

Abstract
Research on ionizing radiation (IR)-induced clonogenic cell death is important for understanding the effect of IR on malignant tumors and normal tissues. Here, we describe a quick and cost-effective one-step assay for simultaneously assessing the major modes of clonogenic cell death induced by IR, i.e., apoptosis, mitotic catastrophe, and cellular senescence. In this method, cells grown on a cover slip are irradiated with X-rays and stained with 4',6-diamidino-2-phenylindole dihydrochloride (DAPI). Using fluorescence microscopy, apoptosis, mitotic catastrophe, and cellular senescence are identified based on the characteristic morphologies of the DAPI-stained nuclei. Apoptosis is determined by the presence of apoptotic bodies (i.e., condensed and fragmented nuclei). Mitotic catastrophe is determined by the presence of nuclei that exhibit two or more distinct lobes and micronuclei. Cellular senescence is determined by the presence of senescence-associated heterochromatic foci (i.e., nuclear DNA containing 30-50 bright, dense foci). This approach allows the experimenter to easily screen for clonogenic cell death modes using various cell lines, treatment settings, and/or time points, with the goal of elucidating the mechanisms of cell death in the target cells and conditions of interest.
Keywords: Cancer Research, Issue 128, Radiation biology, ionizing radiation, clonogenic cell death, apoptosis, mitotic catastrophe, cellular senescence, fluorescence microscopy, DAPI staining
Introduction
Ionizing radiation (IR) induces multiple modes of clonogenic cell death. Research on IR-induced clonogenic cell death is important for understanding the toxicity of IR to normal tissues, as well as for developing methods to increase the treatment efficacy of cancer radiotherapy. Apoptosis, mitotic catastrophe, and cellular senescence are the major modes of clonogenic cell death induced by IR1. Apoptosis is a regulated mode of cell death that is initiated by DNA damage1. Mitotic catastrophe is cell death that occurs due to aberrant mitosis resulting from unrepaired DNA double-strand breaks1. Cellular senescence is defined as a state of irreversible cell growth arrest2; note that cellular senescence is not cell death per se, but it is a mode of clonogenic cell death because it abolishes the clonogenic survival of the senescent cell.
Various cell-based assays have been developed to individually assess apoptosis, mitotic catastrophe, and cellular senescence. Apoptosis can be assessed by terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) staining, annexin V staining, DNA fragmentation assays, and sub-G1 cell-cycle phase determination by flow cytometry1. Mitotic catastrophe can be assessed by immunofluorescence staining for mitotic markers, including MPM2, TUNEL staining, and electron microscopy1. Cellular senescence can be assessed by senescence-associated β-galactosidase (SA-β-Gal) staining, growth-arrest assays, and electron microscopy1. Importantly, the predominant mode of clonogenic cell death differs among cell lines and treatment settings3,4. Therefore, to elucidate the overall profile of clonogenic cell death for a given experimental setting, multiple assays covering all of these modes of death must be conducted together, which is labor- and cost-intensive.
In this article, we describe a quick and cost-effective one-step assay for simultaneously assessing apoptosis, mitotic catastrophe, and cellular senescence induced by IR3,4. In this method, cells grown on a cover slip are irradiated with X-rays and stained with 4',6-diamidino-2-phenylindole dihydrochloride (DAPI). Using fluorescence microscopy, apoptosis, mitotic catastrophe, and cellular senescence are each identified based on the respective characteristic morphologies of the DAPI-stained nuclei. This approach will be useful for applications including screening for clonogenic cell death profiles in various cell lines, treatment settings, and time points, with the goal of investigating the mechanisms of cell death in the target cells and conditions of interest.
Protocol
1. Preparation of Materials
- Autoclave cover slips.
- Place cover slips in a glass beaker.
- Cover the glass beaker with foil.
- Autoclave at 121 °C at 15 psi for 20 min.
- Dry at 50 °C. Store at room temperature in a culture hood.
