

Abstract
The promoter region of the telomerase reverse transcriptase gene (TERT) is mutated in a subpopulation of patients with glioblastoma multiforme (GBM). In the present study, preclinical and clinical implications of the mutation were analyzed in 25 GBMs to evaluate its utility as a therapeutic target. Associations between the TERT promoter mutation and a number of preclinical/clinical characteristics were analyzed. Notably, the TERT promoter mutation was identified in 92.3% of GBMs where dissociated cells revealed in vitro sphere formation capacity; while the TERT promoter mutation was identified in 33.3% of GBMs without in vitro sphere formation capacity (P=0.004). In addition, this significantly increased mutation rate was observed in GBMs with in vivo tumorigenic potential (80% vs. 0%; P=0.004). Furthermore, patients with GBM exhibiting the TERT promoter mutation demonstrated significantly decreased overall survival rate compared with patients lacking this mutation (81.7 vs. 152.6 weeks; P=0.026). The results of the present study indicated that the TERT promoter mutation is associated with the self-renewal capacity of GBM cells and clinical aggressiveness of GBMs, which may be translated to a targeting therapy against TERT to inhibit the self-renewal of GBM cells.
Keywords: glioblastoma multiforme, telomerase reverse transcriptase, telomerase reverse transcriptase promoter mutation, sphere formation, self-renewal capacity
Introduction
Glioblastoma multiforme (GBM; World Health Organization grade IV glioma) is the most common type of primary brain tumor worldwide (1). In spite of radical surgery combined with concomitant chemoradiation therapy based on temozolomide, the median survival rate of patients with GBM remains ~1 year (2). Furthermore, clinical trials have demonstrated only limited benefits of targeted regimens, indicating that the identification of the genetic and molecular characteristics of GBM is required to develop more effective treatment strategies (2–4).
Genetic studies including a large number of patients with GBM have revealed various genetic alterations in GBM, including isocitrate dehydrogenase gene mutations and mutations in the promoter region of telomerase reverse transcriptase gene (TERT) (4,5). Of these, TERT promoter mutations have been identified in a subpopulation of GBM and were revealed to be significantly associated with poor clinical prognosis (2,5,6). In addition, these mutations were associated with increased expression of TERT (7,8). These results indicated the potential for personalized therapy against TERT in GBM on the basis of mutation status.
In vitro and in vivo preclinical models derived from surgical samples of GBMs have revealed the molecular and functional features of the parental tumors, and may represent the GBM population experimentally (9). Therefore, patient-derived preclinical models exhibiting GBMs with or without TERT promoter mutations may enable experimental examination of personalized TERT-targeted treatments. In the present study, a patient-derived GBM preclinical model library, including GBMs with and without TERT promoter mutations, was established, and preclinical and clinical implications were determined.
Materials and methods
GBM patients, primary cell culture and stereotactic transplantation
Surgical specimens and clinical records were obtained from 25 patients with primary GBM from May 2004 to June 2006 at the Samsung Medical Center (Seoul, Korea). All tissue samples were collected with written informed consent under a protocol approved by the Institutional Review Board of the Samsung Medical Center (Seoul, Korea). GBMs were pathologically diagnosed by specialized neuropathologists, on the basis of the World Health Organization criteria (10). For genomic analysis, parts of the specimens were snap-frozen and preserved in liquid nitrogen until use. Genomic DNA and mRNA were extracted using the DNeasy Kit (Qiagen GmbH, Hilden, Germany) and the RNeasy kit (Qiagen GmbH, Hilden, Germany).
Sections of the surgical samples were enzymatically dissociated into single cells as previously described (11). Dissociated GBM cells were cultured in neurobasal media (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) containing 1X N2 and 2X B27 supplements (Invitrogen; Thermo Fisher Scientific, Inc.), human recombinant basic fibroblast growth factor and epidermal growth factor (25 ng/ml each; R&D Systems, Inc., Minneapolis, MN, USA) (12). Because clonogenic growth as neurospheres is an in vitro indicator of the self-renewal of GBM cells, sphere formation (diameter, ≥50 µm), within 4 weeks, was used to identify the in vitro sphere formation capacity of dissociated GBM cells. For neurosphere formation, cells were seeded at a range of 1–200 cells per well. Following 4 weeks, the number of wells without spheres were counted and analyzed.
