

Abstract
Background
The Ras pathway genes KRAS, BRAF, or ERBBs have somatic mutations in ~ 60% of human colorectal carcinomas. At present, it is unknown whether the remaining cases lack mutations activating the Ras pathway or whether they have acquired mutations in genes hitherto unknown to belong to the pathway.
Methods
To address the second possibility and extend the compendium of Ras pathway genes, we used genome-wide transposon mutagenesis of two human colorectal cancer cell systems deprived of their activating KRAS or BRAF allele to identify genes enabling growth in low glucose, a Ras pathway phenotype, when targeted.
Results
Of the 163 recurrently targeted genes in the two different genetic backgrounds, one-third were known cancer genes and one-fifth had links to the EGFR/Ras/MAPK pathway. When compared to cancer genome sequencing datasets, nine genes also mutated in human colorectal cancers were identified. Among these, stable knockdown of FOXO3, NCOA3, and TCF7L2 restored growth in low glucose but reduced MEK/MAPK phosphorylation, reduced anchorage-independent growth, and modulated expressions of GLUT1 and Ras pathway related proteins. Knockdown of NCOA3 and FOXO3 significantly decreased the sensitivity to cetuximab of KRAS mutant but not wild-type cells.
Conclusions
This work establishes a proof-of-concept that human cell-based genome-wide forward genetic screens can assign genes to pathways with clinical importance in human colorectal cancer.
Electronic supplementary material
The online version of this article (doi:10.1186/s13073-017-0511-4) contains supplementary material, which is available to authorized users.
Keywords: Forward genetics, piggyBac transposon, Colorectal cancer, Ras pathway
Background
Large-scale mutational analyses are currently unraveling the somatic genetics of human cancer. Apart from mutations in known components of key cancer pathways, such as the Wnt, Ras, and PI3K pathways, moderate to low somatic mutation prevalences have been observed in a large number of genes in colorectal cancers (CRCs) [1]. Concurrently, the fraction of patient tumors where mutations in a given core cancer pathway can be accounted for often amounts to 60% or less, as exemplified by the Ras, PI3K, and TGFB pathways [2]. This conundrum may be due to the importance of yet unknown processes in tumorigenesis, but also to imperfect knowledge of molecular pathways in human cancer cells. Specifically, KRAS mutations occur in 35–40% of CRC cases, whereas BRAF mutations occur in ~ 10% of cases. Mutations in KRAS and BRAF are mutually exclusive in CRC, suggesting both confer the same phenotype [3]. In the Ras pathway, K-Ras binds to and activates B-Raf, thereby activating mitogen-activated protein kinase (MAPK) signaling, and oncogenic K-Ras activation enables anchorage-independent growth in vitro [4, 5]. Human CRC cells deprived of their mutant KRAS or BRAF oncogenes have lost their transcriptional upregulation of the glucose transporter GLUT1 and the associated ability to grow under low-glucose conditions induced by oncogenic Ras pathway activity [6, 7]. Importantly, a subset of clones arising after low-glucose selection of DLD-1 KRASwt/- and RKO BRAFwt/-/- cells had de novo oncogenic mutations in KRAS or BRAF [6]. This connection between pathway genotype and a distinct phenotype provides a means for classical genetic screens to identify genes in the Ras pathway in CRC. Approaches such as tissue-restricted transposon mutagenesis have identified genes causing CRC and other tumors in transgenic mice [8, 9], some mutated also in human cancers, but only a subset of such experiments provide guidance as to which pathways the genes belong [10]. The piggyBac transposon is effective in a wide range of species [11] and one could envision such transposition in human cancer cells with defined somatic mutations to map cancer pathways as activating as well as inactivating mutations can be introduced.
Here, we prove the feasibility of assigning genes to cancer pathways by genome-wide forward genetics using genome-edited human cell systems, link FOXO3, NCOA3, and TCF7L2 to phenotypes of the EGFR/Ras/MAPK pathway, and implicate FOXO3 and NCOA3 in the response to anti-EGFR therapy.
Methods
Cell lines and cell culture
DLD-1 and RKO parental cell lines and DLD-1 KRASwt/- [12] and RKO BRAFwt/-/- [6] knock out cell lines were obtained from Horizon Discovery Ltd. All cells were maintained in DMEM (Invitrogen) medium supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin (Invitrogen) at 37 °C in 5% CO2.
Genome-wide piggyBac transposon mutagenesis and selection for transposon-mediated low-glucose tolerance
A codon optimized hyperactive piggyBac transposase construct (HyPBase) [13], gene trap transposons (PB-GT) in three reading frames, and a promoter containing transposon (PB-CAG-SD) were generated (Additional file 1: Figure S2A–C). Five million DLD-1 KRASwt/- or RKO BRAFwt/-/- cells were used in lipofectamine-mediated transfection of piggyBac with 12 μg each of transposase and transposon constructs. After 48 h of post-transfection incubation in DMEM, low-glucose selection medium (0.4 mM glucose in DMEM) was applied for approximately three weeks [6]. Next, surviving clones were further selected in regular DMEM with hygromycin (Gibco, 0.2 mg/mL) for 12 days. Clones surviving hygromycin selection were either individually picked or pooled. Trypan Blue staining was used to facilitate clone counting under a microscope.
Quantification of GLUT1 expression
Total RNA was isolated with the QIAamp RNA Blood Mini Kit (Qiagen) and treated with DNase I (Roche). First strand complementary DNA (cDNA) synthesis was carried out with the RevertAid H-minus First Strand cDNA synthesis kit (Fermentas). The quantitative polymerase chain reactions (qPCRs) were set up with Maxima SYBR Green/ROX qPCR Master Mix (2X) SYBR green (Thermo Scientific). For GLUT1 transcript level quantification, qPCRs (fwd 5′-GTC ACC ATC CTG GAG CTG TT-3′, rev 5′-GAA GGC CGT GTT GAC GAT AC-3′) were performed in triplicate with β-actin (fwd 5′-CCAACCGCGAGAAGATGA-3′, rev 5′-TCCATCACGATGCCAGTG-3′) as reference gene.
Splinkerette PCR and sequencing of integration sites
Genomic DNA was isolated from clone pools with the Nucleospin genomic extraction kit (MACHEREY NAGEL). Splinkerette PCR was performed as described [14]. Briefly, genomic DNA was digested with Sau3AI and ligated to adaptors and a primary PCR reaction was performed with a Splink1- 5′-CGAAGAGTAACCGTTGCTAGGAGAGACC-3′ and HMsp1-5′- CGAAGAGTAACCGTTGCTAGGAGAGACC-3′ [15] primer pair. Secondary PCR was carried out with barcoded primers (Additional file 1: Table S6) and the products were treated with ExoSap. The barcoded samples were mixed in equimolar concentrations followed by Illumina sequencing.
Sequence processing and identification of genes with transposon integration sites
The sequenced transposon-genome junction fragments were first trimmed of 3′ bases with quality value below QV30. The initial six bases of the fragments were aligned to barcode reference sequences (maximum one mismatch allowed) and the pair-end reads, which had identical barcodes identified on both fragments, were assigned to the corresponding samples. To identify the 3′piggyBac inverted terminal Repeats (3′ PB) and splinkerette adaptor (SP) on the sequenced fragments, both sequences were aligned on the fragments to find the adaptor locations. The fragments with correct PB adaptor location and orientation having junctional TTAA sequences were selected for downstream analysis. The trimmed fragments were then aligned to the human genome (hg19) using Smalt software (http://www.sanger.ac.uk/science/tools/smalt-0, version 0.7.5.1, with default parameters). The mapping results were parsed to identify the chromosomal locations of transposon integrations, together with the number of fragments supporting each integration. To remove possible artifacts, integrations sites found in multiple barcode libraries were only assigned to the library with the highest fragment count if they had more than tenfold higher fragment numbers than all the other libraries; integration sites that did not fit these criteria were ignored. Activating (PB-CAG-SD) transposon integrations occurring within a gene or 5 kb upstream of the protein-coding sequences were assigned to the gene regardless of strand or direction of integration. Similarly, inactivating (PB-GT) transposons occurring in introns of a gene were assigned to the gene. As integration of promoter-containing as well as inactivating transposon constructs can, in principle, result in either inactivation or activation of gene function, we regarded them as identical for the purpose of identifying targeted genes. Poisson statistics were applied to each gene to identify the gene candidates with a significant number of integration sites [16], whereas the detection of multiple independent integration sites in both DLD-1 KRASwt/- and RKO BRAFwt/-/- libraries was used as criterium for the selection of a set of recurrently targeted genes. The sequencing data were processed in R (version 3.0.1) with several analysis packages (stringr, version 0.6.2; Biostrings, version 2.28.0; Rsamtools, version 1.12.4; BSgenome. Hsapiens. UCSC.hg19, version 1.3.19; plyr, version 1.8, fdrtool, version1.2.11). The raw fastq files from the sequencing were deposited at NCBI Sequence Read Archive (SRA) with BioProject accession number PRJNA419878.
