

Abstract
Dopamine receptor 2 (DR2) may be a biomarker for various types of cancer. Ovarian cancer cells overexpress DR2; therefore, blocking DR2 may be a novel treatment strategy for ovarian cancer. Thioridazine, a DR2 blocker, has antineoplastic activity in a variety of cancer cells. In view of the requirement for novel therapeutic agents in ovarian cancer, the present study aimed to determine the potential effects of thioridazine in vitro and in vivo. It was revealed that the DR2 blocker thioridazine induced cell death in a dose-dependent manner in ovarian cancer cells. Thioridazine treatment induced apoptosis and autophagy, which may be attributed to an increased level of reactive oxygen species and associated DNA damage. Additionally, the expression of various proteins increased with oxidative stress, including nuclear factor E2-related factor 2, which is a pivotal transcriptional factor involved in cellular responses to oxidative stress. Heme oxygenase 1, NAPDH quinone dehydrogenase 1 and hypoxia inducible factor-1α and phosphorylated (p)-protein kinase B expression was significantly decreased, and the expression level of p-extracellular signal-related kinases and p-P38 was increased. Using 3-methyl adenine to inhibit autophagy caused the rate of apoptosis to increase. Thioridazine inhibited the growth of SKOV3 xenografts in nude mice. The present study demonstrated that the DR2 blocker thioridazine exhibited anticancer effects in vitro and in vivo, suggesting that thioridazine may be used as a potential drug in ovarian cancer therapy.
Keywords: dopamine receptor 2 blocker, thioridazine, apoptosis, autophagy, oxidative stress, DNA damage, ovarian cancer
Introduction
Epithelial ovarian cancer is the leading cause of mortality among gynecological types of cancer. Surgical debulking combined with chemotherapy is the standard therapeutic strategy. However, the relapse rate is high, primarily due to the development of chemotherapy resistance (1), therefore, novel modalities must be explored.
Dopamine receptor (DR) expression may be associated with the development of various types of cancer. Patients with schizophrenia who receive DR antagonists have a reduced incidence of cancer of the rectum, colon, prostate and uterine cervix (2,3). Patients with Parkinson's disease, which functionally similar to disease-induced DR antagonism, also have a lower incidence of cancer. It was hypothesized that DR may be a biomarker for cancer (4). Knockdown/blocking of DR2 inhibited the proliferation of cancer cells, including cancer stem cells (5). This suggested that DR2 may be a treatment target for types of cancer that expresses dopamine receptor 2.
Thioridazine is a DR2 antagonist and has been clinically approved to treat schizophrenia and other psychotic disorders (6). Of note, thioridazine exhibits anticancer action in breast cancer, leukemia, hepatoma and cervical carcinoma (5,7–9). Ovarian cancer cells express a number of DRs, with the exception of DR3 (10), suggesting that thioridazine may be used to treat ovarian cancer. In the present study, the effects of thioridazine on ovarian cancer were explored in vitro and in vivo. The findings suggested that thioridazine may be a promising candidate drug for ovarian cancer therapy.
Materials and methods
Reagents
Thioridazine was obtained from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany) and kept as 50 mM stock solutions in water. Autophagy inhibitor 3-methyl adenine (3-MA) was purchased from Sigma-Aldrich (Merck KGaA) and kept as 50 mM stock solutions in dimethyl sulfoxide.
Cell culture
A2780 and SKOV3 human ovarian cancer cell lines (China Center for Type Culture Collection, Wuhan, China) were cultured in RPMI-1640 medium (Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal bovine serum (Zhejiang Hisun Chemical Co., Ltd., Taizhou, China) and 1% penicillin-streptomycin solution at 37°C and 5% CO2.
Cell survival
SKOV3 and A2780 cells were seeded into a 96-well plate at a density of 1×104 cells/well, followed by thioridazine treatment with increasing doses (5, 10, 15, 20 and 25 µm) for 24 h at 37°C. The controls were not exposed to the thioridazine. The cell viability was determined by performing a tetrazolium assay (Cell Counting kit-8; Dojindo Molecular Technologies, Inc., Kumamoto, Japan) according to the protocol of the manufacturer. A concentration of 15 µM thioridazine induced cell inhibition, which was then utilized in subsequent experiments. Cells (5×105) were seeded in a 6-well plate, then 15 µM thioridazine was added and maintained at 37°C for various time periods (6, 12 and 24 h) prior to assay analysis. Following exposure to 15 µM thioridazine for various time periods (6, 12 and 24 h), the morphological changes and the protein expression related to the DR2, apoptosis and autophagy were detected. Experiments were performed in triplicate.
Detection of reactive oxygen species (ROS)
Cells were incubated with 10 µM 2′,7′-dichlorofluorescein diacetate (Sigma-Aldrich; Merck KGaA) for 30 min at 37°C, rinsed with PBS, trypsinized, centrifuged (800 × g for 5 min at room temperature), resuspended in 1 ml PBS and then analyzed by flow cytometry, and 4 fields of view were observed under a fluorescence microscope at magnification, ×200.
