

Abstract
Metformin protects against insulin resistance by restoring insulin sensitivity and may also possess anticancer activity. The aim of the present study was to investigate the effects of metformin alone or combined with cisplatin (DDP) on the cell viability and apoptosis of HO-8910 human ovarian cancer cells, and to investigate metformin as a potential novel therapeutic for treating ovarian cancer. The viability of HO-8910 cells was assessed using a cell proliferation and cytotoxicity assay following treatment with different concentrations of metformin (0.01, 0.5, 1, 5 and 10 mM) and/or 5 µM of DDP. Flow cytometry was performed to examine cell apoptosis, and western blotting was used to measure the expression of extracellular signal-related kinase 1/2 (ERK1/2) phosphorylated (p)-ERK1/2, vascular endothelial growth factor (VEGF), VEGF receptor 2 (VEGFR2), B-cell lymphoma 2 (Bcl-2), Bcl-2-associated X (Bax) and caspase-3. The resultsof the present study revealed that metformin reduced the viability of HO-8910 cells in a time- and concentration-dependent manner and induced cell apoptosis in a concentration-dependent manner. Metformin combined with DDP evidently inhibited cell viability and induced apoptosis. In addition, ERK1/2 and genes associated with apoptosis regulation, such as VEGF, VEGFR2, Bcl-2, Bax and caspase-3, exhibited differential expression in the HO-8910 cells. The present study demonstrated that expression of p-ERK1/2, VEGF, VEGFR2 and Bcl-2 was downregulated by treatment with increasing concentrations of metformin, whereas expression of Bax and caspase-3 was evidently upregulated. Taken together, these data demonstrate that metformin in combination with DDP reduces cell viability and induces apoptosis of human ovarian cancer cells.
Keywords: human ovarian cancer, metformin, cisplatin, apoptosis, extracellular signal-related kinase 1/2
Introduction
Ovarian cancer is the fifth leading cause of cancer-associated mortality in women (1). The majority of patients present with advanced-stage disease, as symptoms are rarely overt at early disease stages. As a result, patients with ovarian cancer generally have poor disease prognosis, with a 5-year survival rate of 45% (2). Recent evidence suggests that chemotherapies could increase the long-term survival of ovarian cancer patients, acting through a multitude of different mechanisms to cause ovarian cancer cell death. Metformin (N,N-dimethylbiguanide) has been regarded as potential drugs for ovarian cancer treatment (3,4). However, the mechanisms by which metformin act son the tumorigenesis of ovarian cancer are not fully understood.
Metformin, an oral anti-diabetic drug, is widely used by overweight and obese people with type-2 diabetes (5). Prior studies have identified metformin as having potential as an anticancer drug (6,7). In addition, previous studies have demonstrated that metformin could improve the survival of diabetic patients and decrease their incidence of cancer (8). The detailed mode of action of metformin in cancer cells is not yet clearly understood; however, several reports have demonstrated that this therapeutic agent induces apoptosis through a serine/threonine kinase 11-mediated 5′-adenosine monophosphate-activated protein kinase (AMPK) signaling pathway (9). Other studies have reported that metformin inhibits proliferation and induces cell death through the phosphoinositide 3-kinase/AKT serine/threonine kinase/mechanistic target of rapamycin and AMPK/acetyl coenzyme A carboxylase pathways (4).
Cisplatin (DDP) is a member of a class of platinum-containing chemotherapy drugs used to treat a variety of malignancies, including ovarian, lung, gastric and head and neck cancer (3,10–12). The predominant anti cancer mechanism of DDP is by binding to DNA, which causes crosslinking of DNA strands and ultimately blocks tumor cell proliferation and induces apoptosis (13). Metformin has been demonstrated to act synergistically with DDP to decrease tumor size and inhibit angiogenesis in mice, indicating that it could bea reasonable candidate for combination with DDP-dependent therapy (3). However, a previous study found that metformin antagonizes the DDP-dependent apoptotic effect by suppressing oxidative stress and inhibiting the caspase-dependent activation of apoptosis in certain cancer cells (14).
The present study examined the combination of metformin and DDP as a novel treatment for ovarian cancer. Metformin and DDP were observed to decrease cell viability and induce apoptosis of HO-8910 cells. Further detailed analysis of HO-8910 cells demonstrated that the anticancer effect of metformin was mediated through the inhibition of extracellular signal-regulated kinase 1/2 (ERK1/2) signaling and an increase in the B-cell lymphoma 2-associated X (Bax)/B-cell lymphoma 2 (Bcl-2) ratio and the expression of vascular endothelial growth factor (VEGF), VEGF receptor 2 (VEGFR2), and caspase-3.