Pathway analyses and intersections with cancer genome datasets
The genes with significant transposon integrations in both DLD-1 KRASwt/- and BRAFwt/-/- cells were subjected to pathway analysis using KEGG profile (version 1.2.0) with KEGG database version 2.9.1. For a curated pathway analysis, KEGG Pathways and PubMed papers (last accessed 30 January 2015) were searched for the 163 genes with recurring integrations in both DLD-1 KRASwt/- and RKO BRAFwt/-/- to support: (1) their cancer gene status, defined as subject to somatic mutations in several patients with any human cancer; (2) their assignment to a known CRC pathway; or (3) their role in intracellular glucose metabolism. Mutual exclusivity to KRAS or BRAF mutation in the TCGA COAD dataset [2] was analyzed in cBIOPORTAL (www.cbioportal.org). Non-synonymous somatic mutations, copy number alterations from GISTIC, and messenger RNA (mRNA) expression Z-scores were considered and the strength of the mutual exclusivity relationship was determined by the odds ratios along with P values from Fisher’s exact test [10]. To generate control gene lists for the literature-based pathway assignment, random samples of 163 genes were drawn from the 32,746 Ensembl genes using the sample function in R. Mutation prevalences were obtained from in cBIOPORTAL (www.cbioportal.org) considering copy number aberrations (CNA) and point mutations in the TCGA COAD dataset [2]. The three validated genes were analyzed in the Candidate Cancer Gene Database (CCGD; http://ccgd-starrlab.oit.umn.edu/about.php), a collection of transposon-based forward genetics studies in the mouse [17].
Validation of target genes by small interfering RNA (siRNA)-mediated knockdown assays
Transient gene knockdown assays were performed by seeding 5000 DLD-1 cells in each well of a 96-well plate followed by siRNA transfections using 100 nM siRNAs (ON TARGET Plus SMART pool siRNAs, GE Healthcare) (Additional file 1: Table S7) and DharmaFECT 2. Next, cDNAs were prepared directly from cell lysates using the Cell-to-Ct kit (Life Technologies). Knockdown efficiency was measured by real-time PCR (RT-PCR) using transcript specific TaqMan assays (Additional file 1: Table S7).
Generation of stable knockdown cell lines by lentiviral transduction of short hairpin RNAs (shRNAs)
Lentiviral transductions were performed with GIPZ lentiviral particles (GE Healthcare) (Additional file 1: Table S8). The day before transduction, 50,000 cells were plated in each well of a 24-well plate. Viruses were diluted in 250 μL of normal growth medium with 7.5 mg/mL Sequa-Brene per well. The plating medium was removed and 250 μL of diluted virus was added to each well. After 24 h of incubation at 37 °C, virus containing media were replaced with fresh medium. After 48 h of incubation, the cells were FACS sorted for high green fluorescent protein (GFP) and the pool of cells containing the shRNA for a gene of interest as well as with high GFP were maintained in Puromycin selection (Gibco, 1 μg/mL). After expanding for 2–3 passages, knockdown efficiencies were determined by qPCR.
Clonogenic survival assays
Two thousand cells were plated in each well of a six-well plate and grown in DMEM supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin (Invitrogen) at 37 °C in 5% CO2 for ten days. The cells were stained in Methylene Blue and scanned in an Epson Perfection V700 photo scanner.
Real-time growth assays
For the real-time growth assays in low-glucose (0.4 mM) and normal glucose (25 mM) DMEM (Invitrogen) tissue culture medium, 30,000 and 15,000 cells were plated in each well of a 24-well plate, respectively. The plates were kept in an IncuCyte instrument placed inside a tissue culture incubator and the cell confluence monitored at indicated time points.
Anchorage-independent growth assays
Five thousand cells per well in 0.3% agarose were seeded on top of a 1% agarose layer in a six-well plate. After three weeks of culture at 37 °C and 5% CO2, the colonies were stained with Crystal Violet, scanned, and counted using ImageJ v2.0.0-rc-30/1.49 t.
Determination of MEK and ERK phosphorylation
The protocol was modified from [6]. Cells were lysed in Bicine/CHAPS buffer containing phosphatase and protease inhibitors (ProteinSimple, Santa Clara, CA, USA). The cell lysates were clarified by centrifugation and protein concentration was measured by using BCA Protein Assay Kit (Pierce, Rockford, IL, USA). Lysates were mixed with ampholyte premix (040-972, G2 pH 5-8) and fluorescent pI standards (040-646, pI Standard Ladder 3) before being loaded into the NanoPro 1000 system (ProteinSimple, Santa Clara, CA, USA) for analysis. Isoelectric focusing was performed in capillaries filled with a mixture of cell lysate (0.15 μg/μL protein), fluorescently labeled pI standards, and ampholytes. The separated proteins were cross-linked onto the capillary wall by ultraviolet irradiation followed by immunoprobing with antibodies to ERK (Cell Signaling Technology, #9102, 1:50), pERK (Cell Signaling Technology, #4377, 1:50), MEK (sc-436, Santa Cruz, 1:100), or pMEK (abcam, ab32088, 1:40). Horse Radish Peroxidase conjugated secondary antibodies were from Jackson ImmunoResearch (Donkey Anti-Rabbit IgG, #711-035-152). The signal was visualized by ECL and captured by a charge-coupled device camera. The digital images were analyzed and peak areas quantified with Compass software (ProteinSimple, Santa Clara, CA, USA).
Sample preparation for proteomic analyses
Cell pellets with 106 cells were lysed in 0.1 M Tris-HCl with pH 7.8, 2% sodium dodecyl sulfate, and 0.05 M dithiothreitol for 5 min at 100 °C. The lysates were sonicated with a Branson-rod-type and then centrifuged at 16100 × g for 10 min to clarify the lysates. Samples were then processed in 30-kDa ultrafiltration units with the MED-FASP [18], using Lys-C and trypsin. Concentrations of proteins and peptides were measured with the tryptophan fluorescence assay [19].
Liquid chromatography-tandem mass spectrometry
Aliquots containing 5 μg of total peptide were chromatographed on a 50-cm column with 75-μm inner diameter packed C18 material. Peptide separation was carried out at 300 nL/min for 75 min using a two-step acetonitrile gradient of 5–40% over the first 60 min and 40–95% for the following 15 min. The temperature of the column oven was 55 °C. Peptide mixtures were then analyzed using a QExactive HF mass spectrometer (Thermo-Fisher Scientific, Palo Alto, CA, USA) in data-dependent mode with survey scans acquired at a resolution of 50,000 at m/z 400 (transient time = 256 ms). Up to the top 15 most abundant isotope patterns with charge ≥ + 2 from the survey scan (300–1650 m/z) were selected with an isolation window of 1.6 m/z and fragmented by HCD with normalized collision energies of 25. The maximum ion injection times for the survey scan and the MS/MS scans were 20 and 60 ms, respectively. The ion target value for MS1 and MS2 scan modes was set to 3 × 106 and 105, respectively. The dynamic exclusion was 25 s and 10 ppm. The MS data were analyzed using MaxQuant software (version 1.5.3.14). Proteins were identified by searching MS and MS/MS data of peptides with a fragment ion mass tolerance of 0.5 Da and parent ion tolerance of 20 ppm against a decoy version of the UniProtKB (August 2015) containing 50,807 sequences. The protein and peptide false discovery rates (FDRs) were set to 1%. Protein abundances were calculated using the “total protein approach” (TPA) method [20]. A two-tailed t-test was performed on the extracted gene products for each cell line. The calculations were performed in Microsoft Excel.
