

Abstract
BACKGROUND:
Sodium thiosulfate (STS) is a potent drug used to treat calcific uremic arteriopathy in dialysis patients and its mode of action is envisaged by calcium chelation and antioxidant potential. STS's action on mitochondrial dysfunction, one of the major players in the pathology of vascular calcification is yet to be explored.
METHODS:
Adenine (0.75%, 28 days)-treated vascular calcified rat kidney was used to isolate mitochondria, where the animal was administered with or without STS for 28 days. Isolated mitochondria were subjected to physiological oxidative stress by nitrogen gas purging (hypoxia/ischemia-reperfusion injury) to assess mitochondrial recovery extent due to STS treatment in vascular calcified rat kidney.
RESULTS:
The results confirmed an elevated oxidative stress and deteriorated mitochondrial enzyme activities in all groups except the drug-treated group.
CONCLUSION:
The STS treatment, besides rendering renal protection against adenine-induced renal failure, also helped to maintain mitochondrial functional integrity in a later insult due to hypoxia/ischemia-reperfusion injury.
Key words: Mitochondria, oxidative stress, sodium thiosulfate, vascular calcification
Introduction
Sodium thiosulfate (STS, Na2S2O3) had been identified as an effective agent to prevent vascular calcification (VC) in adenine-induced rats,[1] where VC is considered to be one of the major contributing factors for the pathological association of cardiovascular mortality and kidney disease.[2] Previous studies have shown that STS mediated protective effect is attributed to its antioxidant and calcium chelation effect.[3] However, STS possesses mitochondria modulating effect as evident from our early study on urolithiasis[4] and VC associated changes in rat brain.[5] Reactive oxygen species are considered to be one of the major causative factors of vascular smooth muscle cell calcification (VSMC) and the mitochondria in VMSC is considered to be one of the major sources for these oxidants.[6] Furthermore, excessive vascular calcification is also associated with VSMC abnormalities that lead to impaired mitochondrial function and adenosine triphosphate production.[7] Accumulated evidence indicate that STS can be a reliable source of hydrogen sulfide, which mediates numerous physiological activities in the heart, brain, kidney by preserving the mitochondrial activity.[5,8,9] Even though the STS treatment is found to be safe in both the preclinical (adenine-induced rat model) and clinical trials (in hemodialysis patients) in decreasing the complications of vascular calcification, its molecular effect in reversing the altered mitochondrial function is not determined. Hence, we designed an in vitro hypoxia/ischemia reoxygenation (I/R) experiment on the isolated mitochondria from VC rat kidney, where the animals were pretreated with or without STS for 28 days and their mitochondrial ability to withstand the oxidative stress due to I/R were evaluated.
Materials and Methods
All reagents used were of analytical grade. Prior approval was obtained for conducting animal experiments from the Institutional Animal Ethical Committee (IAEC) at SASTRA University, Thanjavur, India, and were conducted according to guidelines given by the Committee for the purpose of Control and Supervision of Experiments on Animals vide the approval number 258/SASTRA/IAEC/RPP. The rats were housed in polycarbonate cages and maintained at a temperature of 25°C ± 2°C with a relative humidity of 65% ± 2% and 12 h light/dark cycle.
Male Wistar rats (4–6-week-old weighing around 200–220 g) were randomly assigned to four groups with 6 each: normal, adenine-induced, STS preventive, and STS curative. Normal group rats were fed with normal chow diet whereas adenine-induced rats were fed with 0.75% adenine mixed with regular feed for 28 days. Thiosulfate (STS = 400 mg/kg) was orally co-administered with adenine for 28 days in STS preventive treated the group as described previously,[10] whereas the STS curative rats given adenine for 28 days and thiosulfate (STS = 400 mg/kg) was administered orally for another 28 days. All the rats were euthanized using carbon dioxide inhalation at the end of the study, and kidney tissue was collected from each group and preserved in liquid nitrogen for further analysis.
