

Abstract
Salmonella typhimurium is a facultative intracellular bacterium that causes gastroenteritis in humans. After invasion of the lamina propria, S.typhimurium bacteria are quickly detected and phagocytized by macrophages, and contained in vesicles known as phagosomes in order to be degraded. Isolation of S.typhimurium-containing phagosomes have been widely used to study how S.typhimurium infection alters the process of phagosome maturation to prevent bacterial degradation. Classically, the isolation of bacteria-containing phagosomes has been performed by sucrose gradient centrifugation. However, this process is time-consuming, and requires specialized equipment and a certain degree of dexterity. Described here is a simple and quick method for the isolation of S.typhimurium-containing phagosomes from macrophages by coating the bacteria with biotin-streptavidin-conjugated magnetic beads. Phagosomes obtained by this method can be suspended in any buffer of choice, allowing the utilization of isolated phagosomes for a broad range of assays, such as protein, metabolite, and lipid analysis. In summary, this method for the isolation of S.typhimurium-containing phagosomes is specific, efficient, rapid, requires minimum equipment, and is more versatile than the classical method of isolation by sucrose gradient-ultracentrifugation.
Keywords: Immunology, Issue 128, Phagosome, endosome, lysosome, antigen-processing, phagosome-isolation, Salmonella, biotin-streptavidin labeling
Introduction
Macrophages are circulating specialized phagocytic cells that detect, engulf, and degrade any foreign particle present in peripheral tissues, ranging from apoptotic cells to invading microorganisms such as bacteria. Upon surface receptor-mediated recognition of pathogen specific markers commonly present on the surface of microorganisms (known as pathogen-associated molecular patterns or PAMPs), macrophages initiate a complex reorganization of cellular membrane in order to surround and phagocytize the pathogen1.
The engulfed pathogen is then contained by the macrophage in an intracellular vesicle known as phagosome. Through a series of fusion and fission events with other vesicles such as endosomes and lysosomes, the pathogen-containing phagosome acquires a set of proteins required for the elimination of the phagosomal content. Therefore, the enzymatic composition of the phagosome is highly variable during the course of this process, known as phagosome maturation2.
Shortly after phagocytosis, the multimeric complex vacuolar ATPase (v-ATPase) is incorporated into the phagosome membrane by fusion with endosomes3. This complex utilizes ATP to pump protons from the cytosol to the lumen of the phagosome4. Acidification of the phagosome is essential for the fusion events with other vesicles5 and for the activation of a great number of pH-dependent degradative enzymes6. Another multimeric enzymatic complex that is quickly assembled on the phagosome membrane is the NADPH-oxidase (NOX) complex. NOX complex oxidizes NADPH in order to produce reactive oxygen species (ROS) that are secreted into the phagosome lumen and that significantly contribute to the killing of the engulfed microorganisms7.
During the initial steps of maturation, phagosomes present markers typically such as Rab5 and Rab7 of early and late endosomes respectively along with the V0 subunit of the v-ATPase8. Fusion of phagosomes with lysosomes and late endosomes results in the exposure of the phagocytized pathogen to a wide variety of hydrolytic enzymes such as cathepsin proteases, lipases, and β-galactosidase9. Acidification of the lumen is also required for the activation of these enzymes. For example, the cleavage of cathepsin D to produce the active short form is pH-dependent10. These enzymes degrade the pathogen and mediate the production of pathogen-derived short peptides that are presented by the macrophage major histocompatibility complex (MHC) class II molecules to T cells to trigger an adaptive immune response11.
Hence, phagosome maturation is crucial for the innate immune response and links the innate and adaptive arms of the immune system. It is no surprise that the pathogens have evolved strategies to overcome elimination by macrophages through the above-described process of phagosome maturation. For example, the intracellular bacteria Mycobacterium tuberculosis and Legionella pneumophila prevent phagosome maturation by inhibiting v-ATPase assembly and consequent lumen acidification12,13. Other bacteria, such as Listeria monocytogenes or Shigella flexneri induce pore formation in the phagosome membrane to escape into the cytosol14,15. On the other hand, Salmonella enterica serovar typhimurium (S. typhimurium) is able to modify the properties of the phagosome within the vacuole to transform it into a suitable location for its replication16. This ability makes S. typhimurium a very interesting model to study pathogen-mediated interference of phagosome maturation.
S.typhimurium is a facultative intracellular bacterium that causes gastroenteritis in humans. After invasion of the lamina propria, S. Typhimurium bacteria are quickly detected and phagocytized by macrophages, and contained within phagosomes17. Some reports have previously described that S. typhimurium-containing phagosomes present makers for both endosomes and lysosomes18, and other studies have found phagosome-lysosome fusion prevented upon S. Typhimurium infection19.
Initially, phagosome maturation upon S. typhimurium infection has been investigated by immunofluorescence microscopy. The development of techniques for the isolation of bacteria-containing phagosomes enabled a more accurate study of the phagosome content in terms of endosome and lysosome markers. To date, the main method used for the isolation of bacteria-containing phagosomes is the subcellular fractionation on sucrose step gradients18,20. However, this method requires multiple centrifugation steps that can cause mechanical damage to phagosomes, can affect the stability of phagosomal components (proteins and lipids), and is time consuming. Moreover, it requires the use of an ultracentrifuge: a piece of specialized equipment that is not accessible for every laboratory.
Recently, a new approach has been applied to the isolation of bacterial-containing phagosomes, in which bacterial pathogens are labeled with biotinylated lipopetide (Lipobiotin) and later extracted using streptavidin-conjugated magnetic beads21. We propose an alternative complementary method by labeling bacterial surface amine-containing macromolecules with NHS-Biotin followed by streptavidin-conjugated magnetic beads. Phagosomes obtained by this method are highly enriched in endosome and lysosome markers and can be used for a broad range of assays, from protein analysis to omics analysis. Additionally, it does not require specialized equipment such as ultracentrifuges. Moreover, by eliminating the centrifugation steps, both the mechanical damage to phagosomes and the amount of time employed are considerably reduced. This method can be easily adapted for the isolation of phagosomes containing other bacteria, such as the Gram-positive Staphylococcus aureus, also included in this manuscript. In summary, this method for the isolation of S. typhimurium-containing phagosomes is a simple, cost-effective, and less time consuming than the classical isolation by sucrose gradient-ultracentrifugation, rendering highly enriched bacteria-containing phagosomes.