Materials and methods
Cell culture
The human ovarian HO-8910 cancer cell line was purchased from the American Type Culture Collection (Rockville, MD, USA), and cultured in 4,500 mg/l D-glucose Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS) (Thermo Fisher Scientific, Inc., Waltham, MA, USA) and 1% penicillin-streptomycin (Beijing Solarbio Science & Technology Co., Ltd., Beijing, China). All cells were incubated in a humidified incubator at 37°C in 5% CO2.
Cell viability assay
Cell viability was analyzed using the Cell Counting Kit-8 (CCK-8; Signalway Antibody LLC, College Park, MD, USA), as previously described (15). HO-8910 cells were cultured in 96-well plates and to 80% confluence. HO-8910 cells were treated with metformin (0.01, 0.5, 1, 5 and 10 mM) or/and DDP (5 mM). The CCK-8 solution (10 ml) at a 1:10 dilution with FBS-free DMEM (100 ml) was added to each wellat 0, 24, 48 and 72 h according to the manufacturer's protocol, followed by a further 1-h incubation under the same incubator conditions. Following this, the absorbance was read at 450 nm using a microplate reader (Molecular Devices, LLC, Sunnyvale, CA, USA). The mean optical density in the indicated groups was used to calculate the percentage of cell viability.
Cell apoptosis assay
Flow cytometric analysis was performed to determine the number of apoptotic cells. Following treatment with metformin (0.5, 1 and 5 mM) and/or DDP (5 µM) for 72 h, the HO-8910 cells were collected by trypsinization, washed twice with PBS, and resuspended in trypsin-EDTA solution (containing 0.25% trypsin and 0.02% EDTA; Beyotime Institute of Biotechnology, Haimen, China) binding buffer (1×106 cells/ml). Next, 195 µl fluorescein isothiocyanate (FITC)-conjugated Annexin V and 5 µl propidium iodide (PI) were added to HO-8910 cells, followed by incubation for 15 min at room temperature in the dark. The stained HO-8910 cells were analyzed by flow cytometry (BD Biosciences, Franklin Lakes, NJ, USA).
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted using TRIzol reagent (Thermo Fisher Scientific, Inc.), and single-stranded cDNA was synthesized from 1 µg of total RNA using the Moloney Murine Leukemia Virus Reverse Transcriptase reagent kit (Thermo Fisher Scientific, Inc.), following the manufacturer's instructions. qPCR was performed with the SYBR-Green PCR Master Mix on an Applied Biosystems 7500 Fast Real-Time PCR system (both from Thermo Fisher Scientific, Inc.). Sequences of primers used are listed in Table I. Data were normalized to GAPDH mRNA content, using the 2−ΔΔCq method (16). The PCR cycling conditions were as follows: 95°C for 10 min; followed by 40 cycles at 95°C for 15 sec and 60°C for 45 sec; and a final extension step of 95°C for 15 sec, 60°C for 1 min, 95°C for 15 sec and 60°C for 15 sec. The experiments were performed at least three times in triplicates.
Table I.
Primers used in the present study.