Dissociated GBM cells were stereotactically (2 mm left and 1 mm anterior to the bregma, 2 mm deep from the dura) transplanted into the brains of immuno-deficient NOG mice (between 2.5×104 and 1.0×105 cells/10 µl Hank's Balanced Salt solution for each mice, n=4–9 for each sample) (13). For the intracranial injection, 250 6–8 week-old female immune-deficient NOG mice (4–9 mice per group) were obtained from Orient Bio, Inc. (Seongnam, Korea). Mice were housed under controlled temperature (22±2°C) and a 12 h light/dark cycle in laminar flow cabinets with filtered air, and handled using aseptic procedures and food and water were provided ad libitum. The average weight of the mice prior to the procedure was 20 g. Mice were sacrificed and their brains were processed for pathological diagnosis, following the procedures previous reported (9). Animal experiments were approved by the Institutional Review Boards of the Samsung Medical Center and conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
TERT promoter mutation analysis
Genomic DNA was isolated from tumor samples a using QIAamp DNA mini kit (Qiagen GmbH). The TERT promoter region was amplified from isolated genomic DNA using the polymerase chain reaction (PCR), as described previously (14). PCR was performed using AmpliTaq Gold DNA polymerase (Applied Biosystems; Thermo Fisher Scientific, Inc.). PCR products were electrophoresed on 1.5% agarose gel and stained with ethidium bromide to validate the sizes of the bands. Subsequently, direct DNA sequencing for the TERT promoter region was performed using an ABI 3730 DNA sequencer by Bionics Co., Ltd. (Seoul, Korea). The sequences for TERT were as follows: Sense, 5′-CACCCGTCCTGCCCCTTCACCTT-3′ and antisense, 5′-GGCTTCCCACGTGCGCAGCAGGA-3′.
Relative telomere length determination
Telomere length was analyzed using quantitative PCR (qPCR) (7990HT Fast Real-time, Applied Biosystems; Thermo Fisher Scientific, Inc.). For telomere length determination, DNA were extracted from cells. To quantitatively determine telomere length relative to nuclear DNA, specific primers for telomere (T) and nuclear DNA-encoded β-globin (S) were selected according to a previous study (14). The β-globin gene was used as the reference gene. qPCR was performed using a LightCycler 480 II system (Roche Diagnostics, Basel, Switzerland) with SYBR PCR master mix (Toyobo Life Science, Osaka, Japan). For telomere PCR, there were 18 cycles at 95°C for 15 sec and at 54°C for 2 min. Relative telomere length was determined by calculating T/S values using the formula T/S=2−∆Cq, where ∆Cq=average Cqtelomere-average Cqβ-globin. Each measurement was repeated in triplicate and 5 serially diluted control samples were included in each experiment. The primer sequences used in this experiment were follows: β-globin sense, 5′-TGTGCTGGCCCATCACTTTG-3; β-globin antisense, 5′-ACCAGCCACCACTTTCTGATAGG-3′; telomere sense, 5′-CGGTTTGTTTGGGTTTGGGTTTGGGTTTGGGTTTGGGTT-3′; and telomere antisense, 5′-GGCTTGCCTTACCCTTACCCTTACCCTTACCCTTACCCT-3′.
GBM subtype prediction
According to The Cancer Genome Atlas data of the mRNA expression of 840 genes in 173 patients with GBM, the subtypes (proneural, neural, classical and mesenchymal) of the 25 GBMs used in the present study were determined (15). The Cancer Genome Atlas data were downloaded from tcga-data.nci.nih.gov/docs/publications/gbm_exp. The Nearest Template Prediction algorithm was used to predict the class of a sample with statistical significance (false discovery rate, <0.2) using a predefined set of markers specific to multiple classes (GenePattern Modules; version 2; Broad Institute) (15). For the dataset in the present study, the 788 overlapping genes, out of the 840 genes, were used to predict the subtype.
Genetic alteration analysis
Microarray-based comparative genomic hybridization (aCGH) was performed using the Agilent SurePrint G3 Human CGH 4×180K arrays (Agilent Technologies, Inc., Santa Clara, CA, USA). aCGH feature extraction files were processed and normalized fold changed of matched normal samples using Agilent Genomic WorkBench 7.0.4.0 (Agilent Technologies, Inc.). The DNAcopy R package (version 1.48.0; performed in Bioconductor) was used to estimate DNA copy numbers (16). From the copy numbers at the segment level, the copy number for each gene was measured using the mean value of the copy numbers of all exonic segments of the gene (9).