Protocol
All the steps involving the use of pathogenic S. typhimurium must be carried out in a BSL-2 or higher biological security level facility. The culture and coating of S. Typhimurium, as well as the infection of bone marrow-derived macrophages (BMDMs) must be performed under a laminar flow hood to prevent contamination. The isolation of S. typhimurium-containing phagosomes can be performed on any BSL-2 laboratory bench.
The extraction of bone marrow from mice for its differentiation into macrophages was performed in accordance with institutional guidelines on animal welfare and approved by the North Rhine-Westphalian State Agency for Nature, Environment, and Consumer Protection [Landesamt für Natur, Umwelt and Verbraucherschutz (LANUV) Nordrhein-Westfalen; File no: 84-02.05.40.14.082 and 84-02.04.2015.A443] and the University of Cologne.
1. Culturing S. typhimurium
Inoculate S. typhimurium (SL1344 strain) from a single bacterial colony into 5 mL of brain heart infusion (BHI) broth using a bacterial loop.
Incubate the bacterial suspension in an incubator at 37 °C overnight with shaking.
On the following day, transfer 1 mL of the bacterial suspension into 19 mL of BHI broth in a conical flask and incubate at 37 °C in an incubator with shaking.
Monitor the S. typhimurium growth by measuring the optical density (OD) at 600 nm (OD600) with a spectrophotometer. Measurements should be taken approximately every 30 min.
When OD600 reaches 1.0, remove bacterial suspension from the incubator and transfer into 50 mL tube. Centrifuge bacteria at 5,400 x g for 15 min at 4 °C.
Remove supernatant and resuspend in 10 mL of sterile phosphate-buffered saline (PBS).
Centrifuge at 5,400 x g for 15 min at 4 °C.
Remove the supernatant and resuspend the bacteria in 4.9 mL of sterile PBS.
2. Coating of S. typhimurium with NHS-Biotin/Streptavidin-conjugated Magnetic Beads
Add 100 µL of 10 mg/mL biotin linking solution (NHS-Biotin dissolved in dimethyl sulfoxide, DMSO) freshly prepared, to the bacterial suspension prepared in the previous step. For example, dilute 5 mg of NHS-Biotin into 0.5 mL of sterile DMSO. Mix properly by pipetting up and down several times.
Split the mix into five 1.5 mL tubes.
Incubate for 2 h at room temperature (RT) on a thermoblock with constant shaking at 350 rpm.
Centrifuge tubes at 15,000 x g for 10 min at RT and discard the supernatant.
Resuspend in 1 mL of sterile PBS, centrifuge at 15,000 x g for 10 min at RT and discard the supernatant.
Repeat step 2.5 two more times to completely remove the excess biotin linking solution.
After the last wash, resuspend the pellet in 1 mL of sterile PBS.
Transfer 100 µL of 10 mg/mL streptavidin-conjugated magnetic beads solution into a 1.5 mL tube and leave it on the magnetic rack for 5 min.
Remove the solvent with a pipette, take the streptavidin- conjugated magnetic beads solution from the magnetic rack, and resuspend it with 1 mL of biotin coated-bacterial suspension prepared in step 2.7.
Incubate for 1 h at RT on a thermoblock with constant shaking at 350 rpm.
Place the solution on the magnetic rack; wait for 5 min and then remove with a pipette the bacteria that did not adhere onto the wall (keep it in a tube labeled as "non-coated bacteria"). The fraction adhering to the wall of the tube in contact with the magnet is the biotin/streptavidin-coated bacteria.
Remove the tube containing the labeled bacteria from the magnetic rack and resuspend in 1 mL of sterile PBS.
Place it again on the magnetic rack, wait for 5 min, and use a pipette to remove the PBS.
Repeat steps 2.13 and 2.14 two more times to remove all the bacteria that are not coated with the streptavidin-conjugated magnetic beads.
After the last wash, resuspend the coated-bacteria in 500 µL of sterile PBS and label the tube as "coated bacteria".
Count the colony forming units (cfu) of both "non-coated bacteria" and "coated bacteria" solutions by plating serial dilutions on BHI agar plates. Approximately 2 x 108 cfu/mL of coated-bacteria are obtained from 20 mL of initial bacterial suspension with OD600 of 1.0. Keep the coated-bacterial suspension at 4 °C to be used on the following day to infect macrophages.
3. Infection of BDMDs
On the same day when the bacteria are coated with biotin and streptavidin-conjugated magnetic beads, plate fully the differentiated BMDMs22 in 6 cm dishes at a density of 5 x 106 cells per dish.
On the next day, centrifuge the coated-bacterial suspension at 15,000 x g for 5 min at 4 °C.
Remove the supernatant and resuspend in Roswell Park Memorial Institute (RPMI) medium supplemented with 10% fetal bovine serum (FBS) at a concentration of 10 x 106 cfu/mL.
To infect macrophages at a multiplicity of infection (MOI) of 10, remove the medium from the BMDMs and add 5 mL of coated-bacterial suspension prepared. Optimal MOI may be assessed for every cell type or experimental purpose of the isolated phagosomes.
Incubate at RT for 10 min to synchronize phagocytosis.
Incubate at 37 °C in a 5% CO2 incubator for 30 min to initiate phagocytosis. Other cell types may require different incubation times to ensure internalization of the bacteria.
Remove the bacteria-containing medium from the dishes and wash the cells with RPMI three times to remove non-internalized bacteria.
Finally add 10 mL of RPMI containing 10% FBS and 50 µg/mL gentamycin to kill any non-phagocytized bacteria.
Incubate the infected BMDMs at 37 °C with 5% CO2 until the desired time point. The desired time point must be experimentally determined according to the bacteria and cell type used and the purpose of analysis. For the study of early phagosome-endosome fusion events in S. Typhimurium-infected BMDMs, a 30 min incubation is recommended. For the analysis of later events, phagosomes can be extracted after 2 h or 4 h of incubation. Prolonged incubation of macrophages with S. Typhimurium beyond 24 h results in death of the macrophages. Extended intracellular infection before the phagosome extraction could probably lead to degradation of biotin molecules. However, we did not observe loss of biotin on coated-bacteria until 24 h.