Detection of light chain 3 (LC3) using immunofluorescence
Cells were rinsed with PBS three times, fixed in 4% paraformaldehyde for 15 min, blocked with 5% bovine serum albumin (Beyotime Institute of Biotechnnology, Haimen, China) for 30 min and permeabilized with 1% Triton X-100 for 10 min, all at room temperature. Subsequently, cells were incubated with a rabbit anti-LC3 antibody (1:100) (Sigma-Aldrich; Merck KgaA; cat. no. L8918) at 4°C overnight. Following incubation with a secondary antibody (goat anti-rat IgG (H+L), FITC conjugate, 1:1,000 (cat. no. SA00003-11); ProteinTech Group, Inc., Chicago, IL, USA) for 1 h at room temperature and nucleus staining using DAPI (Beyotime Institute of Biotechnnology), the cells in 4 fields of view were visualized using a fluorescence microscope at magnification, ×200.
Detection of apoptosis using Annexin V. SKOV3 and A2780 cells were pre-treated with 5 mM 3-MA (3-Methyladenine) (Sigma-Aldrich; Merck KGaA; cat. no. M9281) or not, followed by a 24-h thioridazine treatment. Cells were stained using fluorescein isothiocyanate-labelled Annexin V (Nanjing KeyGen Biotech Co., Ltd., Nanjing, China) according to the protocol of the manufacturer, and analyzed using flow cytometry (FACSCanto II) according to the protocol of the manufacturer.
Western blotting
Nuclear factor erythroid 2-related 2 (Nrf2), phosphorylated Nuclear factor erythroid 2-related 2 (p-Nrf2), NAD(P)H quinone dehydrogenase 1 (NQO1), heme oxygenase 1 (HO-1), c-Jun N-terminal kinase (JNK), p-JNK, p38, p-p38, extracellular signal-related kinase (ERK), p-ERK, protein kinase B (AKT), p-AKT, hypoxia inducible factor-1 (HIF-1), vascular endothelial growth factor (VEGF), P62, LC3 and cleaved caspase-3 were detected using western blotting. Total proteins were harvested by lysing cells in a buffer supplemented with protease inhibitor for 10 min in an ice bath. Equal amounts (25 µg/µl) of protein were separated using 8–12% gradient SDS-PAGE (Bio-Rad Laboratories, Inc., Hercules, CA, USA) and then transferred to polyvinylidene difluoride membranes. Following blocking 5% skimmed milk powder for 2 h at room temperature, the membranes were incubated with various primary antibodies (1:1,000) overnight. Antibodies against Nrf2 (cat. no. 12721s), NQO1 (cat. no. 62262S), HO-1 (cat. no. 1:5853S), JNK (cat. no. 4672S), p-JNK (cat. no. 4668S), p38 (cat. no. 8690S), p-p38 (cat. no. 9216S), ERK (cat. no. 9102S), p-ERK (cat. no. 4377T), AKT (cat. no. 4685S), p-AKT (cat. no. 13038S), HIF-1 (cat. no. 14179S), VEGF (cat. no. 2463S), P62 (cat. no. 8025S) and cleaved caspase-3 (cat. no. 9664S) were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA), the antibody against LC3 was obtained from Sigma-Aldrich (cat. no. L8918; Merck KGaA), and the antibody against P-Nrf2 (cat. no. ab76026) was purchased from Abcam (USA). Subsequently, the blots were rinsed 3 times with PBS, incubated with a goat anti-rat IgG (H+L) horseradish peroxidase-conjugated secondary antibody (cat. no. SA00001-15; dilution, 1:1,000; ProteinTech Group, Inc., Chicago, IL, USA) at 4°C overnight and observed using an ECL reagents kit (KeyGEN BioTECH, Nanjing, China) according to the protocol of the manufacturer. GAPDH served as the reference. Results were analyzed using Image Lab software from Bio-Rad Laboratories, Inc. (Hercules, CA, USA; v.4.1.0.0). Experiments were performed in triplicate.
Detection of DNA damage using an alkaline comet assay
DNA damage was detected using the alkaline Comet Assay (11), using normal-(cat. no. A9414) and low-melting point agarose (A9414; Sigma-Aldrich; Merck KGaA). In brief, a slide was pre-coated with 150 µl 1% normal-melting-point agarose and mounted on a coverslip for 20 min at 4°C. Subsequently, 70 µl (containing 1×104 cells) 1% low-melting-point agarose was spread on the agarose pre-coated slide and then mounted using a coverslip for 20 min at 4°C. The slide was immersed in 40 ml 176.55 g/l freshly prepared alkaline lysis solution (2.5 M NaCl, 100 mM EDTA, 10 mM Tris, 34 mM N-lauroylsarcosine sodium at pH 10.0–10.5 with freshly added 1% Triton X-100) at 4°C overnight. Following rinsing with PBS, the slide was placed in an electrophoresis chamber filled with freshly prepared alkaline electrophoresis buffer (300 mM NaOH, 1 mM EDTA, pH 13) for 20 min at 4°C in the dark and electrophoresed at 300 mA and 25 V for 20 min. Subsequently, the slide was washed with 0.4 M Tris buffer (pH 7.5) for 10 min and dried at room temperature for 1 h. The slide was stained with 5 µg/ml DAPI (Beyotime Institute of Biotechnnology) for 15 min at room temperature, rinsed with cold distilled water, and 4 fields of view were observed under a fluorescence microscope at microscope, ×200. The comet formation rate, or the comet quantity/total number of cells was used to evaluate the degree of DNA damage.