- Prepare Fixation Solution: 3% paraformaldehyde + 2% sucrose in phosphate-buffered saline (PBS).
- To a 1,000 mL glass bottle, add 700 mL PBS and 30 g paraformaldehyde. CAUTION: Paraformaldehyde is corrosive, wear appropriate gloves.
- Dissolve paraformaldehyde completely by heating to 80 °C using a microwave. CAUTION: Paraformaldehyde fumes are toxic, wear a mask.
- Leave at room temperature overnight.
- Add 20 g sucrose.
- Add PBS to make the total volume 1 L.
- Aliquot in 50 mL tubes. Fixation Solution can be stored for 3 years at -20 °C or for 3 months at 4 °C.
2. Preparation of Cell Culture
NOTE: These procedures should be performed in a culture hood.
Culture and passage U2OS human osteosarcoma cells to maintain logarithmic growth5. NOTE: Other cell types can be used after determining the optimal irradiation dose.
Wipe a scalpel with paper towels moistened with 70% ethanol. Using the scalpel, place a cover slip on a 35 mm cell culture dish (hereafter simply referred to as "the dish"). NOTE: The number of cover slips in one dish can be increased according to the experimental design (see Discussion).
Add 1 mL culture media: DMEM supplemented with 10% fetal bovine serum. NOTE: Other media can be used according to the chosen cell type.
Wipe forceps with paper towels moistened with 70% ethanol. Using the forceps, gently hold the center of the cover slip. Aspirate the culture media from the dish completely, while maintaining a hold on the cover slip. NOTE: This step promotes immobilization of the cover slip to the dish.
- Detach adherent cultured cells using trypsin5.
- Aspirate culture media from the dish.
- Add 1 mL PBS and shake the dish gently.
- Aspirate PBS from the dish.
- Add 1 mL trypsin [0.25w/v% Trypsin-1mmol/L EDTA] to the dish.
- Incubate for 5 min at 37 °C in an atmosphere containing 5% CO2.
- Add 9 mL culture media to the dish.
- Using a 1000 μL micropipette, prepare a single-cell suspension.
- Count the cells5.
- In a 1.5 mL tube, add 0.9 mL cell suspension and 0.1 mL 0.4% Trypan Blue solution.
- Apply 10 μL to a hemocytometer.
- Examine the number of unstained (i.e., live) cells under an inverted-phase microscope.
In a 15 mL tube, prepare 3 mL cell suspension in culture media at a density of 0.5 x 105 cells/mL. NOTE: Cell density can be modified according to experimental need (see Discussion).
Gently add 2 mL of the cell suspension to the dish.
Incubate overnight at 37 °C in an atmosphere containing 5% CO2.
3. Irradiation
CAUTION: Handle the X-ray irradiator carefully according to the manufacturer's instructions.
Examine the cells under an inverted-phase microscope. Confirm that the cells are attached to the cover slip and alive.
Irradiate the dish with 6 Gy X-rays (1.4 Gy/min, 300 KVP, 20 mA).
Incubate for 72 h at 37 °C in an atmosphere containing 5% CO2. NOTE: Incubation time can be modified according to the experimental design (see Discussion).
4. Fixation
Aspirate culture media from the dish.
Using scissors, cut the tip of a 1,000 μL micropipette tip 5 mm from the end. NOTE: The cut tip helps to smoothly apply the Fixation Solution.
Using the cut tip, add 1 mL Fixation Solution (prepared in step 1.2) to the dish from the side wall of the dish. NOTE: This step is time-sensitive and should therefore be performed without changing micropipette tips when handling multiple samples. Application of Fixation Solution directly to the bottom of the dish can damage the cells.
Place the dish in a square culture dish. Shake the square culture dish gently to distribute the Fixation Solution over the cover slip evenly.
Incubate for 10 min at room temperature.
Aspirate Fixation Solution from the dish.