Proteomic analysis
The quantified proteins were imported to Perseus [21], version 1.5.5.3. Proteins/protein groups with three or more Razor + unique peptides were extracted and used in the downstream analysis. Missing data were imputed with default settings in Perseus. Genes from pathways relevant to either RAS or glucose metabolism were collected from the ConsensusPathDB version 31 [22, 23] from the COAD dataset mentioned above and from the gene family RAB (RAS oncogene GTPases) and RABL (RAB like GTPases) from HGNC [24]. These genes were extracted from the dataset of quantified proteins and subjected to a two-sample two-tailed test within Perseus with the following parameters: Student’s t-test; S0 set to 0.05; permutation-based FDR; FDR set to 0.05; 250 randomizations. Volcano plots with the same parameters were also created in Perseus.
In vitro response to cetuximab
shRNA knockdown cell lines in DLD-1 and isogenic DLD-1 KRASWt/- cells were seeded at 2 × 104 cells per well in 100 μl of complete medium and incubated overnight at 37 °C and 5% CO2. The next day, the culture medium was exchanged for serum-free or complete medium with 20 and 40 μg/mL of Erbitux (cetuximab) (Merck KGaA, Germany) followed by incubation for 4–5 days at 37 °C and 5% CO2. Cell viability was determined by incubation with Alamar Blue for 4 h followed by counting in a Victor2™ 1420 Multilabel counter (Wallac). The viability of treated samples was normalized to that of the untreated samples.
Candidate Cancer Gene Database (CCGD) analysis
Three validated genes (i.e. FOXO3, NCOA3, and TCF7L2) were analyzed in the CCGD (http://ccgd-starrlab.oit.umn.edu/about.php), a database of collections of transposon-based forward genetics studies [17].
Molecular network analysis
The three validated genes (NCOA3, FOXO3, and TCF7L2) were subjected to network analysis with the gene set “Ras-Raf-MEK-Erk/JNK signaling” in cBIOPORTAL (www.cbioportal.org) in the COAD dataset. Copy number aberrations, nucleotide level mutations, and mRNA expression patterns were included in the analysis.
Statistical analysis
All statistical analyses were performed using GraphPad Prism software version 6.0 f. All Students’ t-tests were two-tailed.
Biosafety declaration
The Swedish work environment authority approved the work with genetically modified and replication deficient lentiviral particles (Arbetsmiljöverket ID 202100-2932 v72). All the experiments with lentiviral particles were conducted under Biosafety Level 2.
Results
Genome-wide piggyBac forward genetic screen for genes restoring Ras pathway activity
To discover genes in the Ras pathway, we transposon mutagenized human CRC cells having had their active KRAS or BRAF oncogene removed by genome editing and used a two-step selection procedure where: (1) tolerance to low glucose was selected by culturing three weeks in medium with 0.4 mM L-glucose; and (2) the resulting clones were selected for having productive transposon integrations by culture in hygromycin-containing medium (Additional file 1: Figure S1). The hygromycin selection serves to eliminate background clones generated by transposon-independent mechanisms, such as those having acquired KRAS and BRAF hotspot mutations through the endogenous MMR deficiency [6]. Next, the gene set with recurring transposon integrations is intersected with genes observed mutated in human CRCs to select candidate Ras pathway genes for experimental validation. Mutagenesis of DLD-1 KRASwt/- and RKO BRAFwt/-/- cell lines with transposase and transposon constructs (Additional file 1: Figure S2A–C) followed by selection in low glucose and hygromycin resulted in sixfold to 22-fold more clones than transposon constructs alone (Fig. 1a and b; Additional file 1: Figure S3), demonstrating that transposon mutagenesis could restore growth in low-glucose medium. The majority of selected clones (13/16) and all clone pools had twfold to sixfold higher GLUT1 expression, a biomarker for Ras pathway activation in human CRC cells [6] compared to DLD-1 KRASwt/- and RKO BRAFwt/-/- cells (Fig. 1c and d). Two gene trap libraries (PB-GT) of 1500 and 2000 clones each, as well as two individual activation libraries (PB-CAG-SD) of 3000 and 2480 clones each in DLD-1 KRASwt/- and RKO BRAFwt/-/- cells, respectively (Fig. 1c and d), were subjected to sequencing. The clone pools were analyzed by splinkerette PCR amplification followed by sequencing of transposon integration sites, resulting in 2029 genes in DLD-1 KRASwt/- and 2887 genes in RKO BRAFwt/-/- pools having more frequent integrations than expected given their TTAA site density (P ≤ 0.05, Poisson distribution) on all the chromosomes (Additional file 1: Figure S4; Additional file 1: Table S1 and S2; Additional file 1: Figure S5).
Fig. 1.
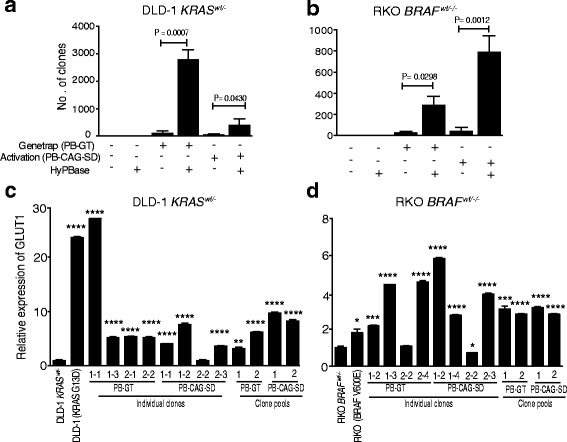
A piggyBac transposon screen in human CRC cells identifies putative Ras pathway genes. a, b Gene trap as well as promoter-containing piggyBac transposons can revert human CRC cells deprived of their oncogenic Ras pathway allele to survive growth in low glucose. Co-transfection of DLD-1 KRASwt/- and RKO BRAFwt/-/- cells with transposase (HyPBase) and gene trapping (PB-GT) or promoter-containing (PB-CAG-SD) transposon constructs was followed by selection in DMEM with 0.4 mM L-glucose for three weeks. Clones emerging after glucose deprivation were selected in hygromycin for 12 days to enrich clones with productive transposon integrations (in-frame gene trap or promoter insertion) and the surviving clones were stained and counted. Mean and standard deviation (SD) from three independent experiments. c, d Transposon mutagenesis and selection in low glucose results in upregulation of GLUT1, a phenotype of Ras pathway activation in human CRC cells. RT-PCR of GLUT1 in parental DLD-1 and RKO cells, their isogenic derivatives DLD-1 KRASwt/- and RKO BRAFwt/-/- with single clones and clone pools derived from mutagenesis with gene trap (PB-GT) or promoter-containing (PB-CAG-SD) transposons. Mean RQs and ΔCt SEs as fold of DLD-1 KRASwt/- and RKO BRAFwt/-/- from three replicates. The P < 0.05 values were calculated using Student’s t-test where ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05
Enrichment of cancer genes and Ras pathway genes
The significantly targeted genes included KRAS (Additional file 1: Table S2) and BRAF (Additional file 1: Table S3). When intersecting the significantly targeted genes in DLD-1 KRASwt/- and RKO BRAFwt/-/- cell systems to pinpoint drivers in both genetic backgrounds, 483 genes were identified (Additional file 1: Table S3) with enrichment in CRC pathways such as Wnt, ErbB, and MAPK (P = 5.23 × 10–5, 0.0045, and 0.046; Additional file 1: Table S4). Next, we identified 623 and 777 genes having at least two unique integration sites in both DLD-1 KRASwt/- and RKO BRAFwt/-/- libraries. After intersecting these two libraries, 163 genes with at least two integration sites were identified (Additional file 1: Figure S6A; P = 1.31 × 10–90). A combined literature and pathway analysis showed that: (1) 31% of the 163 genes were recurrently mutated in human cancers (P = 8.05 × 10–47 under the assumption of 400 cancer genes of 20,000 total protein-encoding genes, hypergeometric distribution); (2) 19% had previously been linked to the EGFR/Ras/MAPK pathway and 6% to the associated Hippo pathway; and (3) 11% had been linked to intracellular glucose metabolism. In comparison, 1–10% of the genes had been associated with other CRC pathways such as the Wnt, TGFB, PIK3CA, or TP53 pathways (Additional file 1: Table S5a). The intersections of these 163 genes with driver genes observed in mutational analyses of human CRC and transposon models of mouse CRC were also larger than expected (Additional file 1: Figure S6B and 6C; P = 5.63 × 10–5 and P = 1.01 × 10–5). The 31 genes known to act within the EGFR/Ras/MAPK pathway included the ERBB receptor ligands NRG1 and NRG3, the tumor suppressor microRNA mir-31, FOXP1, PARD3, PDE4D, PTPRG, RGS6, TRPS1, VMP1, and components of the glutamate transport system. Several genes in intracellular glucose metabolism were also targeted, such as PHLDB1, involved in GLUT translocation to the plasma membrane, HIPK3, KCNMA1, PDP1, SNAP25, TXNRD1, and UBR5, along with DGKB, DIS3L2, ENTPD1, RUNX2, and SIAH1 involved in both Ras signaling and glucose metabolism (Additional file 1: Table S5a). In comparison, three sets of 163 randomly picked genes yielded average 6% cancer genes, 4% EGFR/Ras/MAPK pathway genes, and 0–5% of other CRC pathway genes (Additional file 1: Table S5b–d). Thus, the screen enriches for cancer genes in general and Ras pathway genes in particular.