In vivo therapeutic efficacy
Female BALB/c nude mice aged 5–6-weeks (16–17 g) were obtained from the Peking University Health Science Center (Laboratory Animals Science Department, Beijing, China) and kept in an atmosphere of 21–24°C, with a relative humidity of 40–67%. Air exchange was performed 14.4–17.2 times/h. Food and water were provided ad libitum. The mice were injected subcutaneously with 1×107 SKOV3 cells. Tumors reached 100 mm3 following ~14 days. Nude mice were administered an intraperitoneal injection of 25 mg/kg thioridazine dissolved in physiological saline into the abdominal cavity, and length (L, mm) and width (W, mm) of the tumors were determined every once every three days for 3 weeks, so that each mouse received 7 injections. Tumor volume (V, mm3) was subsequently evaluated using the following equation: V=(LxW2)/2. A total of 3 days following the final injection, the nude mice were sacrificed via cervical dislocation and the tumors were excised. All procedures for animal experiments were approved by the Committee on the Use and Care of Animals (Chongqing Medical University, Chongqing, China) and performed in accordance with the institution's guidelines.
Statistical analysis
Data are presented as the mean ± standard deviation. Statistical analysis was performed using SPSS version 17.0 (SPSS Inc., Chicago, IL, USA) and comparison of groups was analyzed by the Student's t-test. P<0.05 was considered to indicate a statistically significant difference.
Results
Thioridazine blocks DR2 and suppresses cell proliferation
SKOV3 and A2780 cells were exposed to thioridazine for 24 h. The concentration that produced significant cell inhibition was determined to be 15 µM (P<0.05; Fig. 1A). The cell viability values for each cell type at each concentration of thioridazine were as follows: SKOV3: 5 µm, 95.24±3.64%; 10 µm, 90.7±3.48%; 15 µm, 57.00±4.13%; 20 µm, 36.04±4.52%; 25 µm, 21.45±4.23%; and A2780: 5 µm, 92.51±6.36%; 10 µm, 74.21±4.54%; 15 µm, 53.22±7.81%; 20 µm, 21.17±5.74%; 25 µm, 9.97±3.62%. The morphological changes observed included cellular rounding, vacuolation and detachment (Fig. 1B). Western blotting indicated that the expression of DR2 in SKOV3 and A2780 was downregulated following treatment with 15 µM thioridazine (Fig. 1C). These results suggested that the inhibition of cell proliferation by thioridazine may be associated with the suppression of DR2.
Figure 1.
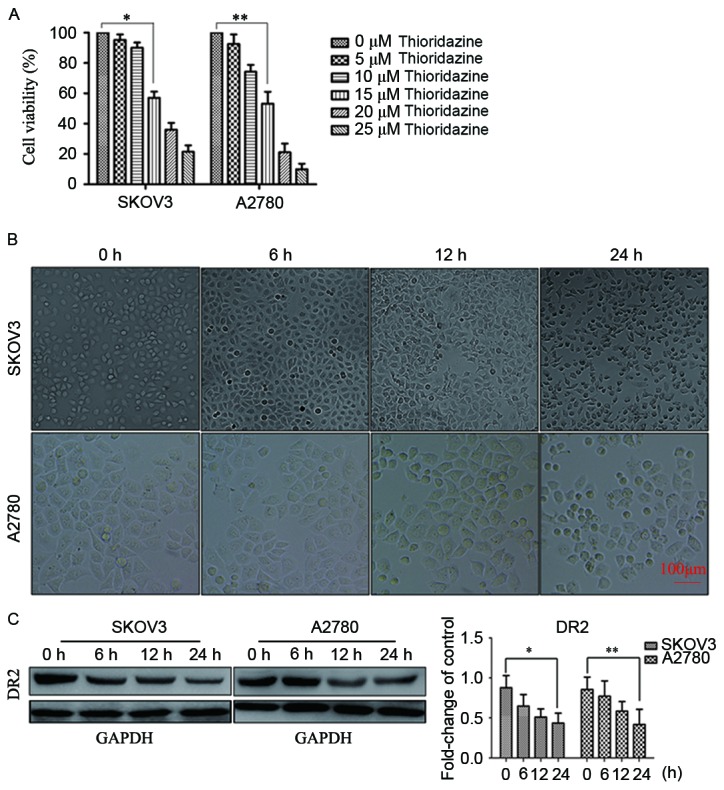
Thioridazine blocked DR2 and suppressed cell proliferation. (A) The effect of thioridazine on cell proliferation; a concentration of 15 µM was the minimum required to induce the inhibition of SKOV3 and A2780 cells. Data are presented as the mean ± standard derivation of three independent experiments. *P=0.003; **P=0.009 vs. control. (B) Light microscopy images of ovarian cancer cell morphology following treatment with 15 µM thioridazine for the designated length of time (0, 6, 12 and 24 h). Scale bar, 100 µm. (C) DR2 expression as determined by western blotting analysis. GAPDH was used as the reference. Bar graph represents the mean ± standard deviation of triplicate experiments. *P=0.019, **P=0.038, compared with the control group.