Calcium and phosphorus content in aorta was estimated using the method described previously[5] and alkaline phosphatase (ALP) activity was estimated in renal tissue using the diagnostic kit purchased from Agappe Diagnostics Ltd.
Mitochondria were isolated from kidney tissues using the method described previously.[11] Isolated mitochondria were suspended in incubation buffer containing 220 mM sucrose, 70 mM mannitol, 10 mM Tris-HCl, pH 7.4 and 1 mM EDTA and stored at 4°C. The mitochondrial protein was determined using Bradford reagent (Bio-Rad, USA) as per the kit instructions.
100% nitrogen gas was infused into a hollow horizontal tube containing four equal diameter holes, to which four needles were attached. To the needles, four Eppendorf tubes were attached firmly by piercing them. The isolated Mitochondria from the above groups were loaded into the Eppendorf tubes and nitrogen gas was allowed to pass through horizontal tubes thereby inducing ischemia for 30 min. After 30 min, 100% oxygen gas was allowed to pass through to induce reperfusion for 30 min.
NADH dehydrogenase, malate dehydrogenase (MDH), and succinate dehydrogenase (SDH) activities in mitochondrial samples were determined according to the standard protocols as described in our previous publication.[12]
Lipid peroxidation was estimated by measuring malondialdehyde chromogen formation at 535 nm as per thiobarbituric acid reactive species assay (TBARS). Activities of antioxidants such as reduced glutathione (GSH), glutathione peroxidase, catalase and superoxide dismutase were assayed by standard methods as mentioned in our previous publication.[12]
All results are expressed as mean ± standard deviation (n = 6). One-way ANOVA was used for comparison of more than two groups, with Bonferroni post hoc analysis. In all cases, P < 0.05 was considered statistically significant. GraphPad Prism 5.0 (GraphPad software, Inc, CA, USA) was used to perform statistical analysis.
Results and Discussion
To confirm the protective effect of STS in completely reversing the mitochondrial perturbations occurring during the vascular calcification, we obtained the mitochondria from rat administered with STS for 28 days prior and after calcification by adenine administration. These mitochondria were subjected to hypoxia/ischemia-reperfusion without the further addition of any STS.
The adenine-induced calcification in rat was confirmed by the elevated calcium-phosphorus product in the aorta by 49% [Figure 1a] and increased ALP activity (1.2 fold) in the renal tissue [Figure 1b]. A strong correlation between calcium-phosphorus product and calcification was reported in previous studies.[13] Earlier investigation in hemodialysis patients[14] and adenine-induced vascular calcified rat demonstrate that STS can be an effective drug in decreasing the rate of progression of vascular calcification. In fact, the mode of action of STS has been attributed to its binding efficiency with calcium ions and antioxidant properties[10] without mentioning the role of mitochondrial dysfunction in the calcification process. Our research group has shown that STS can modulate mitochondria in rendering protection against urolithiasis[4] and adenine-induced vascular calcification.[5] Mitochondrial calcification is considered to be an early step in the calcification process and can be attenuated by reversal of mitochondrial function using drugs affecting mitochondrial function such as α-lipoic acid and fucoidan.[6,15] Moreover, STS is metabolized in the mitochondria through the oxidative mechanism.[16] Hence, it is extremely important that STS, which is used in the clinic to treat vascular calcification in the end-stage renal failure patients, needs to be evaluated for its capacity to completely reverse the mitochondrial dysfunction during vascular calcification.
Figure 1.
