

Abstract
One of the major goals for regenerative medicine is the generation and maintenance of hematopoietic stem cells (HSCs) derived from human pluripotent stem cells (hPSCs). Until recently, efforts to differentiate hPSCs into HSCs have predominantly generated hematopoietic progenitors that lack HSC potential, and instead resemble yolk sac hematopoiesis. These resulting hematopoietic progenitors may have limited utility for in vitro disease modeling of various adult hematopoietic disorders, particularly those of the lymphoid lineages. However, we have recently described methods to generate erythro-myelo-lymphoid multilineage definitive hematopoietic progenitors from hPSCs using a stage-specific directed differentiation protocol, which we outline here. Through enzymatic dissociation of hPSCs on basement membrane matrix-coated plasticware, embryoid bodies (EBs) are formed. EBs are differentiated to mesoderm by recombinant BMP4, which is subsequently specified to the definitive hematopoietic program by the GSK3β inhibitor, CHIR99021. Alternatively, primitive hematopoiesis is specified by the PORCN inhibitor, IWP2. Hematopoiesis is further driven through the addition of recombinant VEGF and supportive hematopoietic cytokines. The resulting hematopoietic progenitors generated using this method have the potential to be used for disease and developmental modeling, in vitro.
Keywords: Developmental Biology, Issue 129, Pluripotent stem cells, human embryonic stem cells, cell culture, embryoid bodies, WNT signaling, definitive hematopoiesis, primitive hematopoiesis, hemogenic endothelium
Introduction
Human pluripotent stem cells (hPSCs) are defined as encompassing both human embryonic stem cells (hESCs) and human induced pluripotent stem cells (hiPSCs), and have the unique capability of not only undergoing self-renewal under appropriate growth conditions, but also, the capacity to differentiate into all cell types derived from the three germ layers: endoderm, mesoderm, and ectoderm1. Due to these unique abilities, hPSCs hold great promise for regenerative medicine, disease modeling, and cell-based therapies2. While multiple cell types have been successfully differentiated from hPSCs, one significant challenge is the in vitro specification of exclusively adult-like hPSC-derived hematopoietic stem cells (HSCs) and definitive hematopoietic progenitors.
One likely barrier to the development of human HSCs from hPSCs is the presence of multiple hematopoietic programs within the human embryo3. The first program which emerges, termed "primitive hematopoiesis," originates within the extraembryonic yolk sac tissue and is best characterized by its transient production of erythroblast progenitors (EryP-CFC), macrophages, and megakaryocytes. Notably, this program does not give rise to HSCs, nor does it give rise to T and B lymphoid progenitors. However, the yolk sac does transiently give rise to restricted definitive hematopoietic progenitors, such as the erythro-myeloid progenitor (EMP4,5,6,7,8) and the erythroid-deficient lymphoid-primed multipotent progenitor (LMPP9). However, neither EMPs nor LMPPs are fully multipotent, or capable of HSC-like engraftment in adult recipients. In contrast, later in development, the classically defined "definitive" hematopoietic program is specified in the aorta-gonad-mesonephros region of the embryo proper, giving rise to all adult hematopoietic lineages, including the HSC. The specification of these intra-embryonic definitive hematopoietic cells occurs in a Notch-dependent fashion, via an endothelial-to-hematopoietic transition from hemogenic endothelium (HE)3,10,11,12,13,14. Aside from reconstitution capacity, the multilineage potential and Notch-dependence of these cells can be used to distinguish these definitive hematopoietic progenitors from the EMP and the LMPP (reviewed in references3,13).
Understanding the mechanism(s) governing primitive and definitive hematopoietic specification from hPSCs is likely critical to the reproducible production of definitive hematopoietic progenitors across a variety of hPSC lines. Until recently, hPSC differentiation protocols that could separate multipotent primitive and definitive hematopoietic progenitors did not exist15,16,17,18,19,20,21,22,23,24,25. Many approaches using fetal bovine serum (FBS) and/or stromal co-culture first outlined the hematopoietic potential of hPSC differentiation, with mixtures of primitive and definitive hematopoietic potential15,16,17,19,22,23,25. Further, many serum-free hematopoietic protocols have described the signal requirements for the specification of mesoderm from hPSCs that harbors hematopoietic potential18,20,21,24. However, as these methods still gave rise to heterogeneous mixtures of both programs, their use in clinical applications and understanding developmental mechanisms may be limited.
We have recently built on these studies, having outlined the stage-specific signal requirements for ACTIVIN/NODAL and WNT signaling in primitive and definitive hematopoietic specification from hPSC-derived mesoderm18,26. The latter was particularly unique, as its use of stage-specific WNT signal manipulation allows for the specification of either exclusively primitive or exclusively definitive hematopoietic progenitors26. During mesoderm specification, the inhibition of canonical WNT signaling with the PORCN inhibitor IWP2 results in the specification of CD43+ EryP-CFC and myeloid progenitors, with no detectable lymphoid potential. In sharp contrast, stimulation of canonical WNT signaling with the GSK3β inhibitor, CHIR99021, during the same stage of differentiation resulted in the complete absence of detectable CD43+ EryP-CFC, while simultaneously leading to the specification of CD34+CD43− HE. This population possessed myeloid, HBG-expressing erythroid, and T-lymphoid potential. Subsequent analyses identified this HE as lacking the expression of CD7327,28 and CD18428, and its hematopoietic potential was NOTCH-dependent28. Further, single-cell clonal analyses demonstrated that these definitive hematopoietic lineages could be derived from multipotent single cells28. Taken together, these studies indicate that stage-specific WNT signaling manipulation can specify either pure primitive hematopoietic progenitors, or multipotent NOTCH-dependent definitive hematopoietic progenitors.