Identification of putative Ras pathway genes that regulate GLUT1 expression
While mutational analyses and genetic screens provide indications of cancer gene status, experimental validation is required for ultimate proof. To explore the most fruitful approaches to enrich true positives, 13 genes from the intersection of 163 genes having at least two unique integration sites were selected for functional validation based on: (1) mutual exclusivity with KRAS or BRAF mutation in the TCGA COAD dataset [2] (Additional file 1: Table S3 and 5a; FOXO3, NCOA3, SEMA5A); (ii) intersection with other human CRC exome datasets (Additional file 1: Table S5a and Additional file 1: Figure S6B; PTPRD, CLSTN2, CSMD3, NAV3, ROBO1, TCF7L2); and (3) intersection with datasets from murine transposon models of CRC [8] (Additional file 1: Table S5a and Additional file 1: Figure S6C; ROCK2, SIPA1L1, SND1, ZCCHC7). By transient siRNA knockdown, significant effects on GLUT1 transcript levels were observed for FOXO3, NCOA3, and TCF7L2 (Additional file 1: Figure S7A–D) but not for the other ten genes (Additional file 1: Figure S7E and F).
Functional validation of the putative Ras pathway genes FOXO3, NCOA3, and TCF7L2 by real-time growth assays in low-glucose medium and GLUT1 expression analysis
As FOXO3, NCOA3, and TCF7L2 were also detected in both transposon data analysis approaches (Additional file 1: Table S3 and 5a) in the gene trap/inactivation library, we generated shRNA-mediated knockdowns in DLD-1 and RKO cells to assess whether these genes were involved in one or several phenotypes associated with the Ras pathway. First, we assessed whether the reduced growth of DLD-1 KRASwt/- cells in low-glucose medium could be reverted. Stable knockdown of FOXO3, NCOA3, and TCF7L2 increased growth of DLD-1 KRASwt/- cells in low glucose without any growth advantage in normal glucose medium (Fig. 2a and b). The altered growth in low glucose is expected to involve increased GLUT1 expression [6]. In agreement with this postulate, the stable knockdown lines for FOXO3 and TCF7L2 displayed upregulation of GLUT1 in DLD-1 KRASwt/- and RKO BRAFwt/-/- (Fig. 2c and e). Whereas the stable knockdown lines for FOXO3 and TCF7L2 displayed reduced and increased expression of GLUT1 in DLD-1 (KRAS G13D) (Additional file 1: Figure S8A), respectively, stable knockdown of TCF7L2 led to upregulation of GLUT1 in RKO (BRAF V600E) (Additional file 1: Figure S8C) cells. However, stable knockdown of NCOA3 led to upregulation of GLUT1 in RKO BRAFwt/-/- and DLD-1 (KRAS G13D) cells (Fig. 2f and Additional file 1: Figure S8B, respectively) but not in DLD-1 KRASwt/- (Fig. 2d). Stable knockdown of FOXO3, NCOA3, and TCF7L2 in SW48 colorectal cell lines devoid of KRAS mutations did not show changes in GLUT1 expression (Additional file 1: Figure S9A) and the phenotype in low glucose was similar to normal medium (Additional file 1: Figure S9B, C). This demonstrated that these two phenotypes were indeed related to Ras pathway activation and that FOXO3, NCOA3, and TCF7L2 were not directly linked to glucose metabolism. To understand whether these genes could directly regulate GLUT1 transcription, we also used JASPAR (http://jaspar.genereg.net) to determine if binding sites for FOXO3, NCOA3, or TCF7L2 exist in the 3-kb upstream sequence of the initiation site of the GLUT1 gene. While the known positive regulator of GLUT1, HIF1A, had 38 binding sites with a median score of 5.27, this analysis revealed 16 and 13 binding sites for TCF7L2 and FOXO3 with a median score of 5.34 and 5.36, but no direct binding site for NCOA3 (Additional file 1: Table S10). Taken together, stable knockdown of FOXO3, NCOA3, and TCF7L2 in CRC cells deprived of their mutant Ras pathway restored growth in low-glucose medium, which was paralleled by increased GLUT1 expression in the case of FOXO3 and TCF7L2.
Fig. 2.