| Gene | Sequences |
|---|---|
| VEGF (forward) | 5′-GACAGATCACAGGTACAG-3′ |
| VEGF (reverse) | 5′-GAAGCAGGTGAGAGTAAG-3′ |
| VEGFR2 (forward) | 5′-GGGAGTCTGTGGCATCTGAAG-3′ |
| VEGFR2 (reverse) | 5′-AATCTGGGCTGTGCTACCG-3′ |
| Bax (forward) | 5′-AGCTGAGCGAGTGTCTCAAG-3′ |
| Bax (reverse) | 5′-TGTCCAGCCCATGATGGTTC-3 |
| Bcl-2 (forward) | 5′-AGACCGAAGTCCGCAGAACC-3′ |
| Bcl-2 (reverse) | 5′-GAGACCACACTGCCCTGTTG-3′ |
| Caspase-3 (forward) | 5′-AACTGGACTGTGGCATTGAG-3′ |
| Caspase-3 (reverse) | 5′-ACAAAGCGACTGGATGAACC-3′ |
| GAPDH (forward) | 5′-CACCCACTCCTCCACCTTTG-3′ |
| GAPDH (reverse) | 5′-CCACCACCCTGTTGCTGTAG-3′ |
VEGF, vascular endothelial growth factor; VEGFR2, VEGF receptor 2; Bcl-2, B-cell lymphoma 2; Bax, Bcl-2 associated X protein; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
SDS-PAGE and western blot analysis
Following metformin and/or DDP treatment for the indicated times, cells were lysed using radioimmunoprecipitation assay buffer containing protease inhibitor (Beyotime Institute of Biotechnology, Inc., Shanghai, China). The protein concentration was determined using a bicinchoninic acid assay (Thermo Fisher Scientific, Inc.). For each lane, 30 µg of protein was separated by 12% SDS-PAGE and transferred onto a nitrocellulose membrane (EMD Millipore, Billerica, MA, USA). Following blocking in fat-free milk over night at 4°C, the membranes were incubated overnight at 4°C with the following primary antibodies: Monoclonal rabbit anti-p-ERK1/2 (1:1,000 dilution; cat no. 4376); monoclonal rabbit anti-ERK1/2 (1:1,000 dilution; cat no. 4695) (both from Cell Signaling Technology, Inc., Danvers, MA, USA); monoclonal rabbit anti-VEGF (1:1,000 dilution; cat no. ab46154); monoclonal rabbit anti-VEGFR2 (1:1,000 dilution; cat no. ab11939); monoclonal rabbit anti-caspase-3 (1:500; cat no. ab44976) (all from Abcam, Cambridge, MA, USA); polyclonal rsabbit anti-Bax (1:1,000 dilution; cat no. sc-493); polyclonal rabbit anti-Bcl-2 (1:1,000 dilution; cat no. sc-492) (both from Santa Cruz Biotechnology, Inc., Dallas, TX, USA) or monoclonal rabbit anti-GAPDH (1:1,500 dilution; cat no. 5174; Cell Signaling Technology, Inc.). Following 3 additional Tris-buffered saline-Tween-20 washes, the membranes were incubated with goat anti-rabbit horseradish peroxidase-conjugated secondary antibodies (1:1,000 dilution; cat no. A0208; Beyotime Institute of Biotechnology, Inc.,) for 1 h at 37°C and detected by enhanced chemiluminescence (Pierce; Thermo Fisher Scientific). Signals were quantified by densitometry (Quantity One software, version 4.62; Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Statistical analysis
Each experimental value was expressed as the mean ± standard deviation. The statistical significance of the difference between the values of control and treatment groups was determined using either unpaired, two-tail Student's t-test or simple one-way analysis of variance and Tukey's post-hoc test using GraphPad Prism 5 software (GraphPad Software, Inc., La Jolla, CA, USA). P<0.05 was considered to indicate a statistically significant difference.
Results
Metformin combined with DDP inhibits ERK1/2 activation
The HO-8910 human ovarian cancer cell line was treated with different concentrations of metformin for 6 h to examine its effects on the ERK1/2 activity. Treatment with metformin (0.5, 1 and 5 mM) reduced the levels of phosphorylated (p)-ERK1/2 in a concentration-dependent manner in HO-8910 cells (Fig. 1). Metformin combined with DDP significantly decreased the p-ERK1/2 level to lower than in cells treated with metformin alone. However, metformin (0.5, 1 and 5 mM) and DDP (5 µM) alone or combination treatment had no effects on the total expression of ERK1/2 in HO-8910 cells. These data suggest that metformin combined with DDP may inhibit ERK1/2 activity in ovarian cancer cells.
Figure 1.