Somatic mutations were detected using the Agilent SureSelect kit (Agilent Technologies, Inc.) to capture exonic DNA fragments. The Illumina HiSeq 2000 was used to generate 2×101 bp paired-end reads (Illumina, Inc., San Diego, CA, USA). The sequenced reads in FASTQ files were mapped to the human reference genome assembly (hg19), using the Burrows-Wheeler Aligner (version 0.6.2) (17). The initial alignment BAM files were subjected to regular preprocessing prior to mutation calling: Sorting reads by genomic coordinates, removing duplicated reads, locally realigning reads around potential small indels and recalibrating base quality scores using SAMtools (18), Picard (version 1.73; Broad Institute), and Genome Analysis ToolKit (GATK version 2.5.2; Broad Institute). For mutation calling, MuTect (GATK version 1.1.4; Broad Institute) and SomaticIndelDetector (GATK version 2.2; Broad Institute) were used to make high-confidence predictions regarding somatic mutations from the tumor and paired blood. Copy number data were obtained using the ngCGH python package (version 0.4.4) to generate aCGH-like data from whole exome sequencing (WES) data. The matched blood WES data were used as a reference to calculate fold-changes in copy numbers in tumors. In cases without matched blood WES, created ‘pseudo-normal’ profile blood WES data were generated using the same sequencing platform and analysis pipeline as were used for the tumor data. Downstream analysis (segmentation and calculation of copy number) were conducted as described for aCGH data.
Using the Illumina TruSeq RNA Sample Prep kit (Illumina, Inc.), RNA-seq libraries were prepared for all cases. The trimmed reads in FASTQ files were aligned with hg19 using GSNAP (version 2012-12-20) with two output formats: GSNAP native format (exon-skipping analysis) and SAM format (point mutation analysis) (19). The resulting GSNAP native format files were analyzed to isolate the ‘split’ reads spanning non-canonical splicing junctions, with a minimal anchor of five nucleotides on each exon. In cases demonstrating plural reading splits between two exons, the event was termed a skipped exon event between the two exons. The SAM format files were sorted using the same preprocessing procedures as those applied for the WES data, with the exception of local realignments were restricted to exonic regions to prevent the mislabeling of normal splicing events as misaligned indels. Potential point mutations were identified using UnifiedGenotyper (GATK version 1.2.0; Broad Institute).
Statistical analysis
The SPSS statistical package, version 19.0, was used for statistical analyses (IBM Corp., Armonk, NY, USA). χ2, Fischer's exact tests, Mann Whitney U tests and Spearman's correlation analysis were used to analyze the associations between variables. Survival curves, estimated using the Kaplan-Meier method (univariate analysis), were compared using the log-rank test. Overall survival was defined as the time between diagnosis and mortality (as a result of any cause). Progression-free survival was defined as the time between diagnosis and disease recurrence. P<0.05 was considered to indicate a statistically significant difference. Data are presented as the mean ± standard deviation.
Results
TERT promoter mutation status of GBMs is associated with sphere formation capacity
First, TERT promoter mutations were investigated in the 13 GBMs with in vitro sphere formation capacity to establish patient-derived GBM preclinical model libraries, including GBMs with and without TERT promoter mutations. In parallel, in vivo tumor formation, gene mutation status and global gene expression were analyzed. Notably, TERT promoter mutations were identified in 92.3% (12/13) GBMs (Table IA). All TERT mutations were revealed to be C228T, but 1 GBM exhibited C228T and C250T mutations. It was revealed that 1 GBM without TERT promoter mutation exhibited α-thalassemia/mental retardation syndrome X-linked (ATRX) amplification, although it was previously demonstrated that ATRX amplification and TERT promoter mutation were mutually exclusive (6). On the basis of this, it was hypothesized that the genetic alteration in ATRX is equivalent to TERT promoter mutation (6).
Table I.
Association between TERT promoter mutation and other gene mutation status in GBMs.