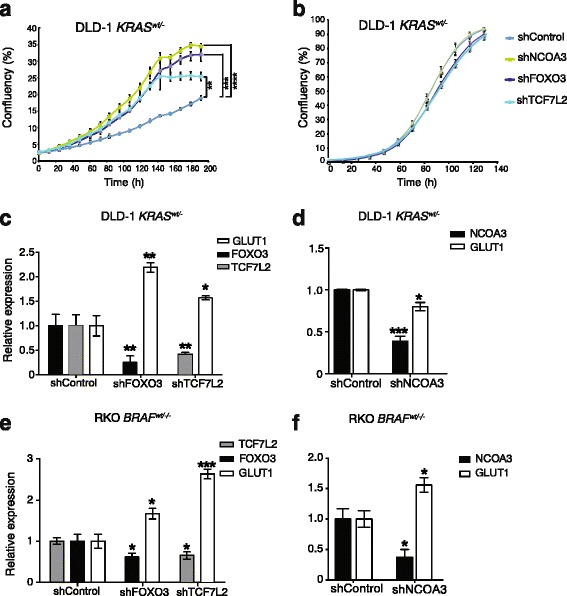
The transposon target genes FOXO3, NCOA3, and TCF7L2 modulate the Ras pathway phenotype of growth in low glucose and regulate GLUT1 expression. Real-time growth curves for stable shRNA knockdown lines of NCOA3, FOXO3, and TCF7L2 in DLD-1 KRASwt/- cells in low-glucose medium (a) and normal medium (b). GLUT1 expression level after stable knockdown of these three genes in DLD-1 KRASwt/- (c, d) and RKO BRAFwt/-/- (e, f). Gene expression levels were measured by qPCR using beta-actin as endogenous control and normalized to shControl. Each experiment was performed three times with three technical replicates. The statistical analysis was performed by Student’s t-test where ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05
Effects of stable shRNA knockdown of FOXO3, NCOA3, and TCF7L2 on anchorage-independent growth and pERK/MEK levels
As oncogenic Ras signaling supports anchorage-independent growth in vitro [5, 12, 14], a hallmark of oncogenic transformation, we investigated the effects of FOXO3, TCF7L2, and NCOA3 shRNA knockdown on anchorage-independent growth by colony formation in soft agar. We observed more than twofold reduced anchorage-independent growth in DLD-1 cells with KRAS G13D mutation and activated oncogenic Ras signaling (Fig. 3a and Additional file 1: Figure S10A) while the rates of growth in normal medium and colony formation on plastic were similar (Fig. 3b, c; Additional file 1: Figure S10B). However, the RKO (BRAF V600E) cells did not form colonies in the same soft agar assay. Mutational or expression changes in genes of the EGFR/Ras/MAPK pathway can alter the cellular ratios of phosphorylated MEK to total MEK and of phosphorylated ERK to total ERK [25, 26]. Stable knockdown of FOXO3 or TCF7L2 reduced pMEK and pERK in DLD-1 cells with KRAS G13D mutation and RKO cells with BRAF V600E mutation, to an extent similar to knockdown of KRAS or BRAF (Fig. 3c–f; Additional file 1: Figure S11A, B and Additional file 1: Figure S12A, B). While knockdown of NCOA3 had little effect on pMEK/MEK and no effect on pERK/ERK ratios in DLD-1 cells with KRAS G13D mutation (Fig. 3c and e; Additional file 1: Figure S11A, B), it had significant effect in RKO (BRAF V600E) cells (Fig. 3d and f; Additional file 1: Figure S12A, B). Hence, stable knockdown of FOXO3, NCOA3, and TCF7L2 significantly reduced anchorage-independent growth and MEK/ERK phosphorylation.
Fig. 3.
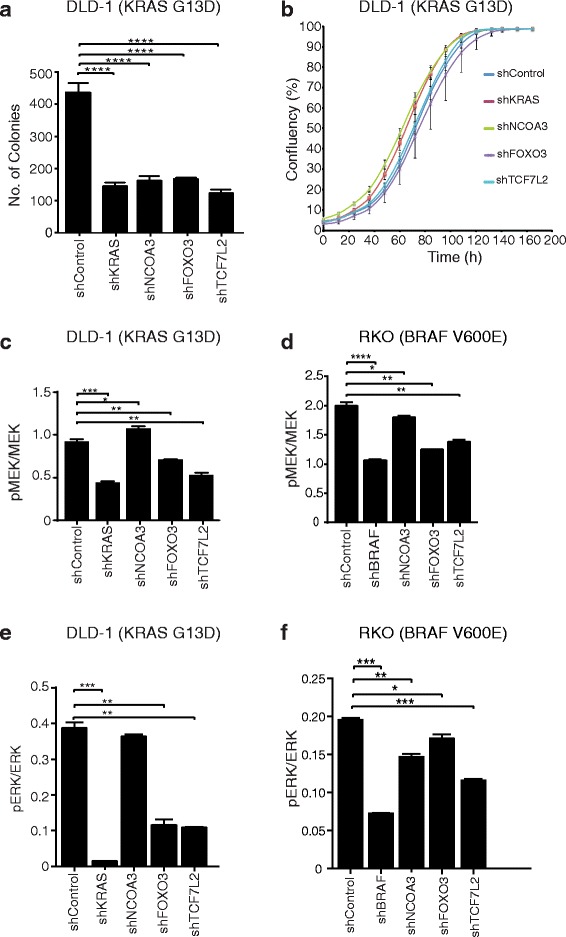
Modulation of the Ras pathway phenotypes of MEK/ERK phosphorylation and anchorage-independent growth by FOXO3, NCOA3, and TCF7L2. Soft agar assays (a) and growth curves in normal medium (b) in stable shRNA lines of NCOA3, FOXO3, and TCF7L2 in DLD-1 cells (KRAS G13D). The impact of NCOA3, FOXO3, and TCF7L2 perturbation on pMEK and pERK in relation to MEK and ERK, respectively, were quantified by NanoPro analysis in DLD-1 (KRAS G13D) (c, e) and RKO (BRAF V600E) (d, f) cells with shKRAS and shBRAF as positive controls. Each experiment was performed at least twice with three technical replicates with statistical analysis by Student’s t-test where ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05
Global expression changes of Ras pathway proteins after knockdown of NCOA3, FOXO3, and TCF7L2
Next, we reasoned that if a gene had a role in a Ras pathway, perturbation of that gene should change expression levels in proteins associated with the Ras pathway. We therefore performed global proteomic analyses of stable shRNA-mediated knockdown lines of NCOA3, FOXO3, and TCF7L2 in both the DLD-1 and isogenic DLD-1 KRASwt/- genetic background. A total of 8556 proteins were identified with concentrations covering seven orders of magnitude (Additional file 1: Figure S13), of which 6905 had three or more unique peptides and were kept for downstream analysis. From the pathway analysis, 210 genes associated with the Ras pathway were extracted (Fig. 4a; Additional file 1: Table S9). Notably, CDKN1A (p21/WAF1) was upregulated in all four knockdown lines in mutant KRAS background. Regulation of p21 by Ras through E2F1 has previously been demonstrated [27]. Further, RAB3A was upregulated in NCOA3 and TCF7L2 knockdowns, whereas EPHA2 was upregulated in NCOA3 but downregulated in TCF7L2 knockdowns. In contrast, there were comparatively fewer regulated protein products in DLD-1 KRASwt/- cells, with RAC1 downregulation in both KRAS and NCOA3 knockdown lines. Together, knockdown of NCOA3, FOXO3, and TCF7L2 modulated Ras pathway-associated proteins primarily in the KRAS-activating mutant background.
Fig 4.
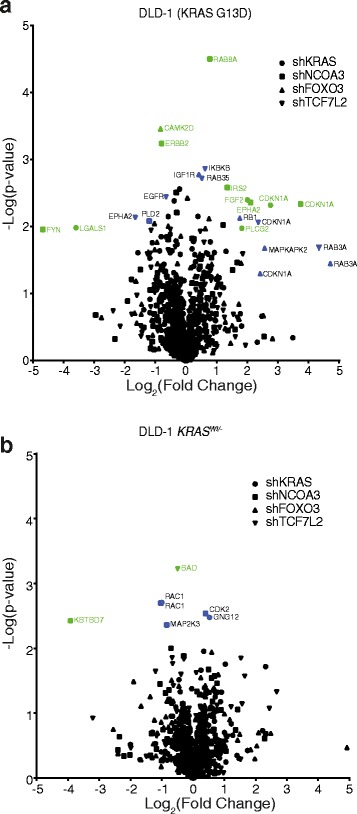
Global proteomic analysis shows expression alterations of Ras-related genes by stable shRNA lines of NCOA3, FOXO3, and TCF7L2. The volcano plot depicts the -log(P values) vs log2(fold change) of Ras-related genes (a, b) for the stable knockdown genes in relation to a shRNA control. Colored symbols indicate genes of interest (display both large fold-change [x-axis] and high statistical significance [y-axis]). Light green symbols, significantly changed (FDR ≤ 0.05); blue symbols, not significant (FDR > 0.05). The full list of genes is presented in Additional file 1: Table S9
Modulation of cetuximab resistance by NCOA3 and FOXO3
The clinical response of CRCs to the EGFR inhibitor cetuximab depends on the KRAS mutation status [28, 29]. To understand whether FOXO3, NCOA3, or TCF7L2 could affect therapy response, we subjected the stable shRNA knockdown cell lines to cetuximab. Knockdown of NCOA3 and FOXO3 caused significantly increased resistance compared to control and KRAS knockdowns (>100% vs 50%) in DLD-1 cells with activating KRAS mutation (Fig. 5a, P < 0.001), but not in DLD-1 KRASWt/- cells (Fig. 5b). The knockdown efficiencies of FOXO3 and NCOA3 were 70–80% (Additional file 1: Figure S8A, B; Fig. 2c and d) and the knockdown efficiency of KRAS was 50% (Additional file 1: Figure S15).