4. Isolation of S. typhimurium-containing Phagosomes
Prepare the volume of required phagosome isolation buffer A (50 mM PIPES, pH.7, 50 mM MgCl2, 5 mM EGTA) by adding dithiothreitol (DTT) to a final concentration of 1 mM, Cytochalasin B to a final concentration of 10 µM, and protease and phosphatase inhibitors as recommended by the manufacturer. NOTE: DTT and Cytochalasin B must be added to the phagosome isolation buffer A always immediately before use. Cytochalasin B disrupts the cytoskeleton, facilitating the rupture of the cell membrane.
At the desired time point, remove the medium from the infected cells and wash with sterile PBS warmed to RT.
Add 750 µL of phagosome isolation buffer A per dish and incubate 20 min on ice. When using different cell numbers, the volume of isolation buffer A should be adjusted proportionally.
Add 250 µL of phagosome isolation buffer B (50 mM PIPES, pH.7, 50 mM MgCl2, 5 mM EGTA, 220 mM Mannitol, and 68 mM sucrose).Rock the plate to ensure that the buffer reaches the complete surface of the dish. When using different cell numbers, the volume of isolation buffer B should be adjusted proportionally.
Remove the cells from the dish by gently scraping using a rubber policeman and transfer them to a pre-chilled 1.5 mL tube.
Pass the cell suspension through a 26G needle using a 1 mL syringe at least 15 times (one aspiration plus one ejection of the cell suspension counts as one time). This is enough to release the cytosolic content of BMDMs. The number of passes through the needle must be optimized for every cell type.
Place the cell suspension on the magnetic rack and wait 5 min. The particles attached to the magnet are the phagosomes containing coated-S. Typhimurium. The suspension contains the rest of the cellular components.
Transfer the suspension into a 1.5 mL tube labeled as "cytosol".
Remove the tube with the isolated phagosomes from the magnetic rack and resuspend them in 1 mL of sterile PBS.
Place the phagosome suspension on the magnetic rack, wait for 5 min, and remove the PBS.
Repeat steps 4.9 and 4.10 to wash the isolated S. typhimurium-containing phagosomes.
Finally remove the PBS and resuspend the phagosomes in the required buffer; for example, in a radioimmunoprecipitation assay (RIPA) buffer for protein analysis. Approximately 50-200 µg of protein is obtained per sample following this protocol, depending on the initial amount of cells and the MOI of infection.
Representative Results
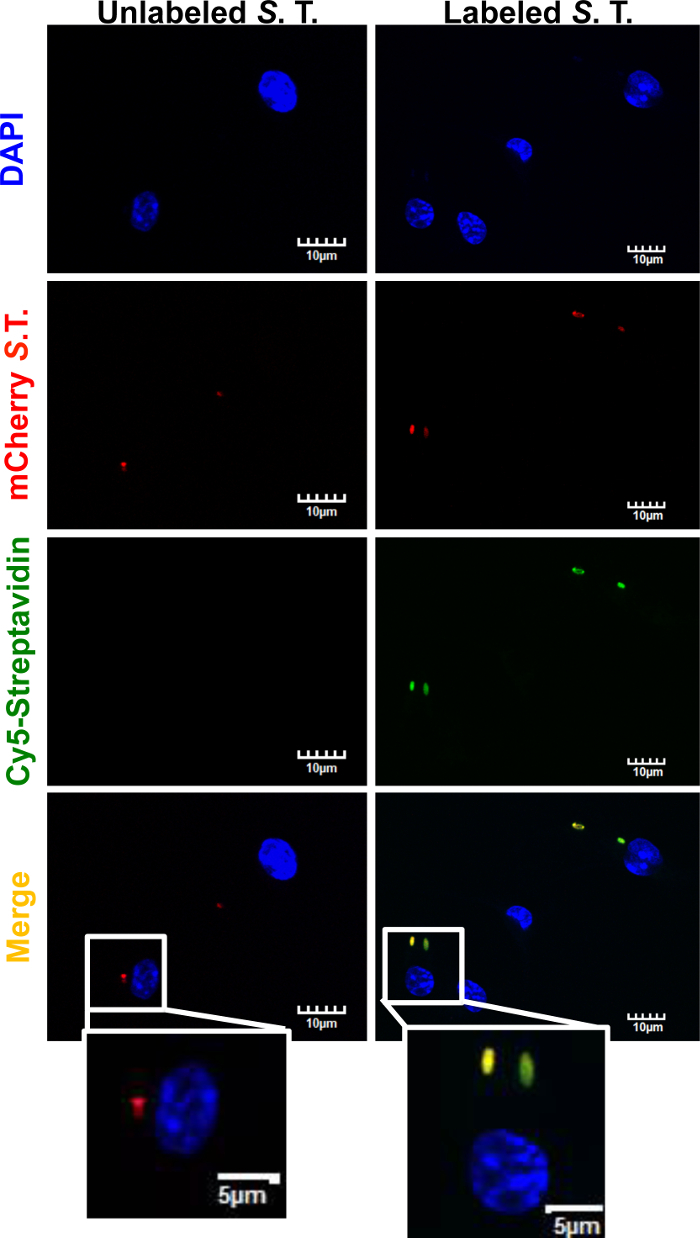
Isolation of bacteria-containing phagosomes by this protocol requires the biotinylation of the bacteria as a first step. We therefore assessed the effectiveness of S. typhimurium biotinylation by confocal microscopy analysis of BMDMs infected with biotinylated mCherry-S. typhimurium labeled with Cy5-Streptavidin. Briefly, BMDMs were infected as described in this protocol with mCherry-S. typhimurium previously biotinylated but not incubated with streptavidin-conjugated magnetic beads. After 30 min incubation at 37 °C, the cells were washed with warm PBS and fixed with 4% formaldehyde for 20 min at RT. Fixation was stopped by extensive washing with PBS. The cells were then permeabilized with 3% Triton in PBS for 5 min at RT. Infected BMDMs were then incubated with Cy5-Streptavidin for 20 min at 4 °C. After extensive washing, the samples were prepared for analysis by confocal microscopy (Figure 1). Not biotinylated mCherry-S. Typhimurium was used as negative control. Fluorescent signal from Cy5-Streptavidin was observed exclusively in biotinylated mCherry-S. typhimurium, confirming that biotinylation of the bacteria was successful. Microscopical analysis was carried out on a confocal microscope using "60X O2 NA:1.40" objective. Excitation wavelengths for the fluorochromes were 405 nm (DAPI), 559 nm (mCherry), and 635 nm (Cy5), and PMT voltage was set at 574v, 565v, and 443v, respectively.