| A, Sphere formation-positive GBMs | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample | |||||||||||||
| Gene mutation status | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 |
| TERT mutation | + | + | + | + | + | + | + | + | + | + | + | + | |
| C228T | + | + | + | + | + | + | + | + | + | + | + | + | |
| C250T | + | ||||||||||||
| Sphere formation | + | + | + | + | + | + | + | + | + | + | + | + | + |
| ATRX amplification | ND | + | ND | ||||||||||
| ATRX mutation | + | + | + | + | + | + | ND | + | |||||
| EGFR mutation | + | + | + | ND | + | + | + | ||||||
| EGFR amplification | + | + | + | + | ND | + | ND | + | + | + | |||
| IDH1 mutation | ND | ||||||||||||
| TP53 mutation | + | + | + | + | ND | ||||||||
| PTEN deletion | + | + | + | + | ND | + | + | ND | + | + | + | ||
| PTEN mutation | + | + | ND | ||||||||||
| CDKN2A deletion | + | ND | + | + | + | ND | + | + | + | ND | + | + | + |
| B, Sphere formation-negative GBMs | |||||||||||||
| Sample | |||||||||||||
| Gene mutation status | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | |
| TERT mutation | + | + | + | + | |||||||||
| C228T | + | + | + | + | |||||||||
| C250T | |||||||||||||
| Sphere formation | |||||||||||||
| ATRX amplification | |||||||||||||
| ATRX mutation | ND | ND | ND | ND | ND | ND | ND | ND | + | ||||
| EGFR mutation | ND | ND | ND | ND | ND | ND | ND | ND | + | ||||
| EGFR amplification | + | + | + | + | + | ||||||||
| IDH1 mutation | ND | ND | ND | ND | ND | ND | ND | ND | |||||
| TP53 mutation | ND | ND | ND | ND | ND | ND | ND | ND | + | ||||
| PTEN deletion | + | + | + | + | |||||||||
| PTEN mutation | ND | ND | ND | ND | ND | ND | + | + | |||||
| CDKN2A deletion | + | + | + | + | + | + | + | ||||||
TERT, telomerase reverse transcriptase gene; GBM, glioblastoma multiforme; ATRX, α-thalassemia/mental retardation syndrome X-linked; EGFR, epidermal growth factor receptor; IDH1, isocitrate dehydrogenase 1; TP53, protein 53; PTEN, phosphatase and tensin homolog; CDKN2A, cyclin dependent kinase inhibitor 2A; ND, Not determined.
The TERT promoter mutation rate (92.3%) in GBMs with in vitro sphere formation capacity was not expected because TERT promoter mutations were observed in between 28–84% of GBMs (20). Accordingly, the present study hypothesized that TERT promoter mutation is associated with the in vitro sphere formation capacity of GBMs. To test the hypothesis, TERT promoter mutations in GBMs without in vitro sphere formation capacity were subsequently analyzed.
TERT promoter mutations are decreased in GMBs without sphere formation capacity
TERT promoter mutations were identified in 33.3% (4/12) GBMs without in vitro sphere formation capacity (Table IB). All mutations were C228T (Table IB). This frequency was significantly decreased compared with that in GBMs with in vitro sphere formation capacity (P=0.004). Other preclinical characteristics and genetic changes were not associated with TERT promoter mutation in GBMs without in vitro sphere formation capacity (data not shown).
TERT promoter mutation is associated with age and sex
Out of 25 GBMs (Table II), TERT promoter mutation was demonstrated in 64.0% (16/25). TERT promoter mutation was significantly associated with increased age (P=0.050) and sex; being more prevalent in male patients (P<0.001). Notably, GBMs with in vivo tumorigenic potential demonstrated a significantly increased TERT promoter mutation rate (P=0.004) compared with those without. Although the values were not statistically significant, TERT promoter mutations were at an increased frequency in GBMs with epidermal growth factor receptor (EGFR) gene mutation (P=0.117), EGFR amplification (P=0.102), cyclin dependent kinase inhibitor 2A (CDKN2A) deletion (P=0.116) and phosphatase and tensin homolog deletion (P=0.102).
Table II.
Clinicopathological and experimental characteristics of GBMs with or without TERT promoter mutation.
| TERT promoter mutation, n (%) | |||
|---|---|---|---|
| Variable | + | − | P-value |
| Age, years | 56.5±8.3 | 47.2±14.6 | 0.050 |
| Sex | <0.001 | ||
| Male | 12 (100) | 0 (0) | |
| Female | 4 (30.8) | 9 (69.2) | |
| Tumor size, cm | 4.70±1.63 | 4.79±0.75 | 0.943 |
| ATRX mutation | 1.00 | ||
| + | 7 (87.5) | 1 (12.5) | |
| − | 6 (75.0) | 2 (25.0) | |
| ATRX amplification | 0.391 | ||
| + | 0 (0) | 1 (100) | |
| − | 14 (63.6) | 8 (36.4) | |
| EGFR mutation | 0.117 | ||
| + | 11 (91.7) | 1 (8.3) | |
| − | 4 (57.1) | 3 (42.9) | |
| EGFR amplification | 0.102 | ||
| + | 10 (76.9) | 3 (23.1) | |
| − | 4 (40.0) | 6 (60.0) | |
| CDKN2A deletion | 0.116 | ||
| + | 12 (70.6) | 5 (29.4) | |
| − | 1 (20.0) | 4 (80.0) | |
| PTEN mutation | 1.00 | ||
| + | 3 (75.0) | 1 (25.0) | |
| − | 10 (83.3) | 2 (16.7) | |
| PTEN deletion | 0.102 | ||
| + | 10 (76.9) | 3 (23.1) | |
| − | 4 (40.0) | 6 (60.0) | |
| Subtype | 0.176 | ||
| Classical | 6 (100) | 0 (0) | |
| Mesenchymal | 2 (40.0) | 3 (60.0) | |
| Proneural | 3 (60.0) | 2 (40.0) | |
| Not determined | 5 (55.6) | 4 (44.4) | |
| In vivo tumor formation | 0.004 | ||
| + | 12 (80.0) | 3 (20.0) | |
| − | 0 (0) | 5 (100) | |
Data are presented as the mean ± standard deviation. TERT, telomerase reverse transcriptase gene; GBM, glioblastoma multiforme; ATRX, α-thalassemia/mental retardation syndrome X-linked; EGFR, epidermal growth factor receptor; PTEN, phosphatase and tensin homolog; CDKN2A, cyclin dependent kinase inhibitor 2A.
