasunna M, et al. Paromomycin for the treatment of Visceral leishmaniasis in sudan: A randomized, open-label, dose-finding study. PLoS Negl Trop Dis. 2010;4:e855. doi: 10.1371/journal.pntd.0000855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hilgers AR, Conradi RA, Burton PS. Caco-2 cell monolayers as a model for drug transport across the intestinal mucosa. Pharm Res. 1990;7:902–10. doi: 10.1023/a:1015937605100. [DOI] [PubMed] [Google Scholar]
- 14.Bansal T, Akhtar N, Jaggi M, Khar RK, Talegaonkar S. Novel formulation approaches for optimising delivery of anticancer drugs based on P-glycoprotein modulation. Drug Discov Today. 2009;14:1067–74. doi: 10.1016/j.drudis.2009.07.010. [DOI] [PubMed] [Google Scholar]
- 15.Davies B, Morris T. Physiological parameters in laboratory animals and humans. Pharm Res. 1993;10:1093–5. doi: 10.1023/a:1018943613122. [DOI] [PubMed] [Google Scholar]
- 16.Davidson RN, den Boer M, Ritmeijer K. Paromomycin. Trans R Soc Trop Med Hyg. 2009;103:653–60. doi: 10.1016/j.trstmh.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 17.Jhingran A, Chawla B, Saxena S, Barrett MP, Madhubala R. Paromomycin: Uptake and resistance in Leishmania donovani. Mol Biochem Parasitol. 2009;164:111–7. doi: 10.1016/j.molbiopara.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parrish KE, Mao J, Chen J, Jaochico A, Ly J, Ho Q, et al. In vitro and in vivo characterization of CYP inhibition by 1-aminobenzotriazole in rats. Biopharm Drug Dispos. 2016;37:200–11. doi: 10.1002/bdd.2000. [DOI] [PubMed] [Google Scholar]
- 19.FDA US. Drug Interaction Studies: Study Design, Data Analysis, Implications for Dosing, and Labeling Recommendations. 2012:1–75. [Google Scholar]
- 20.Boddu SP, Yamsani MR, Potharaju S, Veeraraghavan S, Rajak S, Kuma SV, et al. Influence of grapefruit juice on the pharmacokinetics of diltiazem in Wistar rats upon single and multiple dosage regimens. Pharmazie. 2009;64:525–31. [PMC free article] [PubMed] [Google Scholar]
- 21.Sachs-Barrable K, Thamboo A, Lee SD, Wasan KM. Lipid excipients peceol and gelucire 44/14 decrease P-glycoprotein mediated efflux of rhodamine 123 partially due to modifying P-glycoprotein protein expression within caco-2 cells. J Pharm Pharm Sci. 2007;10:319–31. [PubMed] [Google Scholar]
- 22.Shubber S, Vllasaliu D, Rauch C, Jordan F, Illum L, Stolnik S, et al. Mechanism of mucosal permeability enhancement of CriticalSorb® (Solutol® HS15) investigated in vitro in cell cultures. Pharm Res. 2015;32:516–27. doi: 10.1007/s11095-014-1481-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gupta S, Kesarla R, Omri A. Formulation strategies to improve the bioavailability of poorly absorbed drugs with special emphasis on self-emulsifying systems. ISRN Pharm. 2013;2013:848043. doi: 10.1155/2013/848043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shono Y, Nishihara H, Matsuda Y, Furukawa S, Okada N, Fujita T, et al. Modulation of intestinal P-glycoprotein function by cremophor EL and other surfactants by an in vitro diffusion chamber method using the isolated rat intestinal membranes. J Pharm Sci. 2004;93:877–85. doi: 10.1002/jps.20017. [DOI] [PubMed] [Google Scholar]