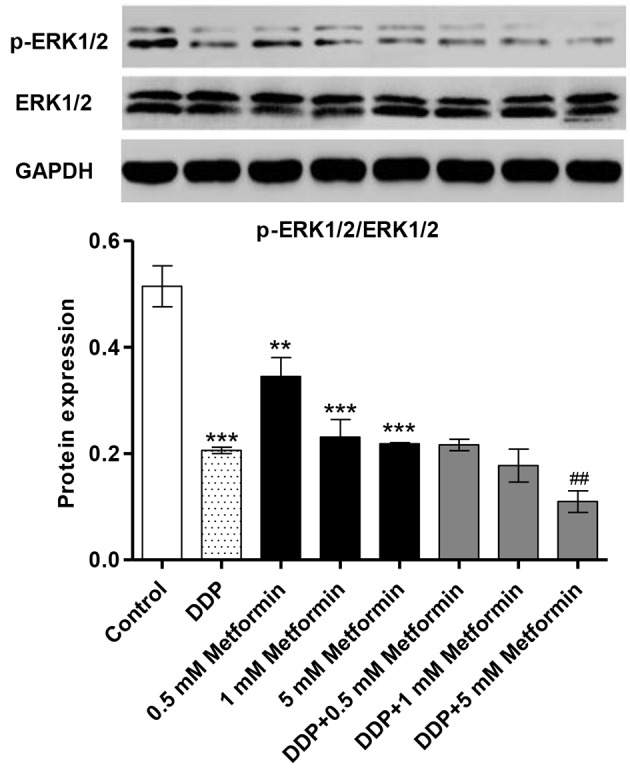
Metformin and DDP inhibit the expression of the ERK1/2 in ovarian cancer cells. HO-8910 cells were treated with metformin (0.5, 1, and 5 mM) or DDP (5 µM) or in combination (0.5, 1, and 5 mM metformin plus 5 µM DDP) for 6 h to detect their effects on the ERK1/2 activity. Western blot analysis was used to examine the levels of ERK1/2 and p-ERK1/2 following treatment with metformin, DDP or both. **P<0.01 and ***P<0.001 vs. control. ##P<0.01 vs. DDP. DDP, cisplatin; p-ERK1/2, phosphorylated ERK1/2; ERK, extracellular signal-regulated kinase.
Metformin combined with DDP reduces the expression of VEGF and VEGFR2 in HO-8910 cells
The expressions of VEGF and VEGFR2 in HO-8910 cells were examined following metformin (0.5, 1 and 5 mM) and DDP (5 µM) treatment using RT-qPCR and western blot analysis. The expression of VEGF and VEGFR2 mRNA in HO-8910 cells was significantly decreased by metformin treatment in a concentration-dependent manner (Fig. 2A). Notably, metformin combined with DDP in significantly decreased the expression of VEGF and VEGFR2 mRNA to even lower levels than with DDP treatment alone (Fig. 2B). Furthermore, the levels of VEGF and VEGFR2 protein were also significantly decreased by metformin or/and DDP treatment in HO-8910 cells (Fig. 2C-E). However, a lower concentration of metformin (0.5 mM) had no effect on VEGF protein expression alone or in combination with DDP in HO-8910 cells (Fig. 2D). Only higher concentrations of metformin enhanced the inhibition of VEGFR2 protein expression induced by DDP treatment in HO-8910 cells (Fig. 2E). These results suggest that metformin combined with DDP effectively reduced the expression of VEGF and VEGFR2, which may reduce angiogenesis.
Figure 2.

Metformin and DDP inhibit the expression of angiogenesis-associated factors in ovarian cancer cells. HO-8910 cellswere treated with metformin (0.5, 1, and 5 mM) or DDP (5 µM) alone or in combination (0.5, 1, or 5 mM metformin plus 5 µM DDP) for 48 h to detect their effects on the expressions of VEGF and VEGFR2. Reverse transcription-quantitative polymerase chain reaction assay was used to examine the (A) VEGF and (B) VEGFR2 mRNA expression following treatment with metformin, DDP or both. (C) Western blot assay was used to examine the protein expressions of (D) VEGF and (E) VEGFR2 following treatment with metformin, DDP or both. *P<0.05, **P<0.01 and ***P<0.001 vs. control. #P<0.05, ##P<0.01 and ###P<0.001 vs. DDP. DDP, cisplatin; VEGF, vascular endothelial growth factor; VEGFR2, VEGF receptor 2.
Metformin combined with DDP induces the expression of Bax, Bcl-2 and caspase-3 in HO-8910 cells
Expression of the apoptosis-associated factors, Bax, Bcl-2 and caspase-3, were also quantified following metformin or/and DDP treatment. Expression of Bax and caspase-3 mRNA was elevated in a dose-dependent manner upon metformin (0.5, 1 and 5 mM) and DDP (5 µM) treatment alone, while combined metformin and DDP treatment enhanced the increase in mRNA expression of Bax and caspase-3 in HO-8910 cells (Fig. 3A and B). However, expression of Bcl-2 mRNA was significantly decreased by metformin treatment in a concentration-dependent manner, with combined metformin and DDP treatment enhancing the effect of treatment with either alone. Expression of Bax and caspase-3 protein was also increased by metformin and DDP alone or combination, whereas expression of Bcl-2 protein was decreased (Fig. 3C-F). However, the lowest concentration (0.5 mM) of metformin had no effect on the expression of caspase-3 protein, either alone or in combination with DDP (Fig. 3F). These results suggest that metformin combined with DDP can effectively induce cell apoptosis by increasing Bax/Bcl-2 ratio and caspase-3 expression.