Telomere length is not associated with TERT promoter mutation status
Relative telomere length (0.92±0.49) was analyzed in the GBMs. However, the length did not differ according to TERT promoter mutation status (0.91±0.42 vs. 0.93±0.77; P=0.598; Fig. 1). When the GBMs were divided into two groups according to in vitro sphere formation capacity, TERT promoter mutation revealed limited association with the relative telomere length (Fig. 1).
Figure 1.
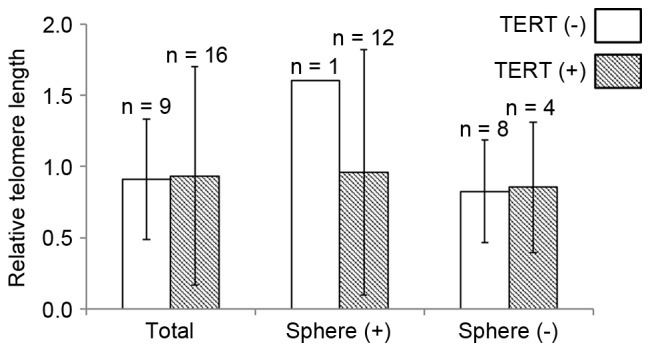
Relative telomere length analysis according to TERT promoter mutation in glioblastoma multiforme cases with and without in vitro sphere formation capacity. TERT, telomerase reverse transcriptase gene.
To explore the association between telomere length and clinicopathological parameters, GBMs were divided into two groups according to the median value (0.92) of relative telomere length. In the analysis, telomere length was not associated with any clinicopathological characteristics or molecular changes (data not shown).
Positive TERT promoter status is associated with poor survival
The clinical prognosis of TERT promoter mutation-positive GBMs (n=16) was compared with that of TERT promoter mutation-negative GBMs (n=9). The median overall survival in GBMs exhibiting TERT promoter mutation was 81.7 [95% confidence interval (CI), 61.71–101.85] weeks, which was significantly decreased compared with that in GBMs without TERT promoter mutation (median, 152.6 weeks; 95% CI, 84.05–221.16; P=0.026; Fig. 2A). According to the median value of telomere length (0.92), GBMs were stratified into longer and shorter groups to analyze the prognostic value of telomere length. Overall survival in patients with GBM with a longer telomere length (median, 75.70 weeks; 95% CI, 40.65–110.75) was reduced compared with those with shorter telomere length (median, 125.04 weeks; 95% CI, 84.02–166.05; P=0.041; Fig. 2B). In contrast, progression-free survival did not differ according to TERT promoter mutation (median, 51.19 vs. 43.32; 95% CI, 27.56–74.82 vs. 29.09–57.54; P=0.463; Fig. 2C) and telomere length (median, 47.43 vs. 43.88; 30.47–64.40 vs. 26.88–60.88; P=0.560; Fig. 2D]. When survival analysis was performed separately according to other variables [in vitro sphere formation capacity, in vivo tumor formation, age (<60 vs. ≥60), sex and subtype], there was no significant prognostic difference in overall survival or progression-free survival (data not shown).
Figure 2.

Prognostic impact of TERT promoter mutation and telomere length in GBMs. OS of patients with GBM, depending on (A) TERT promoter mutation status and (B) TL. PFS of patients with GBM, depending on (C) TERT promoter mutation status and (D) TL. TERT, telomerase reverse transcriptase gene; GBM, glioblastoma multiforme; TL, telomere length; PFS, progression-free survival; Wk, week.