Fig. 5.
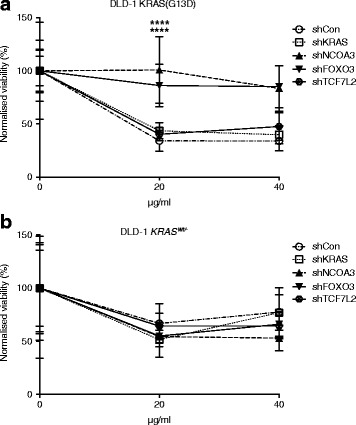
Knockdown of NCOA3 and FOXO3 desensitizes DLD-1 cells to cetuximab. The shRNA knockdown lines of NCOA3, FOXO3, and TCF7L2 in (a) DLD-1 (KRAS G13D) and (b) DLD-1 KRASwt/- genetic backgrounds were treated with cetuximab (20 μg/mL or 40 μg/mL) for 4–5 days. Each experiment was performed three times with eight technical replicates. Mean percentage of Alamar Blue cell viability was normalized to the mean percentage cell viability of untreated control. Mean percentage of viability and SD. Statistical analysis was performed by Student’s t-test where ****P < 0.0001
Discussion
While it is now easy to collect genome sequences from large sets of human cancers, it remains challenging to assign infrequently mutated genes as drivers or passengers in cancer pathways without extensive functional studies. Unbiased pathway mapping in human cells has been hampered by a lack of phenotypes amenable to selection. For example, siRNA screens based on growth in soft agar have discriminated driver from passenger genes mutated in CRC but were performed in a background of activating KRAS and inactivating TP53 mutations and could not assign genes to specific pathways [30]. Here, for the primary screen we exploited previous observations that selection of CRC cells deprived of Ras pathway-activating mutations under glucose-limiting conditions yields reverting clones (Fig. 1a and b), of which a subset contain de novo activating hotspot mutations in KRAS or BRAF [6], followed by subsequent validation of candidate genes in other Ras pathway phenotypes. A challenge in integrating forward genetic screens with human cancer mutational data is that both accumulate passenger mutations in large genes, such as CSMD3 and NAV3. However, 55 of 163 identified genes with multiple integrations in DLD-1 and RKO clone pools were recurrently mutated in human tumors, which is an unbiased indicator of relevance in the cancer context. Thirty-one of the 55 genes had no prior pathway assignment. Whereas transposon integration site biases may explain part of the overlap between the two genetic backgrounds and with murine transposon screens, they are unlikely to selectively affect cancer genes. Therefore, prioritization of genes for validation based on: (1) their degree of mutual exclusivity with mutations in known pathway members in different Ras-driven cancers; (2) gene or transcript size; and (3) status as known cancer gene with unknown function could guide further investigations into this gene set.
Stable shRNA-mediated knockdown of NCOA3, FOXO3, and TCF7L2 in DLD-1 KRASWt/- genetic background increased cell growth in low glucose (Fig. 2a), demonstrating the ability of this forward genetics approach to identify mediators of the Ras pathway. In these knockdown lines, the levels of GLUT1 expression were significantly changed (Fig. 2c–f), which was not the case in corresponding knockdowns in the SW48 colorectal cell line which harbors wild-type KRAS (Additional file 1: Figure S9A). This strengthens the connection between altered GLUT1 expression and Ras pathway activation demonstrated in [6]. Additionally, Ras signaling controls anchorage-independent growth [5] and the level of pERK as well as pMEK [31]. Here, there were significant reductions in anchorage-independent growth and pERK/pMEK level by stable knockdown of NCOA3, FOXO3, and TCF7L2, similar to knockdown of mutant KRAS, in DLD-1 (KRAS G13D) cells. From these validation efforts, FOXO3, NCOA3, and TCF7L2 emerged as mediators of Ras pathway phenotypes. It was noteworthy that these genes acted as negative regulators for low-glucose growth (Fig. 2a) and GLUT1 expression (Fig. 2c–f), but also as positive regulators of anchorage independent growth (Fig. 3a) and pERK/pMEK (Fig. 3c–f). Resistance to cetuximab was reported to be dependent predominantly on the KRAS mutation status [32, 33]. Here, this was not observed in DLD-1 (KRAS G13D) likely due to non-complete knockdown. However, knockdown of FOXO3 and NCOA3 conferred significantly higher cetuximab resistance in DLD-1 (KRAS G13D) cells (Fig. 5a). The significance of the role of NCOA3 and FOXO3 in mediating cetuximab resistance will require further studies in vitro and in clinical materials. Several additional mechanisms for cetuximab resistance, other than the KRAS mutation status, have been identified, including accumulation of stress granules [34] and constitutive activation of EGFR effector molecules [35]. Given that cetuximab resistance is primarily dependent on KRAS mutation status, and that we observed enhanced cetuximab resistance upon stable knockdown of FOXO3 and NCOA3 (Fig. 5a), the data strengthen the links between NCOA3 and FOXO3, and the Ras pathway in CRC. However, the pleiotropic effects of FOXO3, NCOA3, and TCF7L2 observed here can potentially be explained by signaling pathways cross-talk and by the dependency of Ras pathway gene phenotypes on other mutations, genetic background, and the environment [36–38].
These three genes (i.e. FOXO3, NCOA3, and TCF7L2) also play roles in other human cancers. The prevalence of somatic FOXO3 aberrations is 41%, 9%, and 0.5% in breast cancer, prostate cancer, and CRCs in humans, it has been implicated in liver and brain tumors in mice [39] and is a negative regulator of Ras/MAPK [40]. Cancers with activating Ras mutations exhibit enhanced autophagy, where FOXO3 plays a critical role [41]. Copy number alterations and point mutations in NCOA3 (AIB1) have been observed in human breast (31%) and CRCs (14%) [39, 42] and it is a transposon target in murine models of colorectal, liver, pancreas cancer, and melanoma [39, 43]. In vitro phosphorylation of NCOA3 by MAPK has also been demonstrated [44]. The cancer gene TCF7L2 [45] is a component of the Wnt pathway, a recurrent fusion oncogene in human CRC [46, 47], mutated in 14% of CRCs and breast cancers and a transposon target in murine genetic models of CRCs, liver tumors, and brain tumors [39]. Interestingly, aberrations in KRAS and TCF7L2 in human CRC tended to co-occur [2] (P = 0.004, Fisher’s exact test). We observed enrichment of Wnt pathway components among the targeted genes (Additional file 1: Table S3). This suggests that: (1) TCF7L2 is a cross-talk point between Wnt and Ras pathways; and (2) additional aberrations of Wnt signaling may compensate for a lack of canonical Ras pathway mutations in CRC. It is also noteworthy that there were enrichments of integrations in other pathways, other than the Ras pathway (Additional file 1: Table S4). A plausible explanation would be cross-talk between these cancer pathways. Molecular network analyses showed associations of FOXO3, NCOA3, and TCF7L2 with canonical Ras pathway components that had previously been found mutated in human CRC (Additional file 1: Figure S14). TCF7L2 and FOXO3 were directly associated with the c-Myc oncogene, which is overexpressed in subset of human CRCs [48] and regulated by Ras [49]. Collectively, these findings support a role for the three genes FOXO3, NCOA3, and TCF7L2 as actors or modulators of the EGFR/Ras/MAPK pathway in human CRC.