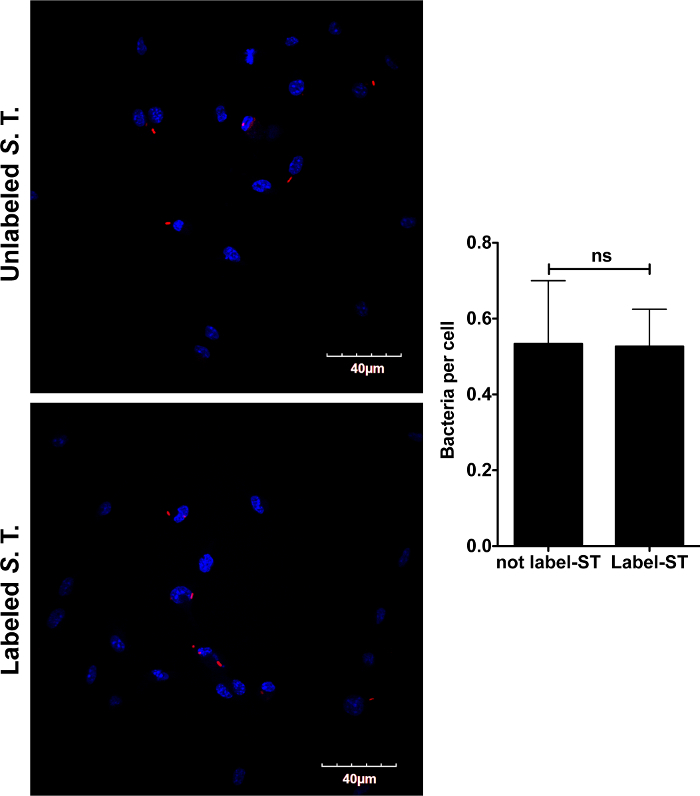
Since the immune response triggered in macrophages by S. typhimurium is largely dependent on the macrophage surface receptor activation by bacterial ligands, we next tested if the coating of S. typhimurium with biotin and streptavidin-conjugated magnetic beads could alter the innate immune response in infected BMDMs. We first assessed whether coating of S. typhimurium could alter the phagocytic capacity of BMDMs by confocal microscopy. BMDMs were infected as described in this protocol with mCherry-S. Typhimurium coated with biotin and streptavidin-conjugated magnetic beads or with uncoated mCherry-S. typhimurium. After 30 min incubation at 37 °C, the cells were washed with warm PBS and fixed with 4% formaldehyde for 20 min at RT. After extensive washing with PBS, the samples were prepared for analysis by confocal microscopy. As shown in Figure 2, macrophages phagocytized coated and uncoated mCherry-S. typhimurium equally.
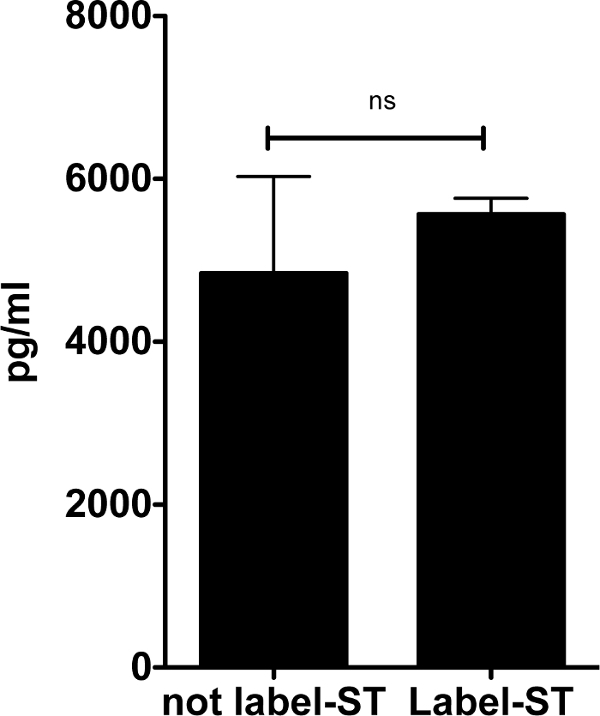
Next, we analyzed the inflammatory response triggered by S. typhimurium coated with biotin and streptavidin-conjugated magnetic beads (Figure 3). Supernatants from the BMDMs infected with coated and uncoated S. typhimurium were collected after 24 h of infection for enzyme-linked immunosorbent assay (ELISA) of secreted interleukin-6 (IL-6). The coating of S. typhimurium with biotin and streptavidin-conjugated magnetic beads did not significantly alter the inflammatory response of macrophages.
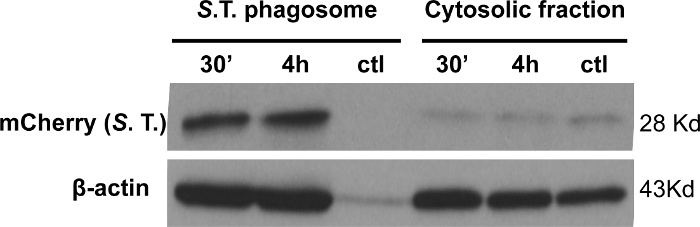
To assess the purity of the bacteria-containing phagosomes obtained by employing this protocol we analyzed isolated mCherry-S. typhimurium-containing phagosomes by immunoblotting. Using specific antibodies against the mCherry tag, we found significant enrichment of mCherry-expressing S. typhimurium in isolated phagosomes in comparison to the cytosolic fraction obtained from the same sample (Figure 4). Isolated phagosomes containing mCherry- S. typhimurium that was not previously coated with biotin were used as a negative control. These results demonstrate that this protocol allows for the purification of phagosomes that are highly enriched in bacteria.