Discussion
The present study revealed that TERT promoter mutations in GBMs are significantly associated with the in vitro sphere formation capacity and in vivo tumorigenic potential of dissociated GBM cells. In vitro and in vivo preclinical models using GBM cells primarily cultured from surgical samples have provided an improved understanding of the biology of the disease (9). Several preclinical characteristics of primary cultured GBM cells, including in vitro sphere formation capacity and in vivo tumorigenic potential, were revealed to be associated with clinical aggressiveness in corresponding patients (9). The associations may be utilized to determine molecular and/or functional mechanisms of clinical aggressiveness of GBMs.
In the present study, in vitro and in vivo preclinical GBM models exhibiting TERT promoter mutation-positive and -negative GBMs were established. As preclinical models may summarize the clinicopathological features of patient with GBMs (9,21–23), the preclinical models may be utilized to predict the treatment effects of TERT-targeting therapies for TERT promoter mutation-positive GBMs, compared with those for TERT promoter mutation-negative GBMs. In the present study, the majority of GBMs with in vitro sphere formation capacity exhibited TERT promoter mutations (92.3%). By contrast, the TERT promoter mutation rate in GBMs without in vitro sphere formation capacity (33.3%) was significantly decreased. This significant difference was observed between GBMs with and without in vivo tumorigenic potential.
The in vitro sphere-forming assay has been widely used in stem cell biology as an experimental method for determining the self-renewal and differentiation potential of stem cells (9,11). Therefore, a significant association between TERT promoter mutation and the in vitro sphere-forming capacity of GBM cells, identified in the present study, suggests that mutations in the TERT promoter region are associated with the biology of GBM cells to enhance self-renewal capacity. Self-renewal capacity is a key feature of GBM cancer stem cells that exert recurrence following anti-cancer treatments (24,25). Therefore, TERT promoter mutations resulting in overexpression of TERT may be associated with treatment resistance of GBMs. In addition, the possible association between TERT promoter mutations and treatment resistance of GBMs is supported by the survival analysis results in the present study, which revealed that GBMs with TERT promoter mutation have significantly decreased overall survival.
Previous studies have demonstrated the negative clinical impacts of TERT promoter mutation or longer telomere length in a number of types of cancer, including GBMs (15,20,21,25,26). TERT promoter mutations may generate novel binding motifs for E26 transformation-specific/T-cell factor transcription factors, and cause two- to four-fold increases in transcriptional activity and telomere length (14,26,27). These data suggested that overexpression of TERT by its promoter mutation may increase the self-renewal capacity of GBM cancer stem cells and induce poor clinical outcomes. The results of the present study did not reveal a significant association between TERT promoter mutation and relative telomere length in the GBM samples. Previous studies demonstrated that the association between telomere length and TERT expression is complex and may be regulated by a number of other factors, including the activities of signaling pathways and alterations of genes (4,28,29).
Previous studies have demonstrated that TERT promoter is the most common type of mutation in GBMs, suggesting that it may be an early event in GBM carcinogenesis (2,4,5,30–32). In the present study, TERT promoter mutations were identified in 64% of GBMs, and were associated with the age and sex of patients with GBM. These results are consistent with those of previous studies (4,5). The association between TERT promoter mutations and other genetic alterations has been observed in previous studies (4,5). These studies suggested that TERT promoter mutations revealed a significant inverse association with isocitrate dehydrogenase 1 mutation and P53 mutation, but a positive association with EGFR amplification and CDKN2 deletion. Though the results of the present study were not statistically significant due to the limited sample size of primary GBMs, TERT promoter mutations tend to be associated with EGFR amplification and CDKN2A deletion. This result supports the reliability of our results.
The present study revealed the association between TERT promoter mutation and preclinical characteristics of GBM, including in vitro sphere-forming capacity and in vivo tumorigenic potential. As TERT promoter mutation is a prognostic marker of GBM, the identification of preclinical characteristics of TERT promoter mutations may reveal the functions of TERT and telomere length in the self-renewal of GBM cells, and treatment resistance of GBM. Furthermore, the results may provide a foundation for the development of innovative telomerase-based therapeutic strategies for treatment-resistant GBMs.
Acknowledgements
The present study was supported by the Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Education (grant no. NRF-2014R1A6A3A04058057). Additional support was supported by the National Research Foundation of Korea (NRF) Grant funded by the Korean Government (MSIP) (grant no. NRF-2016R1A5A2945889).