Thioridazine induced apoptosis
In order to investigate the mechanisms underlying the cytocidal effects of thioridazine, SKOV3 and A2780 cells were treated with 15 µM thioridazine for various durations (0, 6, 12 or 24 h). Flow cytometry analysis revealed a higher percentage of apoptotic cells following a 24-h thioridazine treatment (Fig. 2A). Apoptosis was confirmed with the activation of caspase 3 and the subsequent production of cleaved caspase-3, which was detected by western blot analysis. The expression levels of cleaved caspase-3 (a marker of apoptosis) were increased after a 24-h treatment (Fig. 2B).
Figure 2.

Thioridazine induced apoptosis. (A) Annexin V/PI double staining analysis of SKOV3 and A2780 cells treated with or without 15 µM thioridazine. Data are representative of the results of triplicate independent experiments. *P=0.01, **P=0.005, compared with the control group. (B) Representative western blotting demonstrating the levels of cleaved caspase-3. GAPDH was used as an internal control. Bar graph represents the mean ± standard deviation of triplicate experiments. *P=0.041, 12 h; **P=0.012, 24 h; ***P=0.044, 12 h; ****P=0.026, 24 h, vs. control group. PI, protease inhibitor; NC, negative control.
Thioridazine induces ROS production and DNA damage
In order to investigate the underlying mechanisms of thioridazine induced apoptosis, the cellular ROS, DNA damage and protein expression associated with oxidative stress were evaluated. The ROS levels and comet formation rates (SKOV3: 8.33±1.53 vs. 30.33±3.21%; P=0.002; A2780: 5.67±2.08%; P=0.002) were increased following 15 µm thioridazine treatment for 24 h (Fig. 3A and B). The expression levels of p-Nrf2, a pivotal transcriptional factor involved in the cellular responses to oxidative stress, and its downstream targets, HO-1, NQO1 and HIF-1α, were significantly decreased (P<0.05). The expression level of VEGF, a pivotal enabling factor for tumor angiogenesis, was reduced. VEGF is regulated by HIF-1α (Fig. 3C) (10).
Figure 3.

Thioridazine induces ROS production and DNA damage. (A) Fluorescence microscopy images of cells following exposure to 15 µM thioridazine for 24 h (scale bar, 50 µm). Flow cytometry was used to detect the DCFH-DA fluorescence of the cells treated with thioridazine for various lengths of time (0, 6, 12 or 24 h). (B) DNA damage was detected using an alkaline Comet Assay (scale bar, 50 µm). (C) Western blot analysis investigating the expression levels of p-Nrf2/Nrf2, NQO1, HO-1 HIF-1α and VEGF. GAPDH was used as an internal control.
Thioridazine induces autophagy
To investigate whether thioridazine induces autophagy, the immunofluorescence assay was used to detect the localization of LC3. LC3-green punctate were observed in 15 µm thioridazine-treated cells (Fig. 4A). The expression levels of LC3 (a marker of autophagy) and P62 (a substrate of autophagy) were evaluated. LC3-II was significantly upregulated in a dose- and time-dependent manner in the two cell lines following treatment with 0, 5, 10 and 15 µm thioridazine for 0, 6, 12 and 24 h (Fig. 4B and C; P<0.05). Notably, autophagy occurred with a relatively lower drug concentration (5 µM) and earlier drug processing time (6 h). To investigate the pro-survival or pro-cell death mechanisms underlying thioridazine-induced autophagy, SKOV3 and A2780 cells were pre-treated with 5 mM 3-MA for 2 h to inhibit the formation of autophagosomes, followed by a 24-h treatment with thioridazine (Fig. 4D). The apoptosis rate (SKOV3: 14.43±1.76%; A2780; 29.71±1.98%) was increased in the 3-MA pretreated group (SKOV3: 36.24±3.93%, *P=0.004; A2780: 44.35±3.78%, **P=0.021). These results indicated that thioridazine induces autophagy, which may be a pro-survival mechanism associated with thioridazine-induced cytotoxicity in ovarian cancer cells.