We next performed immunoblot analysis of endosome and lysosome markers in isolated phagosomes containing S. typhimurium (Figure 5). Most of the endosome and lysosome markers were clearly observed in phagosomes isolated at 4 h post-infection in comparison to 30 min. The enrichment of the endosomal markers Rab5 and Rab7, as well as the lysosome markers v-ATPase (both V0 and V1 subunits) and cathepsin D, were observed. Interestingly, only the cleaved form of cathepsin D in isolated phagosomes at 4 h post-infection was observed. The cleavage of pro-cathepsin D (48 kDa) to produce the short active form (33 kDa) only occurs in the phagosomes but not in the cytosol, demonstrating the specificity of this protocol for phagosome isolation.
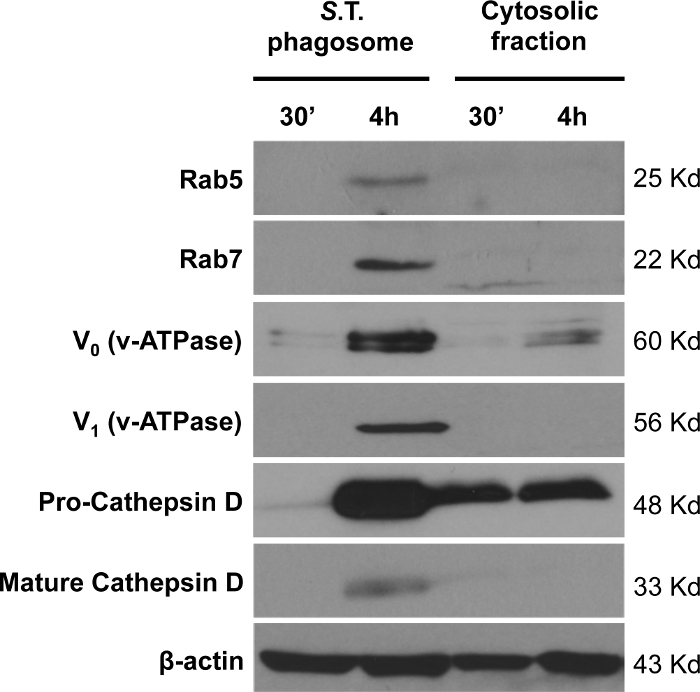
Since markers from organelles such as mitochondria or endoplasmic reticulum (ER) are often detected in preparations of bacteria-containing isolated phagosomes, the S. typhimurium-containing isolated phagosomes were probed with specific antibodies against the mitochondrial transporter Tomm20 and the ER protein calnexin (Figure 6). We also tested them with an antibody against the glycolysis enzyme GAPDH to assess cytosolic contamination in the isolated phagosomes. We found both mitochondria and ER markers in S. typhimurium-isolated phagosomes but no cytosolic contamination.

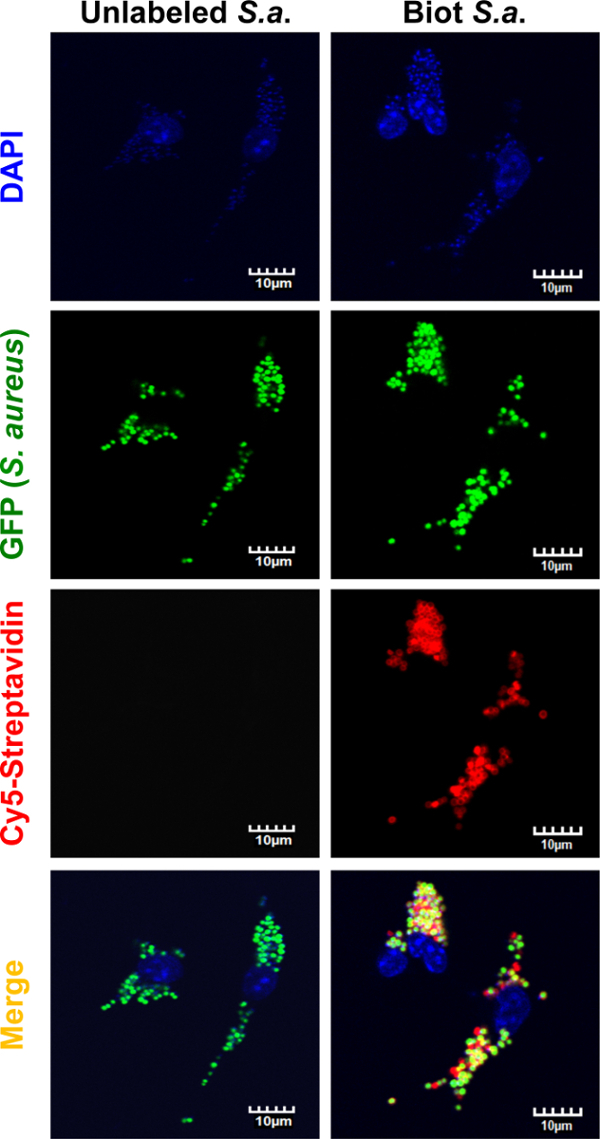
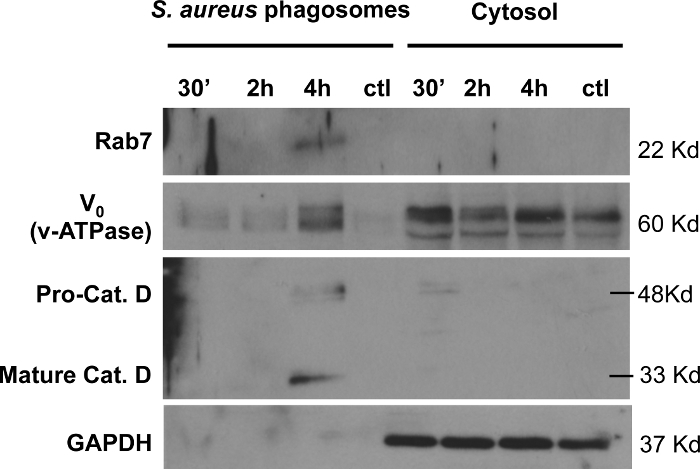
To study the versatility of the protocol for isolation of bacteria-containing phagosomes, the biotin and streptavidin-conjugated magnetic beads coating procedure was applied to the Gram-positive bacterium S. aureus. By employing the same fixation and staining protocol as described for mCherry-S. typhimurium (Figure 1) we confirmed successful biotinylation of green fluorescent protein (GFP) expressing-S. aureus (Figure 7). Moreover, we detected enrichment of the endosome marker Rab7 and the lysosome markers v-ATPase (V0 subunit) and cathepsin D by immunoblot analysis of isolated GFP-S. aureus-containing phagosomes (Figure 8). The cleavage of cathepsin D into its mature form was only detectable after 4 h of infection in the isolated phagosome samples but not in the cytosolic fractions. Isolated S. aureus-containing phagosomes were free of cytosolic contamination as shown by the immunodetection of GAPDH.