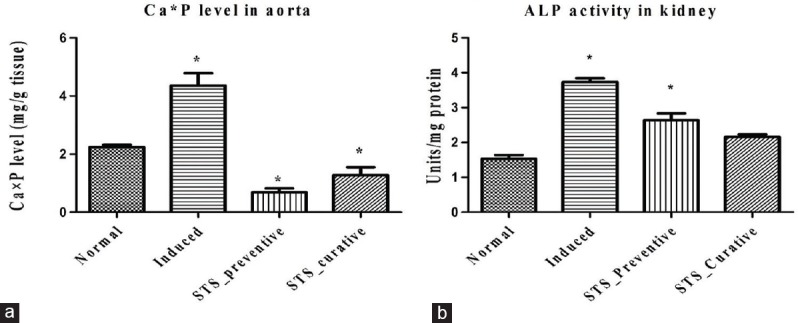
Calcium phosphate product and alkaline phosphate in kidney tissue. Effect of sodium thiosulfate on (a) calcium and phosphorous levels in aorta (b) alkaline phosphatase activity in kidney tissue. Data are expressed in mean ± standard deviation > (n = 6). *Significantly different from normal, (P < 0.05)
The significant decline in Ca × P product (84% decline in STS-preventive and 69% decline in STS-curative groups) and decreased ALP activity with STS administration of adenine treated animal in the present study indicates the efficiency of STS in reducing vascular calcification, as identified by other investigators as well. However, the drug efficacy in recovering mitochondrial function, especially in kidney, where the adenine metabolite gets deposited, leading to renal failure is not known much. In this context, we isolated the mitochondria from the rat kidney subjected to adenine induction followed by STS with or without treatment. To check the functional recovery of renal mitochondria, we subjected the isolated mitochondria from each experimental group to another physiological stress (leading to oxidative stress) namely hypoxia/I/R with the help of inert gas infusion.
The ischemia-reperfusion protocol (30 min ischemia followed by 30 min reperfusion) was implemented in the isolated renal mitochondria from both normal and adenine-induced rats. The significant increase in TBARS by 19% in normal IR and 15% in induced IR and the subsequent decline in GSH concentration by 11% in normal IR and 28% in induced IR with their respective control indicates the efficiency of the model to impart oxidative stress [Figure 2a and b]. Further evaluation of mitochondrial functional activities by measuring NADH and SDH dehydrogenase showed a significant decline in NADH activity by 84% in normal IR and 87% in induced IR, and decreased SDH activity by 35% in normal IR and 82% in induced IR and an increased MDH activity postreperfusion, which support the above findings [Figure 3]. Mitochondria isolated from STS treated rat kidney, when subjected to a secondary stress in the form of hypoxic/ischemic reperfusion injury showed a significant recovery in mitochondrial enzyme activity (99% recovery in SDH activity in STS-preventive IR and 83% in STS-curative IR) and reduced the oxidative stress close to the normal control [Figure 2], while the induced group could not withstand this stress. Among the two modes of STS pretreatment, both showed a similar pattern of protection with respect to preservation of mitochondrial integrity.
Figure 2.

Lipid peroxidation, antioxidant and antioxidant enzyme levels in renal mitochondria. Effect of sodium thiosulfate on isolated renal mitochondria exposed to ischemia reperfusion injury, denoted by lipid peroxidation (a) thiobarbituric acid reactive species, antioxidants (b) reduced glutathione and antioxidant enzymes (c) catalase (d) glutathione peroxidase and (e) superoxide dismutase. Data are expressed in mean ± standard deviation (n = 6). *Significantly different from normal, (P < 0.05)
Figure 3.