Figure 4.

Thioridazine induced autophagy in ovarian cancer cells. (A) LC3 localization following treatment with thioridazine for 12 h (scale bar, 50 µm). (B) Representative western blot analysis demonstrating the expression levels of LC3 following treatment with thioridazine (0, 5, 10 and 15 µM) for 24 h. GAPDH was used as an internal control. (C) Representative western blot analysis demonstrating the expression levels of LC3 and P62 following treatment with 15 µM thioridazine for various lengths of time (0, 6, 12 and 24 h). GAPDH was used as an internal control. (D) Ovarian cancer cells were pre-treated with 5 mM 3-MA for 2 h, and then treated with 15 µM thioridazine for 24 h. Annexin V/PI double staining was used to analyze the effect of thioridazine on ovarian cancer cells. Data are representative of the results of two independent experiments. *P=0.004, **P=0.021 compared with the control group. 3-MA, 3-methyl adenine; NC, negative control; PI, protease inhibitor; LC3, light chain 3.
Thioridazine induces autophagy via the AKT/ERK signaling pathway
It has previously been demonstrated that DR2 expression is associated with the phosphorylation/activation of AKT and ERK [members of the mitogen activated protein kinase (MAPK) family], which serve an essential role in the proliferation, apoptosis and autophagy of cancer cells (12–14). The activity of ERK and AKT was evaluated by western blot analysis, which revealed that p-AKT was downregulated (Fig. 4A) and that the expression of p-ERK was upregulated following treatment with 15 µM thioridazine. Furthermore, the activity of P38 and JNK, which are also members of the MAPK family and share a 40–50% sequence identity to ERK, was analyzed. Consistent with the expression pattern of p-ERK, the expression level of p-P38 was also increased following treatment with 15 µM thioridazine (Fig. 5). However, the expression level of p-JNK was not detected (data not presented).
Figure 5.
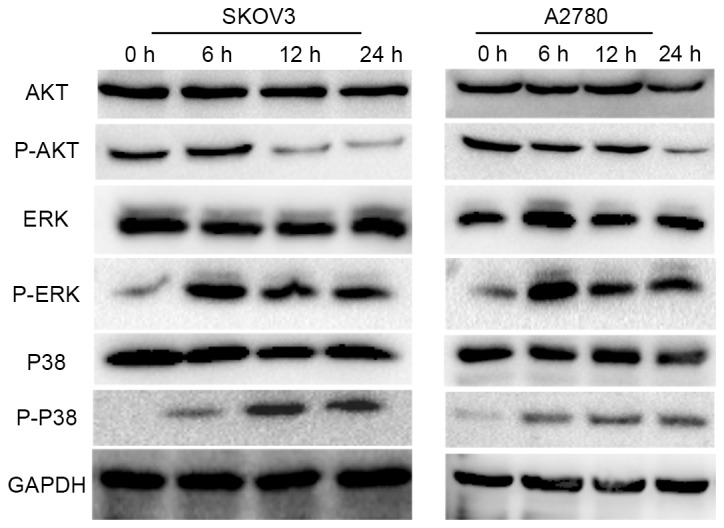
Thioridazine induced autophagy via the AKT/ERK signaling pathway. Representative western blotting demonstrating the expression levels of P-AKT/AKT, P-ERK/ERK, P-P38/P38 following treatment with of 15 µM thioridazine for various lengths of time (0, 6, 12 and 24 h). GAPDH was used as an internal control. AKT, protein kinase B; ERK, extracellular signal-related kinase; P, phosphorylated.
Thioridazine inhibits the growth of SKOV3 xenografts in nude mice
The effect of thioridazine on the growth of ovarian cancer cells in vivo was investigated using a xenograft mouse model. The tumor size was smaller in the thioridazine treatment group (mice treated with 25 mg/kg for 3 weeks) on the day 21 (tumor volume: 1094.74±127.31 mm3) compared with the control group (tumor volume: 478.07±72.63 mm3; P=0.004; Fig. 6A). The morphology of tumors with or without treatment of 25 mg/kg thioridazine on day 21 is demonstrated in Fig. 6B.
Figure 6.
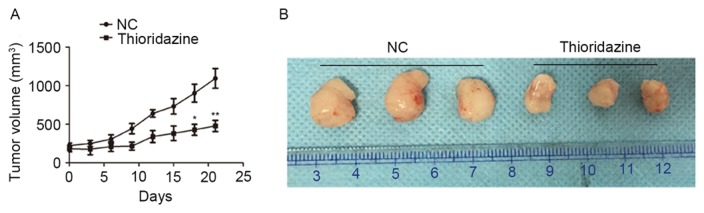
Thioridazine inhibited tumor growth in vivo. (A) Growth curve derived from SKOV3 xenografts in nude mice treated with or without 25 mg/kg thioridazine. *P=0.007, **P=0.004, compared with the control group. (B) Morphology of tumors with or without treatment of 25 mg/kg thioridazine on day 21. NC, negative control.