In summary, we have demonstrated that our phagosome isolation protocol enables the isolation of highly enriched bacteria-containing phagosomes, free of cytosolic contamination. The isolated phagosome preparations can be used for the analysis of endosome and lysosome markers to study phagosome maturation. Moreover, it was shown that this protocol can be used for both Gram-negative and Gram-positive bacteria and that the labeling procedure does not alter the immune response of macrophages.
Figure 1: Fluorescence microscopy analysis of biotinylated S.typhimurium using Cy5-Streptavidin. BMDMs were infected for 30 min with mCherry-S. typhimurium that was biotinylated ("Biotinylated S. T."), as described in this protocol. Not biotinylated mCherry-S. typhimurium ("Not biotinylated S. T.") was used as a negative control. After fixation and permeabilization of the cells, samples were incubated with Cy5-Streptavidin for 20 min at 4 °C. Fluorescence signal from the nucleus (DAPI), mCherry-S. typhimurium, and Cy5-Streptavidin were analyzed using confocal microscopy. Signal from the Cy5-Streptavidin could only be detected in the bacteria previously labeled with biotin, thus confirming efficient biotinylation. Scale bar = 10 µm. Please click here to view a larger version of this figure.
Figure 2: Microscopical analysis of BMDMs infected with mCherry-S. typhimurium coated with biotin and streptavidin-conjugated magnetic beads. BMDMs were infected with mCherry-S.typhimurium labeled with biotin and streptavidin-conjugated magnetic beads as described in this protocol. After allowing the macrophages to phagocytize the bacteria for 30 min, cells were fixed with 4% formaldehyde for 20 min at RT. The coated bacteria phagocytized by macrophages were then analyzed by confocal microscopy. Analysis showed that the intake of coated bacteria ("Coated S.T.") is comparable to the intake of unlabeled mCherry bacteria ("Not coated S.T."), demonstrating that labeling with biotin and streptavidin-conjugated magnetic beads does not alter phagocytosis of S. typhimurium by macrophages. Nuclei were stained with DAPI for microscopy visualization. Data are shown as mean ± S.E.M., and the statistical significance calculated using student t-test is represented as * = p <0.05; ** = p <0.01; *** = p <0.001. Scale bar = 40 µm. Please click here to view a larger version of this figure.
Figure 3: IL-6 secretion by BMDMs infected with biotin-and-streptavidin-conjugated magnetic beads, labeled S. typhimurium compared to unlabeled S. typhimurium. BMDMs were infected with S. typhimurium coated with biotin and streptavidin-conjugated magnetic beads as described in this protocol. After 24 h of infection, supernatants were collected for further analysis of IL-6 secretion by ELISA. Supernatants form BMDMs infected with uncoated S. typhimurium were used as a control. The induction of IL-6 secretion by coated S. typhimurium was not significantly different compared to that of uncoated S. typhimurium. Data are shown as mean ± S.E.M., and the statistical significance calculated using student t-test is represented as * = p <0.05; ** = p <0.01; *** = p <0.001. Please click here to view a larger version of this figure.
Figure 4: Bacterial enrichment in isolated phagosomes. Enrichment of mCherry-tagged S. typhimurium in isolated phagosomes collected after 30 min and 4 h of infection was compared to the cytosolic fraction. β-actin was used as a loading control. The negative control refers to phagosomes isolated using uncoated S. typhimurium at 30 min post-infection. As expected, the final output of phagosome isolation using unlabeled S. typhimurium does not present significant amounts of mCherry or β-actin, demonstrating the specificity of the protocol. 20 µg of protein was loaded per sample. Please click here to view a larger version of this figure.
Figure 5: Analysis of endosome and lysosome markers in isolated S. typhimurium-containing phagosomes. The enrichment of the endosome markers Rab5 and Rab7, and the lysosome markers v-ATPase (V0 and V1 subunits) and cathepsin D was analyzed in isolated S. typhimurium-containing phagosomes extracted after 30 min and 4 h of infection. β-actin was used as a loading control. 20 µg of protein was loaded per sample. Please click here to view a larger version of this figure.
Figure 6: Analysis of mitochondria, ER, and cytosolic markers in isolated S. typhimurium-containing phagosomes. The cytosolic marker GAPDH, the ER marker calnexin, and the mitochondrial marker Tomm20 in S. typhimurium phagosomes isolated at 30 min and 4 h post-infection were analyzed by immunoblotting and compared to the corresponding cytosolic fractions. 20 µg of protein were loaded per sample. Please click here to view a larger version of this figure.