Add 2 mL of PBS to the dish from the side wall of the dish. NOTE: Application of PBS directly to the bottom of the dish can damage the cells.
Aspirate PBS from the dish.
Repeat steps 4.7 and 4.8 twice. NOTE: The dish can be stored for 1 week at 4 °C with the cover slips bathed in 2 mL PBS.
5. DAPI Staining
To a slide glass, apply 5 μL 1 μg/mL DAPI staining reagent. NOTE: The DAPI staining reagent is stable for 6 months when stored protected from light at or below -20 °C.
Using a scalpel, remove the cover slip from the dish.
Draw off the excess PBS on the cover slip by touching the edge of the cover slip with a paper towel.
Mount the cover slip upside-down onto the drop of DAPI staining reagent on the slide glass so that the cells are exposed to DAPI staining reagent. NOTE: This step is time-sensitive. Drying of the DAPI staining reagent can lead to suboptimal staining.
The samples can be stored for 1 year at -20 °C.
6. Image Acquisition
Examine the sample on a fluorescence microscope. Nuclei are visualized using the DAPI filter. NOTE: Either a 20X or a 60X oil lens can be used, according to the researcher's interest. When using a 60X oil lens, add one drop of oil onto the cover slip. Be careful that the samples do not touch the lens; otherwise, the lens can be damaged.
Acquire images of nuclei using a CCD camera and digital image acquisition software with the following settings and parameters: DAPI filter, monolayer images, gain of 1X, and automatic exposure.
Finish image acquisition: Wipe the 20X lens with paper towels moistened with lens cleaner. Wipe the 60X oil lens with paper towels moistened with chloroform.
7. Evaluation of Clonogenic Cell Death Mode
- In randomly selected images, count the number of nuclei that meet the criteria for apoptosis, mitotic catastrophe, and cellular senescence until the total number of counted nuclei reaches 300. Conduct this step in triplicate for each experimental setting.
Representative Results
As an example, U2OS human osteosarcoma cells were treated with 6 Gy X-rays, incubated for 72 h, and subjected to the DAPI staining assay according to the protocol.Figure 1 shows zoomed images for the typical nuclear morphologies associated with apoptosis, mitotic catastrophe, and cellular senescence obtained using a 60× oil lens. Refer to steps 7.1.1-7.1.3 for the characteristic morphologies for each mode of clonogenic cell death.Figure 2shows overview images of nuclei obtained using a 20X lens. Irradiation with 6 Gy X-rays induced mitotic catastrophe frequently, apoptosis less frequently, and cellular senescence rarely. In this manner, examination at a low-magnification field allows one to apprehend at a glance the overall picture of the clonogenic cell death profile in a given experimental setting.Figure 3 shows a graphical representation of the quantified data.
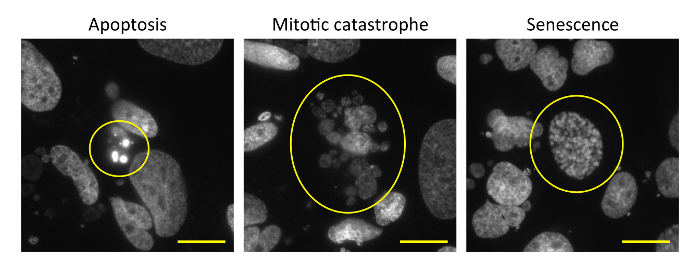
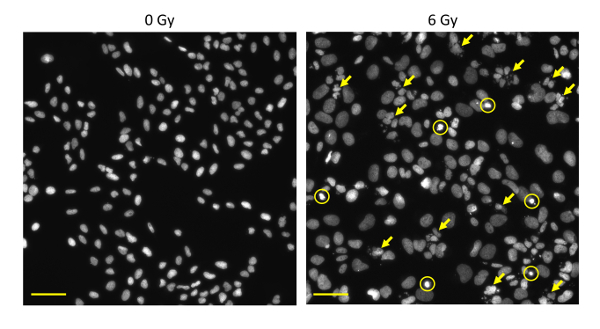
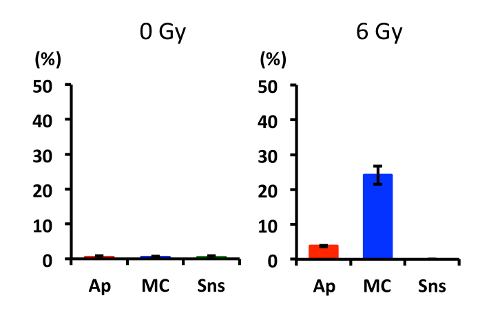
Figure 1: Zoomed images of DAPI-stained nuclei showing typical morphologies of apoptosis, mitotic catastrophe, and cellular senescence. U2OS cells were irradiated with 6 Gy X-rays. After 72 h, the cells were fixed and stained with DAPI. Nuclear images were acquired using a 60X oil lens. Scale bar = 20 μm. Please click here to view a larger version of this figure.