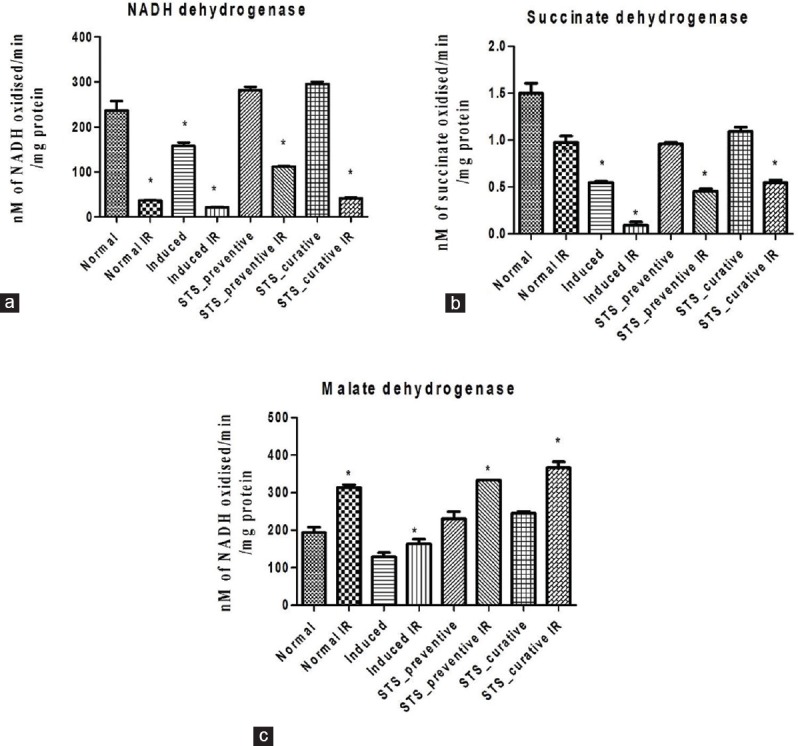
Renal mitochondrial enzyme activity. Effect of sodium thiosulfate on isolated renal mitochondria exposed to ischemia reperfusion injury, represented by the mitochondrial enzymes (a) NADH dehydrogenase (b) Succinate dehydrogenase and (c) malate dehydrogenase. Data are expressed in mean ± standard deviation (n = 6). *Significantly different from normal, (P < 0.05)
Our results suggest that STS treatment not only renders renal-protection against adenine-induced renal failure but also helped to maintain mitochondrial functional integrity in a subsequent insult due to hypoxia/ischemia-reperfusion injury.
Based on these observations, the present study confirms the efficacy of STS in preserving the renal mitochondrial function. In fact, this recovery in mitochondrial activity is not as efficient as compared to the normal renal mitochondria treated with STS, subjected to I/R. Thus, the difference in STS action in an isolated mitochondrial experiment from VC rats and those obtained from STS pretreated VC rats raises the question, whether the STS therapeutic efficacy is systemic or localized. Toward this direction, O'Neill and his groups showed that thiosulfate's ability to ameliorate the vascular calcification is not specific, as sulfate has similar properties.[17]
Conclusion
Based on the above observations, we conclude that STS may be an effective therapeutic drug against vascular calcification, as demonstrated by other investigators, but is not so effective in preserving the diseased renal mitochondria as compared to normal renal mitochondria.
Financial support and sponsorship
The authors would like to acknowledge the honorable Vice-Chancellor of SASTRA University, Thanjavur, Tamil Nadu-India, for providing support and facilities for conduct of this study.
Conflicts of interest
There are no conflicts of interest.
Acknowledgement
The authors would like to acknowledge the honorable Vice-Chancellor of SASTRA University, Thanjavur, Tamil Nadu-India, for providing support and facilities for conduct of this study.