References
- 1.Mosrati MA, Malmström A, Lysiak M, Krysztofiak A, Hallbeck M, Milos P, Hallbeck AL, Bratthäll C, Strandéus M, Stenmark-Askmalm M, Söderkvist P. TERT promoter mutations and polymorphisms as prognostic factors in primary glioblastoma. Oncotarget. 2015;6:16663–16673. doi: 10.18632/oncotarget.4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reitman ZJ, Pirozzi CJ, Yan H. Promoting a new brain tumor mutation: TERT promoter mutations in CNS tumors. Acta Neuropathol. 2013;126:789–792. doi: 10.1007/s00401-013-1207-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Labussière M, Boisselier B, Mokhtari K, Di Stefano AL, Rahimian A, Rossetto M, Ciccarino P, Saulnier O, Paterra R, Marie Y, et al. Combined analysis of TERT, EGFR, and IDH status defines distinct prognostic glioblastoma classes. Neurology. 2014;83:1200–1206. doi: 10.1212/WNL.0000000000000814. [DOI] [PubMed] [Google Scholar]
- 5.Nonoguchi N, Ohta T, Oh JE, Kim YH, Kleihues P, Ohgaki H. TERT promoter mutations in primary and secondary glioblastomas. Acta Neuropathol. 2013;126:931–937. doi: 10.1007/s00401-013-1163-0. [DOI] [PubMed] [Google Scholar]
- 6.Killela PJ, Reitman ZJ, Jiao Y, Bettegowda C, Agrawal N, Diaz LA, Jr, Friedman AH, Friedman H, Gallia GL, Giovanella BC, et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci USA. 2013;110:6021–6026. doi: 10.1073/pnas.1303607110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.George J, Banik NL, Ray SK. Knockdown of hTERT and concurrent treatment with interferon-gamma inhibited proliferation and invasion of human glioblastoma cell lines. Int J Biochem Cell Biol. 2010;42:1164–1173. doi: 10.1016/j.biocel.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beck S, Jin X, Sohn YW, Kim JK, Kim SH, Yin J, Pian X, Kim SC, Nam DH, Choi YJ, Kim H. Telomerase activity-independent function of TERT allows glioma cells to attain cancer stem cell characteristics by inducing EGFR expression. Mol Cells. 2011;31:9–15. doi: 10.1007/s10059-011-0008-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Joo KM, Kim J, Jin J, Kim M, Seol HJ, Muradov J, Yang H, Choi YL, Park WY, Kong DS, et al. Patient-specific orthotopic glioblastoma xenograft models recapitulate the histopathology and biology of human glioblastomas in situ. Cell Rep. 2013;3:260–273. doi: 10.1016/j.celrep.2012.12.013. [DOI] [PubMed] [Google Scholar]
- 10.Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97–109. doi: 10.1007/s00401-007-0278-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Joo KM, Kim SY, Jin X, Song SY, Kong DS, Lee JI, Jeon JW, Kim MH, Kang BG, Jung Y, et al. Clinical and biological implications of CD133-positive and CD133-negative cells in glioblastomas. Lab Invest. 2008;88:808–815. doi: 10.1038/labinvest.2008.57. [DOI] [PubMed] [Google Scholar]
- 12.Pollard SM, Yoshikawa K, Clarke ID, Danovi D, Stricker S, Russell R, Bayani J, Head R, Lee M, Bernstein M, et al. Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell. 2009;4:568–580. doi: 10.1016/j.stem.2009.03.014. [DOI] [PubMed] [Google Scholar]
- 13.Ito M, Hiramatsu H, Kobayashi K, Suzue K, Kawahata M, Hioki K, Ueyama Y, Koyanagi Y, Sugamura K, Tsuji K, et al. NOD/SCID/gamma(c)(null) mouse: An excellent recipient mouse model for engraftment of human cells. Blood. 2002;100:3175–3182. doi: 10.1182/blood-2001-12-0207. [DOI] [PubMed] [Google Scholar]
- 14.Liu T, Wang N, Cao J, Sofiadis A, Dinets A, Zedenius J, Larsson C, Xu D. The age- and shorter telomere-dependent TERT promoter mutation in follicular thyroid cell-derived carcinomas. Oncogene. 2014;33:4978–4984. doi: 10.1038/onc.2013.446. [DOI] [PubMed] [Google Scholar]
- 15.Oh YT, Cho HJ, Kim J, Lee JH, Rho K, Seo YJ, Choi YS, Jung HJ, Song HS, Kong DS, et al. Translational validation of personalized treatment strategy based on genetic characteristics of glioblastoma. PLoS One. 2014;9:e103327. doi: 10.1371/journal.pone.0103327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Olshen AB, Venkatraman ES, Lucito R, Wigler M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004;5:557–572. doi: 10.1093/biostatistics/kxh008. [DOI] [PubMed] [Google Scholar]