Figure 2: Overview images of DAPI-stained nuclei of cells treated with X-rays. U2OS cells were irradiated with 6 Gy X-rays or mock-irradiated. After 72 h, the cells were fixed and stained with DAPI. Images of nuclei were acquired using a 20X lens. Circles, apoptosis; arrows, mitotic catastrophe. Scale bar = 50 μm. Please click here to view a larger version of this figure.
Figure 3: Quantified data for apoptosis, mitotic catastrophe, and cellular senescence induced in cells treated with X-rays. U2OS cells were irradiated with 6 Gy X-rays or mock-irradiated. After 72 h, the cells were fixed and stained with DAPI. Images of nuclei were acquired using a 20X lens. From random fields, a total of 300 nuclei were evaluated for apoptosis (Ap), mitotic catastrophe (MC), and cellular senescence (Sns). The evaluations were performed in triplicate. Average values are shown, and error bars indicate standard deviations. Please click here to view a larger version of this figure.
Discussion
The critical steps within the protocol are as follows. First, cells should be grown on the culture dish as a monolayer because conducting a morphological assessment of DAPI-stained nuclei is difficult for multilayered cells. To this end, in step 2.9, careful transfer of the culture dish to an incubator is recommended; shaking of the culture dish generates a swirl of suspended cells that leads to the concentration of cells at the center of the culture dish. In addition, overconfluence leading to multilayered cells should be avoided. To this end, in step 2.7, the number of cells seeded on the culture dish can be modified based on the population doubling time and the interval between irradiation and fixation. A confluence of approximately 80 at the time of fixation is recommended. Second, speed is important in fixation and DAPI staining (i.e., steps 4 and 5). Inter-sample inconsistency in regard to the time taken for these steps can lead to heterogeneity in DAPI signal intensity in the nuclei, which would obscure the morphological assessment.
In step 2.2, the number of cover slips in a single culture dish can be increased; a maximum of four cover slips can be placed in a 35 mm dish, and the number can be further increased using larger dishes. Placing multiple cover slips in each culture dish enables the efficient operation of time course assessment for a given treatment (i.e., the cover slips can be collected from a culture dish one by one at multiple time points of interest).
In step 3.2, the irradiation dose can be modified according to the researchers' interest. The application of a consistent dose to multiple cell lines enables the comparison of the sensitivity to each mode of clonogenic cell death among the cell lines. On the other hand, the use of iso-clonogenic survival doses for each cell line enables comparison of clonogenic cell death profiles among the cell lines. The iso-clonogenic survival dose can be determined by the clonogenic survival assay12. The D10 value, the dose that provides 10% clonogenic survival, is a common endpoint for the iso-clonogenic survival dose.