Discussion
Treatment with the DR2 blocker, thioridazine, inhibited the proliferation of ovarian cancer cells in vivo and in vitro. The concentration of 15 µM is within the range of tolerability in human patients (15,16). This suggests that thioridazine may be a candidate drug for ovarian cancer treatment. In the present study, thioridazine was observed to induce apoptosis and autophagy. Apoptosis is the primary mechanism underlying the action of anticancer drugs (17). Autophagy, the intracellular degradation of cytoplasmic components, serves an intricate and paradoxical role in tumor chemotherapy (18,19). The results of the present study indicated that autophagy may act as a pro-survival mechanism in the anticancer action of thioridazine. Conversely, a previous study reported that it was autophagy, not apoptosis, induced by thioridazine in glioblastoma cells that appeared to be the principal pro-cell death mechanism (20). In certain cell systems with apoptotic dysfunction, autophagy has acted as a cell death mechanism (21). The role of autophagy in apoptotic competent cells may depend on the type of stimulus (22). In other cases, the role of autophagy in initiating cell death has been identified: In response to hypoxia, ER stress, chemotherapeutic, virus infection and toxin (23–26). The diverse role of autophagy in different cells appears complex, and further research is required.
Thioridazine induced a higher level of cellular ROS and DNA damage in comparison with the control group. Simultaneously, antioxidative stress-associated proteins were downregulated, including p-Nrf2 and its downstream targets, HO-1, NQO1 and HIF1α. This inhibition of the expression of certain cellular antioxidant stress-associated proteins, and consequent increase in intracellular levels of ROS, may explain the mechanism by which thioridazine induces apoptosis and autophagy. HIF1α binds to the promoter of VEGF (27,28), which activated the proliferation and migration of endothelial cells during microvessel formation (29). Consistent with the expression of HIF1α, VEGF was downregulated. Inhibition of angiogenesis may result in anticancer efficacy (30). Overexpression or hyper-activation of Nrf2 may participate in tumorigenesis. Knockdown of Nrf2 may reverse cisplatin resistance in ovarian cancer (31); the present study demonstrated that thioridazine inhibited the expression of p-Nrf2, and suggested that it may be an potential adjuvant for cisplatin therapy. A previous study demonstrated that the co-delivery of thioridazine and doxorubicin (DOX) using polymeric micelles eradicated cancer cells and DOX-resistant cancer stem cells (32).
DR may be associated with cancer chemotherapy (33,34) Thioridazine and its analogs have exhibited antitumor effects in melanoma (35). The development of certain small molecules with a high specificity for DR2 or knockdown of DR2 may confer an improved therapeutic prognosis (34,36). However, the DR2 agonist bromocriptin has inhibited proliferation in MCF-7 breast cancer cells (37). Thus, a strategy specifically targeting DR2 must be developed dependent on the type of cancer.
In conclusion, DR2 may be a therapeutic target for ovarian cancer. The DR2 blocker thioridazine exhibited anticancer effects in vivo and in vitro by inducing oxidative stress and apoptosis, inhibiting tumor angiogenesis and interacting with the AKT and ERK signaling pathways. Thus, thioridazine may be a promising candidate drug for ovarian cancer.
Acknowledgements
The present study was supported by the Natural Science Foundation of Chongqing (grant no. CSTC 2012JJB10030) and The Natural Science Foundation of China (grant no. 81172492).
References
- 1.Shih KK, Chi DS. Maximal cytoreductive effort in epithelial ovarian cancer surgery. J Gynecol Oncol. 2010;21:75–80. doi: 10.3802/jgo.2010.21.2.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dalton SO, Mellemkjaer L, Thomassen L, Mortensen PB, Johansen C. Risk for cancer in a cohort of patients hospitalized for schizophrenia in Denmark, 1969–1993. Schizophr Res. 2005;75:315–324. doi: 10.1016/j.schres.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 3.Mortensen PB. The incidence of cancer in schizophrenic patients. J Epidemiol Community Health. 1989;43:43–47. doi: 10.1136/jech.43.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Driver JA, Logroscino G, Buring JE, Gaziano JM, Kurth T. A prospective cohort study of cancer incidence following the diagnosis of Parkinson's disease. Cancer Epidemiol Biomarkers Prev. 2007;16:1260–1265. doi: 10.1158/1055-9965.EPI-07-0038. [DOI] [PubMed] [Google Scholar]