- 17.Li H, Durbin R. Fast and accurate short read alignment with Burrow-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 1000 Genome Project Data Processing Subgroup: The sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu TD, Nacu S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics. 2010;26:873–881. doi: 10.1093/bioinformatics/btq057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vinagre J, Pinto V, Celestino R, Reis M, Pópulo H, Boaventura P, Melo M, Catarino T, Lima J, Lopes JM, et al. Telomerase promoter mutations in cancer: An emerging molecular biomarker? Virchows Arch. 2014;465:119–133. doi: 10.1007/s00428-014-1608-4. [DOI] [PubMed] [Google Scholar]
- 21.Fichtner I, Rolff J, Soong R, Hoffmann J, Hammer S, Sommer A, Becker M, Merk J. Establishment of patient-derived non-small cell lung cancer xenografts as models for the identification of predictive biomarkers. Clin Cancer Res. 2008;14:6456–6468. doi: 10.1158/1078-0432.CCR-08-0138. [DOI] [PubMed] [Google Scholar]
- 22.John T, Kohler D, Pintilie M, Yanagawa N, Pham NA, Li M, Panchal D, Hui F, Meng F, Shepherd FA, Tsao MS. The ability to form primary tumor xenografts is predictive of increased risk of disease recurrence in early-stage non-small cell lung cancer. Clin Cancer Res. 2011;17:134–141. doi: 10.1158/1078-0432.CCR-10-2224. [DOI] [PubMed] [Google Scholar]
- 23.Lee HW, Lee JI, Lee SJ, Cho HJ, Song HJ, Jeong DE, Seo YJ, Shin S, Joung JG, Kwon YJ, et al. Patient-derived xenografts from non-small cell lung cancer brain metastases are valuable translational platforms for the development of personalized targeted therapy. Clin Cancer Res. 2015;21:1172–1182. doi: 10.1158/1078-0432.CCR-14-1589. [DOI] [PubMed] [Google Scholar]
- 24.Hale JS, Otvos B, Sinyuk M, Alvarado AG, Hitomi M, Stoltz K, Wu Q, Flavahan W, Levison B, Johansen ML, et al. Cancer stem cell-specific scavenger receptor CD36 drives glioblastoma progression. Stem Cells. 2014;32:1746–1758. doi: 10.1002/stem.1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Borah A, Raveendran S, Rochani A, Maekawa T, Kumar DS. Targeting self-renewal pathways in cancer stem cells: Clinical implications for cancer therapy. Oncogenesis. 2015;4:e177. doi: 10.1038/oncsis.2015.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, Kadel S, Moll I, Nagore E, Hemminki K, et al. TERT promoter mutations in familial and sporadic melanoma. Science. 2013;339:959–961. doi: 10.1126/science.1230062. [DOI] [PubMed] [Google Scholar]
- 27.Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA. Highly recurrent TERT promoter mutations in human melanoma. Science. 2013;339:957–959. doi: 10.1126/science.1229259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chiba K, Johnson JZ, Vogan JM, Wagner T, Boyle JM, Hockemeyer D. Cancer-associated TERT promoter mutations abrogate telomerase silencing. Elife. 2015;4 doi: 10.7554/eLife.07918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ko E, Seo HW, Jung ES, Kim BH, Jung G. The TERT promoter SNP rs2853669 decreases E2F1 transcription factor binding and increases mortality and recurrence risks in liver cancer. Oncotarget. 2016;7:684–699. doi: 10.18632/oncotarget.6331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huse JT. TERT promoter mutation designates biologically aggressive primary glioblastoma. Neuro Oncol. 2015;17:5–6. doi: 10.1093/neuonc/nou318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Simon M, Hosen I, Gousias K, Rachakonda S, Heidenreich B, Gessi M, Schramm J, Hemminki K, Waha A, Kumar R. TERT promoter mutations: A novel independent prognostic factor in primary glioblastomas. Neuro Oncol. 2015;17:45–52. doi: 10.1093/neuonc/nou158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Spiegl-Kreinecker S, Lötsch D, Ghanim B, Pirker C, Mohr T, Laaber M, Weis S, Olschowski A, Webersinke G, Pichler J, Berger W. Prognostic quality of activating TERT promoter mutations in glioblastoma: Interaction with the rs2853669 polymorphism and patient age at diagnosis. Neuro Oncol. 2015;17:1231–1240. doi: 10.1093/neuonc/nov010. [DOI] [PMC free article] [PubMed] [Google Scholar]