Figure 3.

Effect of metformin and DDP on the expression of apoptosis-associated factors in ovarian cancer cells. HO-8910 cells were treated with metformin (0.5, 1, and 5 mM), DDP (5 µM) or the two in combination (0.5, 1, and 5 mM metformin plus 5 µM DDP) for 48 h to detect their effects on the expression of Bax, Bcl-2 and caspase-3. Reverse transcription-quantitative polymerase chain reaction was used to examine the mRNA expression of Bax, Bcl-2 and caspase-3 following treatment with (A) metformin or (B) DDP alone or DDP and metformin in combination. (C) Western blot analysis was used to examine the protein expression of (D) Bax, (E) Bcl-2 and (F) caspase-3 following treatment with metformin, DDP or the two combined. *P<0.05, **P<0.01 and ***P<0.001 vs. control. #P<0.05, ##P<0.01 and ###P<0.001 vs. DDP. DDP, cisplatin; Bcl-2, B-cell lymphoma 2; Bax, Bcl-2 associated X.
Metformin combined with DDP inhibits the growth of HO-8910 cells
Having determined the effect of metformin and DDP on ERK1/2 signaling, a CCK-8 cell viability and cytotoxicity assay was performed to investigate the biological effect of the drug son the phenotypes of HO-8910 cells. Metformin (0.01, 0.5, 1, 5 and 10 mM) inhibited the cell viability of HO-8910 cells in a time- and concentration-dependent manner (Fig. 4A). When combined, metformin (0.5, 1, and 5 mM) and DDP (5 µM) inhibited cell growth in a concentration-dependent manner compared with DDP treatment alone (Fig. 4B). These findings suggested that metformin combined with DDP effectively inhibited the cell viability of ovarian cancer cells.
Figure 4.

Metformin and DDP inhibit the cell viability of ovarian cancer HO-8910 cells. Cell viability was measured using a cell proliferation and cytotoxicity assay. HO-8910 cells were treated with (A) metformin (0.01, 0.5, 1, 5, and 10 mM) or (B) DDP (5 µM) alone or in combination with metformin (0.5, 1, and 5 mM metformin plus 5 µM DDP) (B) for 0, 24, 48 and 72 h to examine their effects on cell proliferation. **P<0.01 and ***P<0.001 vs. control. #P<0.05 and ###P<0.001 vs. DDP. DDP, cisplatin.
Metformin combined with DDP induces cell apoptosis of HO-8910 cells
Due to the inhibitory effect of metformin and DDP on ovarian cancer cell proliferation, the effects of metformin and DDP on cell apoptosis of ovarian cancer cells were further investigated. HO-8910 cells treated with metformin and/or DDP treatment were stained with FITC-labeled annexinV/PI and subsequently subjected to flow cytometry analysis. The results revealed that metformin (0.5,1 and 5 mM) significantly increased the number of apoptotic HO-8910 cells in a concentration-dependent manner (Fig. 5A). Additionally, metformin (0.5, 1, and 5 mM) treatment enhanced the apoptotic effect of DDP (5 µM) in HO-8910 cells in a concentration-dependent manner (Fig. 5B). These findings suggest that metformin combined with DDP effectively induces the apoptosis of ovarian cancer cells.
Figure 5.

Metformin and DDP induce apoptosis in ovarian cancer cells. Cell apoptosis was measured by flow cytometry analysis. HO-8910 cells were treated with (A) metformin (0.01, 0.5, 1, 5, or 10 mM) or (B) DDP (5 µM) or a combination of the two (0.5, 1, or 5 µM metformin plus 5 µM DDP) for 48 h to examine their effects on apoptosis. ***P<0.001 compared with control. ##P<0.01 and ###P<0.001 vs. DDP. DDP, cisplatin.
Discussion
The present study demonstrated that metformin alone or combined with DDP inhibited the proliferation of HO-8910 ovarian cancer cells and induced apoptosis via ERK1/2 deactivation. Previous studies have observed that metformin inhibited tumor growth, including in prostate and breast cancer (6,17). The ERK pathway has a critical role in cell proliferation and survival, and represents a conserved signaling system throughout evolution. Activation of the ERK1/2 pathway has been documented in colorectal (18), breast (19) and prostate cancer (20), and in several ovarian cancer cell lines (21). Metformin treatment of human mammary endothelial cells resulted in decreased migration and invasion via inhibition of the ERK1/2 pathway, resulting in the upregulation of thrombospondin-1 expression, which has been shown to be involved in platelet aggregation, angiogenesis and tumorigenesis (22). However, metformin treatment had no effect on the phosphorylation of ERK in U251 human glioma cells (23) and treatment with DDP led to the strong activation of ERK1/2 in SKOV3 human ovarian carcinoma cells (24), findings that are contrary to those of the present study.