Figure 7: Microscopical analysis of GFP-S. aureus infected macrophages. BMDMs were infected with S. aureus expressing green fluorescent protein (GFP) labeled with biotin as described in methodology for labeling S. typhimurium. After allowing macrophages to phagocytize the bacteria for 30 min, the cells were fixed. Uncoated S. aureus ("Not biotinylated S. A.") were used as negative control. After fixation and permeabilization of the cells, samples were incubated with Cy5-Streptavidin for 20 min at 4 °C. The fluorescent signal from the nucleus (DAPI), GFP-S. aureus, and Cy5-Streptavidin were analyzed by confocal microscopy. The signal from Cy5-Streptavidin could only be seen in the bacteria previously labeled with biotin, confirming efficient biotinylation. Scale bars = 10 µm. Please click here to view a larger version of this figure.
Figure 8: Analysis of endosome and lysosome markers in isolated S. aureus-containing phagosomes.S. aureus-containing phagosomes isolated at 30 min, 2 h, and 4 h post-infection, were analyzed by immunoblotting for phagosomal markers Rab5, Rab7, and v-ATPase compared to the corresponding cytosolic fractions. 20 µg of protein were loaded per sample. Samples were also tested with a specific antibody against GAPDH, demonstrating that isolated S. aureus-containing phagosomes are free of cytosolic contamination. Please click here to view a larger version of this figure.
Discussion
A new method for the isolation of S. typhimurium-containing phagosomes by coating the bacteria with biotin and streptavidin-conjugated magnetic beads is described here. After gentle disruption of the cell membrane, bacteria-containing phagosomes can be easily extracted using a magnetic rack. We show that labeling of the bacteria preserves the capacity of the pathogen to induce inflammation and does not alter the phagocytic property of the host cell. Importantly, phagosomes obtained by this method are enriched in bacteria and endosome/lysosome markers and devoid of cytosolic contamination.
This method presents several advantages when compared to the traditional phagosome isolation method that employs sucrose gradient separation. It eliminates the time-consuming multiple centrifugation steps, allowing the researcher to handle more samples in less time and obtain purer phagosome samples by easily increasing the number of washing steps. The use of this protocol eliminates the need for specialized and sensitive equipment, such as ultracentrifugation. High speed centrifugation could alter the characteristics of vesicles and reduce the final yield22, which is avoided in this protocol. We have also demonstrated that the coating of S. typhimurium with biotin and streptavidin-conjugated magnetic beads does not alter the phagocytic capacity of macrophages. Moreover, the induction of IL-6 by macrophages infected with labeled S. typhimurium was unaltered compared to macrophages infected with unlabeled bacteria. Therefore, coating of S. Typhimurium does not induce significant changes in their pathogenicity. To our knowledge, coating of S. typhimurium does not represent an impediment for the use of the isolated phagosomes for any of the common laboratory analysis techniques.
S.typhimurium-containing phagosomes isolated by this method are enriched in bacteria and present typical endosomal and lysosomal markers, such as the v-ATPase subunits V0 and V1 and the cleaved mature protease cathepsin D, which can be detected only in the isolated phagosome fractions. Additionally, we found mitochondria and ER markers in isolated S. typhimurium-containing phagosomes. Interaction of phagosomes with the ER has been widely studied although the amount of proteins from the ER present in pathogen-containing phagosomes may vary with the pathogen studied and the cell type. For example, phagosomes containing Brucella abortus isolated from nonprofessional phagocytes are rich in ER markers23 but not the ones isolated from macrophages24. Calnexin has been previously detected in S. Typhimurium-containing phagosomes isolated by sucrose gradient18. Close interaction of inert bead-containing phagosomes and mitochondria has also been previously reported25, explaining the presence of Tomm20 in isolated phagosomes. Mitochondrial proteins have been found in isolated phagosomes containing L. pneumophila26 or Mycobacterium27. However, the detection of mitochondria components in preparations of isolated pathogen-containing phagosomes by using the sucrose gradient centrifugation method has often been believed to be due to the similar density of this organelle and the pathogen-containing phagosome and, therefore, unspecific28. Our method avoids such a contamination issue and provides more reliable information suggesting that mitochondrial membranes might mingle with the phagosomal membranes. In summary, it will be difficult to avoid ER and mitochondrial markers in pathogen-containing isolated phagosomes preparations, depending on the host cell type and the pathogen. The recommended method for the verification of the purity of the isolated phagosomes is to use tagged-bacteria and specific tag antibodies for the detection of bacteria by Western Blot. We also show that this isolation method can be adapted for other microorganisms with available surface amine groups that can be biotinylated. As an example, we have successfully applied our protocol to isolate S. aureus-containing phagosomes. Phagosomes containing S. aureus isolated using this protocol are enriched in lysosome and endosome markers such as v-ATPase (V0 subunit) and Rab7, respectively, but do not present cytosolic markers such as GAPDH.
Endosomal/phagosomal vesicles provide unique environments for the degradation of pathogens, but preserve the antigens for signaling the adaptive immune system. Although, phagosomes and the maturation process have been extensively investigated, the microenvironment in the phagosome upon hosting various pathogens remains elusive. It is also unclear how metabolic changes in cells alter phagosome maturation and its content. Therefore, the protocol detailed here gives more opportunities to study phagosome biogenesis and its function in the context of various pathologies.
Disclosures
The authors have nothing to disclose.
Acknowledgments
Research in the Robinson's lab is supported by funding from Cologne Excellence Cluster on Cellular Stress Responses in Aging-Associated Diseases, University of Cologne, Germany (CECAD; funded by the DFG within the Excellence Initiative of the German federal and state governments) and grants from Deutsche Forschungsgemeinschaft (SFB 670), Köln Fortune, and Maria-Pesch Foundation of the University of Cologne, Germany.