In step 3.3, the time from irradiation to fixation can be modified according to the researchers' interest; this is important because the peak time for IR-induced apoptosis, mitotic catastrophe, and cellular senescence varies according to cell line and treatment. In this article, we used 72 h after irradiation as the time point that would be most useful for the initial screening of clonogenic cell death profiles, based on multiple studies by our group and others described as follows1,2,3,4,8,9: (i) X-ray-induced apoptosis in cells established from solid tumors mostly occurs a few days after irradiation. (ii) X-ray-induced mitotic catastrophe in cancer cells occurs most prominently at the second or third mitosis after release from the temporary cell-cycle arrest induced by irradiation. The release usually occurs approximately 24 h after irradiation, followed by repeated mitoses at intervals of approximately 24 h. (iii) X-ray–induced cellular senescence becomes evident after an interval dependent upon the cell line in question: 2 days after irradiation for some early cases, and 7 days after irradiation for most cell lines. After obtaining an overall picture of the clonogenic cell death profiles from the initial screening, time course experiments will provide a more detailed elucidation of the peak time for each mode of clonogenic cell death in the specific cell line and/or condition of interest4.
It should be noted that the DAPI staining assay and the clonogenic survival assay, a gold standard method for radiation sensitivity assessment, are not interchangeable. Apoptosis and mitotic catastrophe only last for hours. Thus, the DAPI staining assay for a given time point detects apoptosis and mitotic catastrophe that occurs at the time point of the assessment. On the other hand, the results of the clonogenic survival assay at a given time point include the total amount of apoptosis and mitotic catastrophe that had occurred during the incubation period for typically 10 - 14 days after irradiation. Different from apoptosis and mitotic catastrophe, senescent cells remain on the culture dish; they accumulate gradually over time after irradiation. Therefore, the results of both the DAPI staining assay and the clonogenic survival assay reflect the total amount of senescence that occurred during the incubation period. Importantly, the proportion of apoptosis, mitotic catastrophe, and senescence induced by irradiation varies widely according to cell line and irradiation dose. Taken together, theoretically, the results of one assay cannot be translated directly into those of the other assay.
The DAPI staining assay has a few limitations. First, it remains controversial whether mitotic catastrophe is a distinct mode of cell death. In the field of radiation biology, mitotic catastrophe is considered a major mode of IR-induced cell death that is distinct from other mechanisms of clonogenic cell death1. On the other hand, others argue that mitotic catastrophe is not a distinct mode of cell death but rather a process that precedes cell death including apoptosis and necrosis13,14. Thus, apoptosis and mitotic catastrophe may overlap to some extent. Second, previous studies suggest that cellular senescence can occur in the absence of SAHF in some cell lines and treatment settings2. At present, other assays specifically designed for each clonogenic cell death mode should be used to increase the robustness of the conclusions of a given experiment. Third, the DAPI staining assay cannot assess modes of clonogenic cell death other than apoptosis, mitotic catastrophe, and cellular senescence (e.g., necrosis and autophagy). Fourth, the utility of the DAPI staining assay as a predictor of tumor response to radiotherapy has not been elucidated in the clinic. From this point of view, the clonogenic survival assay, which assesses the total amount of clonogenic cell death, is superior to the DAPI staining assay because a correlation has been established between SF2, the surviving fraction of cells irradiated with 2 Gy X-rays, and the tumor response to radiotherapy15. Nevertheless, it is noteworthy that the clonogenic survival assay is not utilized widely in the clinic, mainly due to the requirement for a high degree of expertise and a long period of time (i.e., 14 days) for data acquisition. By comparison, the procedure for the DAPI staining assay is simpler and takes significantly less time, approximately 3 - 4 days, to generate results. The utility of the DAPI staining assay as a predictor of tumor response to radiotherapy will be tested in the clinic in the near future.