References
- 1.O'Neill WC, Lomashvili KA. Recent progress in the treatment of vascular calcification. Kidney Int. 2010;78:1232–9. doi: 10.1038/ki.2010.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lu KC, Wu CC, Yen JF, Liu WC. Vascular calcification and renal bone disorders. ScientificWorldJournal. 2014;2014:637065. doi: 10.1155/2014/637065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Araya CE, Fennell RS, Neiberger RE, Dharnidharka VR. Sodium thiosulfate treatment for calcific uremic arteriolopathy in children and young adults. Clin J Am Soc Nephrol. 2006;1:1161–6. doi: 10.2215/CJN.01520506. [DOI] [PubMed] [Google Scholar]
- 4.Baldev N, Sriram R, Prabu PC, Kurian Gino A. Effect of mitochondrial potassium channel on the renal protection mediated by sodium thiosulfate against ethylene glycol induced nephrolithiasis in rat model. Int Braz J Urol. 2015;41:1116–25. doi: 10.1590/S1677-5538.IBJU.2014.0585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Subhash N, Sriram R, Kurian GA. Sodium thiosulfate protects brain in rat model of adenine induced vascular calcification. Neurochem Int. 2015;90:193–203. doi: 10.1016/j.neuint.2015.09.004. [DOI] [PubMed] [Google Scholar]
- 6.Kim H, Kim HJ, Lee K, Kim JM, Kim HS, Kim JR, et al. α-lipoic acid attenuates vascular calcification via reversal of mitochondrial function and restoration of gas6/Axl/Akt survival pathway. J Cell Mol Med. 2012;16:273–86. doi: 10.1111/j.1582-4934.2011.01294.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Villa-Bellosta R, Rivera-Torres J, Osorio FG, Acín-Pérez R, Enriquez JA, López-Otín C, et al. Defective extracellular pyrophosphate metabolism promotes vascular calcification in a mouse model of Hutchinson-Gilford progeria syndrome that is ameliorated on pyrophosphate treatment. Circulation. 2013;127:2442–51. doi: 10.1161/CIRCULATIONAHA.112.000571. [DOI] [PubMed] [Google Scholar]
- 8.Sen U, Vacek TP, Hughes WM, Kumar M, Moshal KS, Tyagi N, et al. Cardioprotective role of sodium thiosulfate on chronic heart failure by modulating endogenous H2S generation. Pharmacology. 2008;82:201–13. doi: 10.1159/000156486. [DOI] [PubMed] [Google Scholar]
- 9.Bijarnia RK, Bachtler M, Chandak PG, van Goor H, Pasch A. Sodium thiosulfate ameliorates oxidative stress and preserves renal function in hyperoxaluric rats. PLoS One. 2015;10:e0124881. doi: 10.1371/journal.pone.0124881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pasch A, Schaffner T, Huynh-Do U, Frey BM, Frey FJ, Farese S, et al. Sodium thiosulfate prevents vascular calcifications in uremic rats. Kidney Int. 2008;74:1444–53. doi: 10.1038/ki.2008.455. [DOI] [PubMed] [Google Scholar]
- 11.Weinbach EC. A procedure for isolating stable mitochondria from rat liver and kidney. Anal Biochem. 1961;2:335–43. doi: 10.1016/0003-2697(61)90006-9. [DOI] [PubMed] [Google Scholar]
- 12.Krishnaraj P, Ravindran S, Kurian GA. The renal mitochondrial dysfunction in patients with vascular calcification is prevented by sodium thiosulfate. Int Urol Nephrol. 2016;48:1927–35. doi: 10.1007/s11255-016-1375-z. [DOI] [PubMed] [Google Scholar]
- 13.Llach F. Cardiac calcification: Dealing with another risk factor in patients with kidney failure. Semin Dial. 1999;12:3. [Google Scholar]
- 14.Mathews SJ, de Las Fuentes L, Podaralla P, Cabellon A, Zheng S, Bierhals A, et al. Effects of sodium thiosulfate on vascular calcification in end-stage renal disease: A pilot study of feasibility, safety and efficacy. Am J Nephrol. 2011;33:131–8. doi: 10.1159/000323550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Veena CK, Josephine A, Preetha SP, Rajesh NG, Varalakshmi P. Mitochondrial dysfunction in an animal model of hyperoxaluria: A prophylactic approach with fucoidan. Eur J Pharmacol. 2008;579:330–6. doi: 10.1016/j.ejphar.2007.09.044. [DOI] [PubMed] [Google Scholar]
- 16.Chen NX, O'Neill K, Akl NK, Moe SM. Adipocyte induced arterial calcification is prevented with sodium thiosulfate. Biochem Biophys Res Commun. 2014;449:151–6. doi: 10.1016/j.bbrc.2014.05.005. [DOI] [PubMed] [Google Scholar]
- 17.O'Neill WC, Hardcastle KI. The chemistry of thiosulfate and vascular calcification. Nephrol Dial Transplant. 2012;27:521–6. doi: 10.1093/ndt/gfr375. [DOI] [PMC free article] [PubMed] [Google Scholar]