References
- Freeman SA, Grinstein S. Phagocytosis: receptors, signal integration, and the cytoskeleton. Immunol Rev. 2014;262(1):193–215. doi: 10.1111/imr.12212. [DOI] [PubMed] [Google Scholar]
- Kinchen JM, Ravichandran KS. Phagosome maturation: going through the acid test. Nat Rev Mol Cell Bio. 2008;9(10):781–795. doi: 10.1038/nrm2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafourcade C, Sobo K, Kieffer-Jaquinod S, Garin J, van der Goot FG. Regulation of the V-ATPase along the Endocytic Pathway Occurs through Reversible Subunit Association and Membrane Localization. Plos One. 2008;3(7):e2758. doi: 10.1371/journal.pone.0002758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forgac M. Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat Rev Mol Cell Bio. 2007;8(11):917–929. doi: 10.1038/nrm2272. [DOI] [PubMed] [Google Scholar]
- Marshansky V, Futai M. The V-type H+-ATPase in vesicular trafficking: targeting, regulation and function. Curr Opin Cell Biol. 2008;20(4):415–426. doi: 10.1016/j.ceb.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich HJ, Beatty WL, Russell DG. Direct delivery of procathepsin D to phagosomes: Implications for phagosome biogenesis and parasitism by Mycobacterium. Eur J Cell Biol. 1999;78(10):739–748. doi: 10.1016/S0171-9335(99)80042-9. [DOI] [PubMed] [Google Scholar]
- Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol Rev. 2007;87(1):245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- Haas A. The phagosome: Compartment with a license to kill. Traffic. 2007;8(4):311–330. doi: 10.1111/j.1600-0854.2006.00531.x. [DOI] [PubMed] [Google Scholar]
- Scott CC, Botelho RJ, Grinstein S. Phagosome maturation: A few bugs in the system. J Membrane Biol. 2003;193(3):137–152. doi: 10.1007/s00232-002-2008-2. [DOI] [PubMed] [Google Scholar]
- Turk V, et al. Cysteine cathepsins: From structure, function and regulation to new frontiers. Bba-Proteins Proteom. 2012;1824(1):68–88. doi: 10.1016/j.bbapap.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandra L, Song R, Harding CV. Class II MHC molecules and peptide complexes appear in phagosomes during phagocytic antigen processing. Faseb J. 1998;12(4):A589–A589. [Google Scholar]
- Robinson N, et al. Mycobacterial Phenolic Glycolipid Inhibits Phagosome Maturation and Subverts the Pro-inflammatory Cytokine Response. Traffic. 2008;9(11):1936–1947. doi: 10.1111/j.1600-0854.2008.00804.x. [DOI] [PubMed] [Google Scholar]
- Clemens DL, Lee BY, Horwitz MA. Mycobacterium tuberculosis and Legionella pneumophila phagosomes exhibit arrested maturation despite acquisition of Rab7. Infect Immun. 2000;68(9):5154–5166. doi: 10.1128/iai.68.9.5154-5166.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GA, et al. The two distinct phospholipases C of Listeria monocytogenes have overlapping roles in escape from a vacuole and cell-to-cell spread. Infect Immun. 1995;63(11):4231–4237. doi: 10.1128/iai.63.11.4231-4237.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolard MD, Frelinger JA. Outsmarting the host: bacteria modulating the immune response. Immunol Res. 2008;41(3):188–202. doi: 10.1007/s12026-008-8021-5. [DOI] [PubMed] [Google Scholar]
- Gallois A, Klein JR, Allen LAH, Jones BD, Nauseef WM. Salmonella pathogenicity island 2-encoded type III secretion system mediates exclusion of NADPH oxidase assembly from the phagosomal membrane. J Immunol. 2001;166(9):5741–5748. doi: 10.4049/jimmunol.166.9.5741. [DOI] [PubMed] [Google Scholar]
- Ohl ME, Miller SI. Salmonella: A model for bacterial pathogenesis. Annu Rev Med. 2001;52:259–274. doi: 10.1146/annurev.med.52.1.259. [DOI] [PubMed] [Google Scholar]
- Mills SD, Finlay BB. Isolation and characterization of Salmonella typhimurium and Yersinia pseudotuberculosis-containing phagosomes from infected mouse macrophages: Y-pseudotuberculosis traffics to terminal lysosomes where they are degraded. Eur J Cell Biol. 1998;77(1):35–47. doi: 10.1016/S0171-9335(98)80100-3. [DOI] [PubMed] [Google Scholar]
- Buchmeier NA, Heffron F. Inhibition of Macrophage Phagosome-Lysosome Fusion by Salmonella-Typhimurium. Infect Immun. 1991;59(7):2232–2238. doi: 10.1128/iai.59.7.2232-2238.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luhrmann A, Haas A. A method to purify bacteria-containing phagosomes from infected macrophages. Methods Cell Sci. 2000;22(4):329–341. doi: 10.1023/a:1017963401560. [DOI] [PubMed] [Google Scholar]
- Steinhauser C, et al. Lipid-labeling facilitates a novel magnetic isolation procedure to characterize pathogen-containing phagosomes. Traffic. 2013;14(3):321–336. doi: 10.1111/tra.12031. [DOI] [PubMed] [Google Scholar]
- Weischenfeldt J, Porse B. Bone Marrow-Derived Macrophages (BMM): Isolation and Applications. CSH Protoc. 2008;2008 doi: 10.1101/pdb.prot5080. [DOI] [PubMed] [Google Scholar]
- Detilleux PG, Deyoe BL, Cheville NF. Entry and intracellular localization of Brucella spp. in Vero cells: fluorescence and electron microscopy. Vet Pathol. 1990;27(5):317–328. doi: 10.1177/030098589002700503. [DOI] [PubMed] [Google Scholar]
- Arenas GN, Staskevich AS, Aballay A, Mayorga LS. Intracellular trafficking of Brucella abortus in J774 macrophages. Infect Immun. 2000;68(7):4255–4263. doi: 10.1128/iai.68.7.4255-4263.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, et al. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature. 2011;472(7344):476–480. doi: 10.1038/nature09973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Jagannath C, Rao PK, Singh CR, Lostumbo G. Analysis of phagosomal proteomes: from latex-bead to bacterial phagosomes. Proteomics. 2010;10(22):4098–4116. doi: 10.1002/pmic.201000210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao PK, Singh CR, Jagannath C, Li Q. A systems biology approach to study the phagosomal proteome modulated by mycobacterial infections. Int J Clin Exp Med. 2009;2(3):233–247. [PMC free article] [PubMed] [Google Scholar]
- Rogers LD, Foster LJ. The dynamic phagosomal proteome and the contribution of the endoplasmic reticulum. P Natl Acad Sci USA. 2007;104(47):18520–18525. doi: 10.1073/pnas.0705801104. [DOI] [PMC free article] [PubMed] [Google Scholar]