In summary, the DAPI staining assay is a cost-effective one-step assay to simultaneously assess the three major modes of IR-induced clonogenic cell death. This approach allows one to easily screen for the modes of clonogenic cell death for various cell lines, treatment settings, and time points, with the goal of elucidating the mechanisms of cell death in the target cells and conditions of interest.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
We thank Mrs. Akiko Shibata for technical assistance. This work was supported by Grants-in-Aid from the Ministry of Education, Culture, Sports, Science, and Technology of Japan for programs for Leading Graduate Schools, Cultivating Global Leaders in Heavy Ion Therapeutics and Engineering.
References
- Wouters BG. Cell death after irradiation: how, when and why cells die. In: Joiner MC, van der Kogel AJ, editors. Basic Clinical Radiobiology. 4th ed. London: Hodder Education; 2009. pp. 27–40. [Google Scholar]
- Aird KM, Zhang R. Detection of senescence-associated heterochromatin foci (SAHF) Methods Mol. Biol. 2013;965:185–196. doi: 10.1007/978-1-62703-239-1_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi D, et al. Mitotic catastrophe is a putative mechanism underlying the weak correlation between sensitivity to carbon ions and cisplatin. Sci. Rep. 2017;7:40588. doi: 10.1038/srep40588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amornwichet N, et al. Carbon-ion beam irradiation kills X-ray-resistant p53-null cancer cells by inducing mitotic catastrophe. PLoS One. 2014. p. 115121. [DOI] [PMC free article] [PubMed]
- Masters JR, Stacey GN. Changing medium and passaging cell lines. Nat. Protoc. 2007;2:2276–2284. doi: 10.1038/nprot.2007.319. [DOI] [PubMed] [Google Scholar]
- Sawai Y, et al. Effectiveness of sulforaphane as a radiosensitizer for murine osteosarcoma cells. Oncol. Rep. 2013;29:941–945. doi: 10.3892/or.2012.2195. [DOI] [PubMed] [Google Scholar]
- Cummings BS, Wills LP, Schnellmann RG. Measurement of cell death in mammalian cells. Curr. Protoc. Pharmacol. 2012. Suppl. 56, Chapter 12: Unit 12.8. [DOI] [PubMed]
- Oike T, et al. C646, a selective small molecule inhibitor of histone acetyltransferase p300, radiosensitizes lung cancer cells by enhancing mitotic catastrophe. Radiother. Oncol. 2014;111:222–227. doi: 10.1016/j.radonc.2014.03.015. [DOI] [PubMed] [Google Scholar]
- Oike T, et al. A synthetic lethality-based strategy to treat cancers harboring a genetic deficiency in the chromatin remodeling factor BRG1. Cancer Res. 2013;73:5508–5518. doi: 10.1158/0008-5472.CAN-12-4593. [DOI] [PubMed] [Google Scholar]
- Russo AL, et al. In vitro and in vivo radiosensitization of glioblastoma cells by the poly (ADP-ribose) polymerase inhibitor E7016. Clin. Cancer Res. 2009;15:607–612. doi: 10.1158/1078-0432.CCR-08-2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Micco R, et al. Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer. Nat. Cell Biol. 2011;13:292–302. doi: 10.1038/ncb2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006;1:2315–2319. doi: 10.1038/nprot.2006.339. [DOI] [PubMed] [Google Scholar]
- Vakifahmetoglu H, Olsson M, Zhivotovsky B. Death through a tragedy: mitotic catastrophe. Cell Death Differ. 2008;15:1153–1162. doi: 10.1038/cdd.2008.47. [DOI] [PubMed] [Google Scholar]
- Imreh G, Norberg HV, Imreh S, Zhivotovsky B. Chromosomal breaks during mitotic catastrophe trigger γH2AX-ATM-p53-mediated apoptosis. J. Cell Sci. 2016;129:1950. doi: 10.1242/jcs.190132. [DOI] [PubMed] [Google Scholar]
- Torres-Roca JF. A molecular assay of tumor radiosensitivity: a roadmap towards biology-based personalized radiation therapy. Per. Med. 2012;9:547–557. doi: 10.2217/pme.12.55. [DOI] [PMC free article] [PubMed] [Google Scholar]