VEGF is expressed by tumor cells and initiates vasculogenesis and angiogenesis via binding to the tyrosine kinase receptors (VEGFRs) on endothelial cells. VEGFR2 appears to mediate the majority of the known cellular responses to VEGF. In human breast cancer, VEGF stimulation increased the degree of ERK1/2 phosphorylation, indicating the activation of ERK1/2-associated intra cellular pathways (25). A previous study reported that metformin decreased vascular permeability and expression of VEGF, suggesting that metformin normalizes vascular permeability by regulating VEGF levels (26). Additionally, the effects of metformin and DDP on VEGF modulation have been also described in ovarian cancer in vivo and in Lewis lung cancer rats (3,27). In the present study, metformin alone or in combination with DDP was demonstrated to significantly decrease levels of VEGF and VEGFR2 in HO-8910 cells in a concentration-dependent manner, implicating VEGF/VEGFR2 in the ERK1/2-dependent anticancer effects of metformin and DDP in ovarian cancer.
Furthermore, reduction of ERK1/2 activity causes G1 cell cycle arrest, downregulation of Bcl-2 expression levels, increased activity of caspase-3, −6, −8, and −9, and induces apoptotic cell death (28). In the present study, inhibition of ERK1/2 by treatment with metformin in ovarian cancer cells increased the expression of Bax, with the amount of apoptosis further increased by DDP, which is in line with previous reports (29). In the present study, the effect of metformin and DDP on cell viability and apoptosis was assessed using CCK-8 and flow cytometry analysis, respectively. The results of these assays demonstrated that metformin alone, or in combination with DDP, inhibited cell viability and promoted apoptosis in a concentration-dependent manner. Notably, a previous study reported that metformin treatment inhibited the growth of metastatic nodules and enhanced DDP-dependent cytotoxicity in ovarian cancer, resulting in the inhibition of tumor cell growth by ~90% (3). However, Lesan et al (30) reported that metformin suppressed the anticancer efficacy of DDP and reduced DDP-mediated apoptosis in the MKN-45 cancer cell line. Further in vitro and in vivo studies are therefore required to elucidate the mechanism of action of these therapeutic agents in combination on ovarian cancer cells, as well as other cancer types.
In conclusion, the results of the present study support the use of metformin as an anticancer drug, either alone or in combination with DDP, as it inhibited cell viability and angiogenesis, and induced apoptosis. Therefore, these results provide a strong rationale for the use of metformin as a therapeutic drug in combination with DDP in the treatment of patients with ovarian cancer.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin6. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 2.Smith HO, Arias-Pulido H, Kuo DY, Howard T, Qualls CR, Lee SJ, Verschraegen CF, Hathaway HJ, Joste NE, Prossnitz ER. GPR30 predicts poor survival for ovarian cancer. Gynecol Oncol. 2009;114:465–471. doi: 10.1016/j.ygyno.2009.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rattan R, Graham RP, Maguire JL, Giri S, Shridhar V. Metformin suppresses ovarian cancer growth and metastasis with enhancement of cisplatin cytotoxicity in vivo. Neoplasia. 2011;13:483–491. doi: 10.1593/neo.11148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li C, Liu VW, Chan DW, Yao KM, Ngan HY. LY294002 and metformin cooperatively enhance the inhibition of growth and the induction of apoptosis of ovarian cancer cells. Int J Gynecol Cancer. 2012;22:15–22. doi: 10.1097/IGC.0b013e3182322834. [DOI] [PubMed] [Google Scholar]