To further strengthen the evidence for NCOA3, FOXO3, and TCF7L2 as mediators of the Ras pathway, we performed LC-MS-based global proteomics and observed several significant changes in Ras pathway proteins. These included FGF2, LGALS1, PLCG2, CDKN1A, ERBB2, FYN, EPHA2, RAB8A, MAPKAPK2, RB1, IKBKB, RAB35, RAB3A, and EGFR, with CDKN1A upregulated in all four knockdown lines. In contrast, fewer Ras-related proteins were modulated in the three stable knockdown lines in DLD-1 KRASWt/- genetic background. This is in agreement with NCOA3, FOXO3, and TCF7L2 having roles in the activated Ras pathway.
Conclusions
In summary, we have established proof-of-concept that a pathway-specific forward genetic screen help chart the Ras system in human CRC by identifying known as well as novel pathway members of potential clinical relevance. The gene sets provided here harbor priority candidates for future functional evaluation of their role in Ras pathway phenotypes in CRC and analogous approaches could be envisioned in dissecting other cancer phenotypes and pathways.
Acknowledgements
Illumina sequencing was performed by the SNP & SEQ Technology Platform in Uppsala, supported by Uppsala University, Uppsala University Hospital, Science for Life Laboratory, and the Swedish Research Council (Contracts 80576801 and 70374401). The proteomic work was supported by Max-Planck Society for the Advancement of Science and by the German Research Foundation (DFG/Gottfried Wilhelm Leibniz Prize). The authors are grateful to Prof. Matthias Mann (Max Planck Institute) for continuous support and Katharina Zettl for technical support. We thank Christopher Stroh, Merck Global R&D Translational Innovation Platform Oncology, for the kind gift of cetuximab.
Funding
Grant support to TS from the Swedish Cancer Foundation (CAN 2012/834), The Swedish Foundation for Strategic Research (F06-0050 and RBa08-0114), the Kjell and Märta Beijer Foundation, and to SK and TS from the Olle Engkvist Byggmästare Foundation.
Availability of data and materials
The sequencing datasets generated during the current study are available in the NCBI Sequence Read Archive (SRA) repository, BioProject accession number PRJNA419878. The datasets for mass-spec analysis (Table S9) have been deposited with the ProteomeXchange Consortium via the PRIDE [50] partner repository with the dataset identifier PXD008382.
Abbreviations
- CCGD
Candidate Cancer Gene Database
- CRC
Colorectal cancer
- DMEM
Dulbecco’s Modified Eagle Medium
- HCD
Higher-energy collisional dissociation
- HGNC
HUGO Gene Nomenclature Committee
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- PB-CAG-SD
piggyBac-CAG promoter-splice donor
- PB-GT
piggyBac-gene trap
- qPCR
Quantitative PCR
- RT-PCR
Real-time polymerase chain reaction
- TCGA
The Cancer Genome Atlas
Additional file
and Tables S1–S10 with legends. (PDF 9090 kb)
Authors’ contributions
TS, MH, and SK designed the study. SK, MAA, NP, JL, and MK performed experiments. KB and LH performed bioinformatics analyses. NH, JW, and PA performed the proteomics analyses. TS and SK wrote the paper. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s13073-017-0511-4) contains supplementary material, which is available to authorized users.
References
- 1.Wood LD, Parsons DW, Jones S, Lin J, Sjöblom T, Leary RJ, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–13. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 2.Network CGA. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–7. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature. 2002;418:934. doi: 10.1038/418934a. [DOI] [PubMed] [Google Scholar]
- 4.Ke N, Albers A, Claassen G, Yu DH, Chatterton JE, Hu X, et al. One-week 96-well soft agar growth assay for cancer target validation. Biotechniques. 2004;36:826–8. doi: 10.2144/04365ST07. [DOI] [PubMed] [Google Scholar]
- 5.Yang JJ, Kang JS, Krauss RS. Ras signals to the cell cycle machinery via multiple pathways to induce anchorage-independent growth. Mol Cell Biol. 1998;18:2586–95. doi: 10.1128/MCB.18.5.2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yun J, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagopalan H, et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science. 2009;325:1555–9. doi: 10.1126/science.1174229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Flier JS, Mueckler MM, Usher P, Lodish HF. Elevated levels of glucose transport and transporter messenger RNA are induced by ras or src oncogenes. Science. 1987;235:1492–5. doi: 10.1126/science.3103217. [DOI] [PubMed] [Google Scholar]
- 8.Starr TK, Allaei R, Silverstein KA, Staggs RA, Sarver AL, Bergemann TL, et al. A transposon-based genetic screen in mice identifies genes altered in colorectal cancer. Science. 2009;323:1747–50. doi: 10.1126/science.1163040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.March HN, Rust AG, Wright NA, ten Hoeve J, de Ridder J, Eldridge M, et al. Insertional mutagenesis identifies multiple networks of cooperating genes driving intestinal tumorigenesis. Nat Genet. 2011;43:1202–9. doi: 10.1038/ng.990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takeda H, Wei Z, Koso H, Rust AG, Yew CC, Mann MB, et al. Transposon mutagenesis identifies genes and evolutionary forces driving gastrointestinal tract tumor progression. Nat Genet. 2015;47:142–50. doi: 10.1038/ng.3175. [DOI] [PubMed] [Google Scholar]
- 11.Wu SC, Meir YJ, Coates CJ, Handler AM, Pelczar P, Moisyadi S, et al. piggyBac is a flexible and highly active transposon as compared to sleeping beauty, Tol2, and Mos1 in mammalian cells. Proc Natl Acad Sci U S A. 2006;103:15008–13. doi: 10.1073/pnas.0606979103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shirasawa S, Furuse M, Yokoyama N, Sasazuki T. Altered growth of human colon cancer cell lines disrupted at activated Ki-ras. Science. 1993;260:85–8. doi: 10.1126/science.8465203. [DOI] [PubMed] [Google Scholar]
- 13.Yusa K, Zhou L, Li MA, Bradley A, Craig NL. A hyperactive piggyBac transposase for mammalian applications. Proc Natl Acad Sci U S A. 2011;108:1531–6. doi: 10.1073/pnas.1008322108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Uren AG, Mikkers H, Kool J, van der Weyden L, Lund AH, Wilson CH, et al. A high-throughput splinkerette-PCR method for the isolation and sequencing of retroviral insertion sites. Nat Protoc. 2009;4:789–98. doi: 10.1038/nprot.2009.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang W, Lin C, Lu D, Ning Z, Cox T, Melvin D, et al. Chromosomal transposition of PiggyBac in mouse embryonic stem cells. Proc Natl Acad Sci U S A. 2008;105:9290–5. doi: 10.1073/pnas.0801017105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brett BT, Berquam-Vrieze KE, Nannapaneni K, Huang J, Scheetz TE, Dupuy AJ. Novel molecular and computational methods improve the accuracy of insertion site analysis in Sleeping Beauty-induced tumors. PLoS One. 2011;6:e24668. doi: 10.1371/journal.pone.0024668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abbott KL, Nyre ET, Abrahante J, Ho YY, Isaksson Vogel R, Starr TK. The Candidate Cancer Gene Database: a database of cancer driver genes from forward genetic screens in mice. Nucleic Acids Res. 2015;43:D844–848. doi: 10.1093/nar/gku770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wiśniewski JR, Mann M. Consecutive proteolytic digestion in an enzyme reactor increases depth of proteomic and phosphoproteomic analysis. Anal Chem. 2012;84:2631–7. doi: 10.1021/ac300006b. [DOI] [PubMed] [Google Scholar]
- 19.Wiśniewski JR, Gaugaz FZ. Fast and sensitive total protein and Peptide assays for proteomic analysis. Anal Chem. 2015;87:4110–6. doi: 10.1021/ac504689z. [DOI] [PubMed] [Google Scholar]