- 5.Sachlos E, Risueño RM, Laronde S, Shapovalova Z, Lee JH, Russell J, Malig M, McNicol JD, Fiebig-Comyn A, Graham M, et al. Identification of drugs including a dopamine receptor antagonist that selectively target cancer stem cells. Cell. 2012;149:1284–1297. doi: 10.1016/j.cell.2012.03.049. [DOI] [PubMed] [Google Scholar]
- 6.Kilts CD, Knight DL, Mailman RB, Widerlöv E, Breese GR. Effects of thioridazine and its metabolites on dopaminergic function: Drug metabolism as a determinant of the antidopaminergic actions of thioridazine. J Pharmacol Exp Ther. 1984;231:334–342. [PubMed] [Google Scholar]
- 7.Yin T, He S, Shen G, Ye T, Guo F, Wang Y. Dopamine receptor antagonist thioridazine inhibits tumor growth in a murine breast cancer model. Mol Med Rep. 2015;12:4103–4108. doi: 10.3892/mmr.2015.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu M, Li J, Luo Z, Zhang S, Xue S, Wang K, Shi Y, Zhang C, Chen H, Li Z. Roles of dopamine receptors and their antagonist thioridazine in hepatoma metastasis. OncoTargets Ther. 2015;8:1543–1552. doi: 10.2147/OTT.S77373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mao M, Yu T, Hu J, Hu L. Dopamine D2 receptor blocker thioridazine induces cell death in human uterine cervical carcinoma cell line SiHa. J Obstet Gynaecol Res. 2015;41:1240–1245. doi: 10.1111/jog.12691. [DOI] [PubMed] [Google Scholar]
- 10.Moreno-Smith M, Lee SJ, Lu C, Nagaraja AS, He G, Rupaimoole R, Han HD, Jennings NB, Roh JW, Nishimura M, et al. Biologic effects of dopamine on tumor vasculature in ovarian carcinoma. Neoplasia. 2013;15:502–510. doi: 10.1593/neo.121412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Olive PL, Banáth JP. The comet assay: A method to measure DNA damage in individual cells. Nat Protoc. 2006;1:23–29. doi: 10.1038/nprot.2006.5. [DOI] [PubMed] [Google Scholar]
- 12.Pozzi L, Håkansson K, Usiello A, Borgkvist A, Lindskog M, Greengard P, Fisone G. Opposite regulation by typical and atypical anti-psychotics of ERK1/2, CREB and Elk-1 phosphorylation in mouse dorsal striatum. J Neurochem. 2003;86:451–459. doi: 10.1046/j.1471-4159.2003.01851.x. [DOI] [PubMed] [Google Scholar]
- 13.Rodrigues-Ferreira S, Nahmias C. G-protein coupled receptors of the renin-angiotensin system: New targets against breast cancer? Front Pharmacol. 2015;6:24. doi: 10.3389/fphar.2015.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berhow MT, Hiroi N, Nestler EJ. Regulation of ERK (extracellular signal regulated kinase), part of the neurotrophin signal transduction cascade, in the rat mesolimbic dopamine system by chronic exposure to morphine or cocaine. J Neurosci. 1996;16:4707–4715. doi: 10.1523/JNEUROSCI.16-15-04707.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nahata MC, Ford C, Ruymann FB. Pharmacokinetics and safety of prochlorperazine in paediatric patients receiving cancer chemotherapy. J Clin Pharm Ther. 1992;17:121–123. doi: 10.1111/j.1365-2710.1992.tb01279.x. [DOI] [PubMed] [Google Scholar]
- 16.Morgan RJ, Jr, Synold T, Carr BI, Doroshow JH, Womack EP, Shibata S, Somlo G, Raschko J, Leong L, McNamara M, et al. Continuous infusion prochlorperazine: Pharmacokinetics, antiemetic efficacy, and feasibility of high-dose therapy. Cancer Chemother Pharmacol. 2001;47:327–332. doi: 10.1007/s002800000232. [DOI] [PubMed] [Google Scholar]
- 17.Adams JM. Ways of dying: Multiple pathways to apoptosis. Genes Dev. 2003;17:2481–2495. doi: 10.1101/gad.1126903. [DOI] [PubMed] [Google Scholar]
- 18.Wu HM, Jiang ZF, Ding PS, Shao LJ, Liu RY. Hypoxia-induced autophagy mediates cisplatin resistance in lung cancer cells. Sci Rep. 2015;5:12291. doi: 10.1038/srep12291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.García-Cano J, Ambroise G, Pascual-Serra R, Carrión MC, Serrano-Oviedo L, Ortega-Muelas M, Cimas FJ, Sabater S, Ruiz-Hidalgo MJ, Sanchez Perez I, et al. Exploiting the potential of autophagy in cisplatin therapy: A new strategy to overcome resistance. Oncotarget. 2015;6:15551–15565. doi: 10.18632/oncotarget.3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng HW, Liang YH, Kuo YL, Chuu CP, Lin CY, Lee MH, Wu AT, Yeh CT, Chen EI, Whang-Peng J, et al. Identification of thioridazine, an antipsychotic drug, as an antiglioblastoma and anticancer stem cell agent using public gene expression data. Cell Death Dis. 2015;6:e1753. doi: 10.1038/cddis.2015.