- 5.Correia S, Carvalho C, Santos MS, Proença T, Nunes E, Duarte AI, Monteiro P, Seiça R, Oliveira CR, Moreira PI. Metformin protects the brain against the oxidative imbalance promoted by type 2 diabetes. Med Chem. 2008;4:358–364. doi: 10.2174/157340608784872299. [DOI] [PubMed] [Google Scholar]
- 6.Kourelis TV, Siegel RD. Metformin and cancer: New applications for an old drug. Med Oncol. 2012;29:1314–1327. doi: 10.1007/s12032-011-9846-7. [DOI] [PubMed] [Google Scholar]
- 7.Gonzalez-Angulo AM, Meric-Bernstam F. Metformin: A therapeutic opportunity in breast cancer. Clin Cancer Res. 2010;16:1695–1700. doi: 10.1158/1078-0432.CCR-09-1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiralerspong S, Palla SL, Giordano SH, Meric-Bernstam F, Liedtke C, Barnett CM, Hsu L, Hung MC, Hortobagyi GN, Gonzalez-Angulo AM. Metformin and pathologic complete responses to neoadjuvant chemotherapy in diabetic patients with breast cancer. J Clin Oncol. 2009;27:3297–3302. doi: 10.1200/JCO.2009.19.6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Foretz M, Hébrard S, Leclerc J, Zarrinpashneh E, Soty M, Mithieux G, Sakamoto K, Andreelli F, Viollet B. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J Clin Invest. 2010;120:2355–2369. doi: 10.1172/JCI40671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scagliotti GV, Parikh P, von Pawel J, Biesma B, Vansteenkiste J, Manegold C, Serwatowski P, Gatzemeier U, Digumarti R, Zukin M, et al. Phase III study comparing cisplatin plus gemcitabine with cisplatin plus pemetrexed in chemotherapy-naive patients with advanced-stage non-small-cell lung cancer. J Clin Oncol. 2008;26:3543–3551. doi: 10.1200/JCO.2007.15.0375. [DOI] [PubMed] [Google Scholar]
- 11.Kang YK, Kang WK, Shin DB, Chen J, Xiong J, Wang J, Lichinitser M, Guan Z, Khasanov R, Zheng L, et al. Capecitabine/cisplatin versus 5-fluorouracil/cisplatin as first-line therapy in patients with advanced gastric cancer: A randomised phase III noninferiority trial. Ann Oncol. 2009;20:666–673. doi: 10.1093/annonc/mdn717. [DOI] [PubMed] [Google Scholar]
- 12.Lorch JH, Goloubeva O, Haddad RI, Cullen K, Sarlis N, Tishler R, Tan M, Fasciano J, Sammartino DE, Posner MR. TAX 324 Study Group: Induction chemotherapy with cisplatin and fluorouracil alone or in combination with docetaxel in locally advanced squamous-cell cancer of the head and neck: Long-term results of the TAX 324 randomised phase 3 trial. Lancet Oncol. 2011;12:153–159. doi: 10.1016/S1470-2045(10)70279-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaina B. DNA damage-triggered apoptosis: Critical role of DNA repair, double-strand breaks, cell proliferation and signaling. Biochem Pharmacol. 2003;66:1547–1554. doi: 10.1016/S0006-2952(03)00510-0. [DOI] [PubMed] [Google Scholar]
- 14.Chang J, Jung HH, Yang JY, Lee S, Choi J, Im GJ, Chae SW. Protective effect of metformin against cisplatin-induced ototoxicity in an auditory cell line. J Assoc Res Otolaryngol. 2014;15:149–158. doi: 10.1007/s10162-013-0431-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang YH, Wang Y, Yusufali AH, Ashby F, Zhang D, Yin ZF, Aslanidi GV, Srivastava A, Ling CQ, Ling C. Cytotoxic genes from traditional Chinese medicine inhibit tumor growth both in vitro and in vivo. J Integr Med. 2014;12:483–494. doi: 10.1016/S2095-4964(14)60057-1. [DOI] [PubMed] [Google Scholar]
- 16.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 17.Alimova IN, Liu B, Fan Z, Edgerton SM, Dillon T, Lind SE, Thor AD. Metformin inhibits breast cancer cell growth, colony formation and induces cell cycle arrest in vitro. Cell Cycle. 2009;8:909–915. doi: 10.4161/cc.8.6.7933. [DOI] [PubMed] [Google Scholar]