- 20.Wiśniewski JR, Rakus D. Multi-enzyme digestion FASP and the ‘Total Protein Approach’-based absolute quantification of the Escherichia coli proteome. J Proteomics. 2014;109:322–31. doi: 10.1016/j.jprot.2014.07.012. [DOI] [PubMed] [Google Scholar]
- 21.Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods. 2016;13:731–40. doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- 22.Kamburov A, Wierling C, Lehrach H, Herwig R. ConsensusPathDB--a database for integrating human functional interaction networks. Nucleic Acids Res. 2009;37:D623–628. doi: 10.1093/nar/gkn698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kamburov A, Pentchev K, Galicka H, Wierling C, Lehrach H, Herwig R. ConsensusPathDB: toward a more complete picture of cell biology. Nucleic Acids Res. 2011;39:D712–717. doi: 10.1093/nar/gkq1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gray KA, Yates B, Seal RL, Wright MW, Bruford EA. Genenames.org: the HGNC resources in 2015. Nucleic Acids Res. 2015;43:D1079–1085. doi: 10.1093/nar/gku1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Loboda A, Nebozhyn M, Klinghoffer R, Frazier J, Chastain M, Arthur W, et al. A gene expression signature of RAS pathway dependence predicts response to PI3K and RAS pathway inhibitors and expands the population of RAS pathway activated tumors. BMC Med Genomics. 2010;3:26. doi: 10.1186/1755-8794-3-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shields JM, Pruitt K, McFall A, Shaub A, Der CJ. Understanding Ras: ‘it ain’t over ‘til it’s over’. Trends Cell Biol. 2000;10:147–54. doi: 10.1016/S0962-8924(00)01740-2. [DOI] [PubMed] [Google Scholar]
- 27.Gartel AL, Najmabadi F, Goufman E, Tyner AL. A role for E2F1 in Ras activation of p21(WAF1/CIP1) transcription. Oncogene. 2000;19:961–4. doi: 10.1038/sj.onc.1203411. [DOI] [PubMed] [Google Scholar]
- 28.Benvenuti S, Sartore-Bianchi A, Di Nicolantonio F, Zanon C, Moroni M, Veronese S, et al. Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Res. 2007;67:2643–8. doi: 10.1158/0008-5472.CAN-06-4158. [DOI] [PubMed] [Google Scholar]
- 29.Ibrahim EM, Zekri JM, Bin Sadiq BM. Cetuximab-based therapy for metastatic colorectal cancer: a meta-analysis of the effect of K-ras mutations. Int J Colorectal Dis. 2010;25:713–21. doi: 10.1007/s00384-010-0927-4. [DOI] [PubMed] [Google Scholar]
- 30.Eskiocak U, Kim SB, Ly P, Roig AI, Biglione S, Komurov K, et al. Functional parsing of driver mutations in the colorectal cancer genome reveals numerous suppressors of anchorage-independent growth. Cancer Res. 2011;71:4359–65. doi: 10.1158/0008-5472.CAN-11-0794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Levidou G, Saetta AA, Gigelou F, Karlou M, Papanastasiou P, Stamatelli A, et al. ERK/pERK expression and B-raf mutations in colon adenocarcinomas: correlation with clinicopathological characteristics. World J Surg Oncol. 2012;10:47. doi: 10.1186/1477-7819-10-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lièvre A, Bachet JB, Le Corre D, Boige V, Landi B, Emile JF, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006;66:3992–5. doi: 10.1158/0008-5472.CAN-06-0191. [DOI] [PubMed] [Google Scholar]
- 33.Lièvre A, Blons H, Laurent-Puig P. Oncogenic mutations as predictive factors in colorectal cancer. Oncogene. 2010;29:3033–43. doi: 10.1038/onc.2010.89. [DOI] [PubMed] [Google Scholar]
- 34.Grabocka E, Bar-Sagi D. Mutant KRAS enhances tumor cell fitness by upregulating stress granules. Cell. 2016;167:1803–13. doi: 10.1016/j.cell.2016.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brand TM, Iida M, Wheeler DL. Molecular mechanisms of resistance to the EGFR monoclonal antibody cetuximab. Cancer Biol Ther. 2011;11:777–92. doi: 10.4161/cbt.11.9.15050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stites EC, Trampont PC, Haney LB, Walk SF, Ravichandran KS. Cooperation between Noncanonical Ras Network Mutations. Cell Rep. 2015. doi: 10.1016/j.celrep.2014.12.035. [DOI] [PMC free article] [PubMed]
- 37.Vartanian S, Bentley C, Brauer MJ, Li L, Shirasawa S, Sasazuki T, et al. Identification of mutant K-Ras-dependent phenotypes using a panel of isogenic cell lines. J Biol Chem. 2013;288:2403–13. doi: 10.1074/jbc.M112.394130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schneider G, Schmidt-Supprian M, Rad R, Saur D. Tissue-specific tumorigenesis: context matters. Nat Rev Cancer. 2017;17:239–53. doi: 10.1038/nrc.2017.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wagner M, Koslowski M, Paret C, Schmidt M, Türeci O, Sahin U. NCOA3 is a selective co-activator of estrogen receptor α-mediated transactivation of PLAC1 in MCF-7 breast cancer cells. BMC Cancer. 2013;13:570. doi: 10.1186/1471-2407-13-570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ding S, Wu X, Li G, Han M, Zhuang Y, Xu T. Efficient transposition of the piggyBac (PB) transposon in mammalian cells and mice. Cell. 2005;122:473–83. doi: 10.1016/j.cell.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 41.Starr TK, Largaespada DA. Cancer gene discovery using the Sleeping Beauty transposon. Cell Cycle. 2005;4:1744–8. doi: 10.4161/cc.4.12.2223. [DOI] [PubMed] [Google Scholar]
- 42.Yang JJ, Krauss RS. Extracellular ATP induces anchorage-independent expression of cyclin A and rescues the transformed phenotype of a ras-resistant mutant cell line. J Biol Chem. 1997;272:3103–8. doi: 10.1074/jbc.272.5.3103. [DOI] [PubMed] [Google Scholar]
- 43.Mann MB, Black MA, Jones DJ, Ward JM, Yew CC, Newberg JY, et al. Transposon mutagenesis identifies genetic drivers of Braf(V600E) melanoma. Nat Genet. 2015;47:486–95. doi: 10.1038/ng.3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Font de Mora J, Brown M. AIB1 is a conduit for kinase-mediated growth factor signaling to the estrogen receptor. Mol Cell Biol. 2000;20:5041–7. doi: 10.1128/MCB.20.14.5041-5047.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sjöblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–74. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 46.Nome T, Hoff AM, Bakken AC, Rognum TO, Nesbakken A, Skotheim RI. High frequency of fusion transcripts involving TCF7L2 in colorectal cancer: novel fusion partner and splice variants. PLoS One. 2014;9:e91264. doi: 10.1371/journal.pone.0091264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bass AJ, Lawrence MS, Brace LE, Ramos AH, Drier Y, Cibulskis K, et al. Genomic sequencing of colorectal adenocarcinomas identifies a recurrent VTI1A-TCF7L2 fusion. Nat Genet. 2011;43:964–8. doi: 10.1038/ng.936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Viel A, Maestro R, Toffoli G, Grion G, Boiocchi M. c-myc overexpression is a tumor-specific phenomenon in a subset of human colorectal carcinomas. J Cancer Res Clin Oncol. 1990;116:288–94. doi: 10.1007/BF01612905. [DOI] [PubMed] [Google Scholar]
- 49.Sears R, Leone G, DeGregori J, Nevins JR. Ras enhances Myc protein stability. Mol Cell. 1999;3:169–79. doi: 10.1016/S1097-2765(00)80308-1. [DOI] [PubMed] [Google Scholar]
- 50.Vizcaíno JA, Csordas A, del-Toro N, Dianes JA, Griss J, Lavidas I, et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016;44:D447–456. doi: 10.1093/nar/gkv1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The sequencing datasets generated during the current study are available in the NCBI Sequence Read Archive (SRA) repository, BioProject accession number PRJNA419878. The datasets for mass-spec analysis (Table S9) have been deposited with the ProteomeXchange Consortium via the PRIDE [50] partner repository with the dataset identifier PXD008382.