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jin S, White E. Role of autophagy in cancer: Management of metabolic stress. Autophagy. 2007;3:28–31. doi: 10.4161/auto.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boya P, González-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, Métivier D, Meley D, Souquere S, Yoshimori T, et al. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol. 2005;25:1025–1040. doi: 10.1128/MCB.25.3.1025-1040.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Daido S, Kanzawa T, Yamamoto A, Takeuchi H, Kondo Y, Kondo S. Pivotal role of the cell death factor BNIP3 in ceramide-induced autophagic cell death in malignant glioma cells. Cancer Res. 2004;64:4286–4293. doi: 10.1158/0008-5472.CAN-03-3084. [DOI] [PubMed] [Google Scholar]
- 24.Kanzawa T, Kondo Y, Ito H, Kondo S, Germano I. Induction of autophagic cell death in malignant glioma cells by arsenic trioxide. Cancer Res. 2003;63:2103–2108. [PubMed] [Google Scholar]
- 25.Paglin S, Hollister T, Delohery T, Hackett N, McMahill M, Sphicas E, Domingo D, Yahalom J. A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res. 2001;61:439–444. [PubMed] [Google Scholar]
- 26.Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, Baehrecke EH, Lenardo MJ. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science. 2004;304:1500–1502. doi: 10.1126/science.1096645. [DOI] [PubMed] [Google Scholar]
- 27.Yang Y, Cong H, Han C, Yue L, Dong H, Liu J. 12-Deoxyphorbol 13-palmitate inhibits the expression of VEGF and HIF-1α in MCF-7 cells by blocking the PI3K/Akt/mTOR signaling pathway. Oncol Rep. 2015;34:1755–1760. doi: 10.3892/or.2015.4166. [DOI] [PubMed] [Google Scholar]
- 28.Ioannou M, Paraskeva E, Baxevanidou K, Simos G, Papamichali R, Papacharalambous C, Samara M, Koukoulis G. HIF-1alpha in colorectal carcinoma: Review of the literature. J BUON. 2015;20:680–689. [PubMed] [Google Scholar]
- 29.Risau W. Mechanisms of angiogenesis. Nature. 1997;386:671–674. doi: 10.1038/386671a0. [DOI] [PubMed] [Google Scholar]
- 30.Park MS, Dong SM, Kim BR, Seo SH, Kang S, Lee EJ, Lee SH, Rho SB. Thioridazine inhibits angiogenesis and tumor growth by targeting the VEGFR-2/PI3K/mTOR pathway in ovarian cancer xenografts. Oncotarget. 2014;5:4929–4934. doi: 10.18632/oncotarget.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bao LJ, Jaramillo MC, Zhang ZB, Zheng YX, Yao M, Zhang DD, Yi XF. Nrf2 induces cisplatin resistance through activation of autophagy in ovarian carcinoma. Int J Clin Exp Pathol. 2014;7:1502–1513. [PMC free article] [PubMed] [Google Scholar]
- 32.Ke XY, Lin Ng VW, Gao SJ, Tong YW, Hedrick JL, Yang YY. Co-delivery of thioridazine and doxorubicin using polymeric micelles for targeting both cancer cells and cancer stem cells. Biomaterials. 2014;35:1096–1108. doi: 10.1016/j.biomaterials.2013.10.049. [DOI] [PubMed] [Google Scholar]
- 33.Gabalec F, Beranek M, Netuka D, Masopust V, Nahlovsky J, Cesak T, Marek J, Cap J. Dopamine 2 receptor expression in various pathological types of clinically non-functioning pituitary adenomas. Pituitary. 2012;15:222–226. doi: 10.1007/s11102-011-0316-1. [DOI] [PubMed] [Google Scholar]
- 34.Gatto F, Hofland LJ. The role of somatostatin and dopamine D2 receptors in endocrine tumors. Endocr Relat Cancer. 2011;18:R233–R251. doi: 10.1530/ERC-10-0334. [DOI] [PubMed] [Google Scholar]
- 35.Gil-Ad I, Shtaif B, Levkovitz Y, Nordenberg J, Taler M, Korov I, Weizman A. Phenothiazines induce apoptosis in a B16 mouse melanoma cell line and attenuate in vivo melanoma tumor growth. Oncol Rep. 2006;15:107–112. [PubMed] [Google Scholar]
- 36.Xu HN, Huang WD, Cai Y, Ding M, Gu JF, Wei N, Sun LY, Cao X, Li HG, Zhang KJ, et al. HCCS1-armed, quadruple-regulated oncolytic adenovirus specific for liver cancer as a cancer targeting gene-viro-therapy strategy. Mol Cancer. 2011;10:133. doi: 10.1186/1476-4598-10-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sheikhpour M, Ahangari G, Sadeghizadeh M, Deezagi A. A novel report of apoptosis in human lung carcinoma cells using selective agonist of D2-like dopamine receptors: A new approach for the treatment of human non-small cell lung cancer. Int J Immunopathol Pharmacol. 2013;26:393–402. doi: 10.1177/039463201302600212. [DOI] [PubMed] [Google Scholar]