- 18.Yeh JJ, Routh ED, Rubinas T, Peacock J, Martin TD, Shen XJ, Sandler RS, Kim HJ, Keku TO, Der CJ. KRAS/BRAF mutation status and ERK1/2 activation as biomarkers for MEK1/2 inhibitor therapy in colorectal cancer. Mol Cancer Ther. 2009;8:834–843. doi: 10.1158/1535-7163.MCT-08-0972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sirianni R, Chimento A, De Luca A, Casaburi I, Rizza P, Onofrio A, Lacopetta D, Puoci F, Andò S, Maggiolini M, Pezzi V. Oleuropein and hydroxytyrosol inhibit MCF-7 breast cancer cell proliferation interfering with ERK1/2 activation. Mol Nutr Food Res. 2010;54:833–840. doi: 10.1002/mnfr.200900111. [DOI] [PubMed] [Google Scholar]
- 20.Albrecht DS, Clubbs EA, Ferruzzi M, Bomser JA. Epigallocatechin-3-gallate (EGCG) inhibits PC-3 prostate cancer cell proliferation via MEK-independent ERK1/2 activation. Chem Biol Interact. 2008;171:89–95. doi: 10.1016/j.cbi.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 21.Steinmetz R, Wagoner HA, Zeng P, Hammond JR, Hannon TS, Meyers JL, Pescovitz OH. Mechanisms regulating the constitutive activation of the extracellular signal-regulated kinase (ERK) signaling pathway in ovarian cancer and the effect of ribonucleic acid interference for ERK1/2 on cancer cell proliferation. Mol Endocrinol. 2004;18:2570–2582. doi: 10.1210/me.2004-0082. [DOI] [PubMed] [Google Scholar]
- 22.Tan BK, Adya R, Chen J, Farhatullah S, Heutling D, Mitchell D, Lehnert H, Randeva HS. Metformin decreases angiogenesis via NF-kappaB and Erk1/2/Erk5 pathways by increasing the antiangiogenic thrombospondin-1. Cardiovasc Res. 2009;83:566–574. doi: 10.1093/cvr/cvp131. [DOI] [PubMed] [Google Scholar]
- 23.Janjetovic K, Vucicevic L, Misirkic M, Vilimanovich U, Tovilovic G, Zogovic N, Nikolic Z, Jovanovic S, Bumbasirevic V, Trajkovic V, Harhaji-Trajkovic L. Metformin reduces cisplatin-mediated apoptotic death of cancer cells through AMPK-independent activation of Akt. Eur J Pharmacol. 2011;651:41–50. doi: 10.1016/j.ejphar.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 24.Wei SQ, Sui LH, Zheng JH, Zhang GM, Kao YL. Role of ERK1/2 kinase in cisplatin-induced apoptosis in human ovarian carcinoma cells. Chin Med Sci J. 2004;19:125–129. [PubMed] [Google Scholar]
- 25.Kim EH, Na HK, Surh YJ. Upregulation of VEGF by 15-deoxy-Delta12,14-prostaglandin J2 via heme oxygenase-1 and ERK1/2 signaling in MCF-7 cells. Ann NY Acad Sci. 2006;1090:375–384. doi: 10.1196/annals.1378.041. [DOI] [PubMed] [Google Scholar]
- 26.Elia EM, Quintana R, Carrere C, Bazzano MV, Rey-Valzacchi G, Paz DA, Pustovrh MC. Metformin decreases the incidence of ovarian hyperstimulation syndrome: An experimental study. J Ovarian Res. 2013;6:62. doi: 10.1186/1757-2215-6-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feng P, Zhang ZL, Zhang ZH, Zhang XL, Xiang F, Tang JH, Xiang BL. Effect of endostar combined with cisplatin on expression of VEGF and Sema3A of lewis lung cancer rats. Asian Pac J Trop Med. 2013;6:57–60. doi: 10.1016/S1995-7645(12)60201-6. [DOI] [PubMed] [Google Scholar]
- 28.Boucher MJ, Morisset J, Vachon PH, Reed JC, Lainé J, Rivard N. MEK/ERK signaling pathway regulates the expression of Bcl-2, Bcl-X(L), and Mcl-1 and promotes survival of human pancreatic cancer cells. J Cell Biochem. 2000;79:355–369. doi: 10.1002/1097-4644(20001201)79:3<355::AID-JCB20>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 29.Yasmeen A, Beauchamp MC, Piura E, Segal E, Pollak M, Gotlieb WH. Induction of apoptosis by metformin in epithelial ovarian cancer: Involvement of the Bcl-2 family proteins. Gynecol Oncol. 2011;121:492–498. doi: 10.1016/j.ygyno.2011.02.021. [DOI] [PubMed] [Google Scholar]
- 30.Lesan V, Ghaffari SH, Salaramoli J, Heidari M, Rostami M, Alimoghaddam K, Ghavamzadeh A. Evaluation of antagonistic effects of metformin with Cisplatin in gastric cancer cells. Int J Hematol Oncol Stem Cell Res. 2014;8:12–19. [PMC free article] [PubMed] [Google Scholar]