Here, we outline our differentiation strategy that yields exclusively primitive or definitive hematopoietic progenitors, via manipulation of canonical WNT signaling during mesodermal patterning, and their downstream hematopoietic lineage assays. This protocol is of great value to investigators who are interested in the production of either primitive or definitive hematopoietic progenitors from hPSCs for regenerative medicine applications.
Protocol
1. Reagents
Obtain cell lines; hESCs or hiPSCs1, mouse embryonic fibroblasts (MEFs)29, OP9-DL4 stroma30,31.
- Reagent preparation
- Prepare a 0.1% w/v solution of gelatin in PBS. Sterilize by autoclaving and store at 4 °C after aliquoting.
- Prepare the gelatin-coated plasticware. Coat 6-well dishes with 1.5 mL of 0.1% gelatin solution. Incubate for 15 min at room temperature. Immediately before use, aspirate remaining gelatin solution.
- Prepare the MEF media by making IMDM with 15% v/v FBS and 1x antibiotics. Supplement with 2 mM L-glutamine and 400 µM monothioglycerol (MTG) prior to use and store at 4 °C.
- Prepare the hPSC culture media as a solution of DMEM/F12 with 20% v/v knockout serum replacement (KOSR), 1x non-essential amino acids (NEAA), 1x antibiotics, 55 µM β-mercaptoethanol, 2 mM L-glutamine, and 10 ng/mL bFGF. Filter to sterilize with a 2 µm filter and store in the dark at 4 °C.
- Prepare the serum-free Wash media as a solution of DMEM/F12 with 5% v/v KOSR and 4 mM HCl. Filter to sterilize with a 2 µm filter, aliquot, and store in the dark at 4 °C.
- Make a 1 mg/mL DNase I solution by dissolving DNase I for 3 - 4 h into ice-cold, sterile deionized water on ice. Filter to sterilize with a 2 µm filter and store aliquots at -20 °C.
- Prepare the STOP solution with WASH media: add 10% FBS and 10 µg/mL DNase I. The STOP solution should be prepared immediately prior to use.
- Prepare the basement membrane matrix (e.g., matrigel, referred to as MAT hereon) mixture solution. NOTE: All steps preparing and using the MAT mixture should be performed on ice or at 4 °C. Thaw frozen MAT in ice at 4 °C overnight. Dilute MAT 1:1 by adding 10 mL ice-cold IMDM and chill in ice at 4 °C overnight. Dilute MAT mixture with an additional 20 mL ice-cold IMDM and chill in ice at 4 °C overnight. Aliquot into pre-chilled tubes and store at -20 °C until use. When ready for use, thaw MAT mixture overnight at 4 °C.
- Coat the MAT cell culture plates. NOTE: All steps of coating MAT plates should be performed on ice. Pre-chill 6-well and 24-well plates at -20 °C for at least 1 h. When ready to coat plates, place them on ice. Coat each well with 4x-diluted MAT, removing and re-using any excess solution. Leave on ice for 30 - 60 min, and aspirate excess MAT. Dry the MAT coated plates in a 37 °C 5% CO2 incubator for a minimum of 3 h. Use within 72 h of coating.
- Prepare the solution of 10% Bovine Serum Albumin (BSA) by dissolving 10% w/v BSA in ice-cold, distilled, deionized water at 4 °C for 3 - 4 h. Filter to sterilize with a 2 µm filter into a light-blocking bottle and store at 4 °C.
- Prepare the ascorbic acid solution. Slowly dissolve a 5 mg/mL solution of L-ascorbic acid in ice-cold, distilled, deionized water on ice for 3 - 4 h. Swirl occasionally until dissolved. Filter to sterilize with a 2 µm filter and store aliquots at -20 °C.
- Prepare the serum-free differentiation (SFD) media by making a solution of 75:25 of IMDM:Ham's F-12, then add 0.5% BSA, 1x B27, and 0.5x N2 supplements. Store at 4 °C. Add 1x L-glutamine, 50 µg/mL ascorbic acid, 400 µM MTG, and 150 µg/mL holo-transferrin immediately prior to use.
- Prepare serum-free stem cell culture media (e.g., StemPro, referred to as SP34 hereon) per the manufacturer's instructions. Store at 4 °C. Add 1x L-glutamine, 50 µg/mL ascorbic acid, 400 µM MTG, and 150 µg/mL holo-transferrin immediately prior to use.
- Prepare the 0.2% Collagenase II solution by dissolving Collagenase II to a final concentration of 0.2% w/v in 1:4 FBS:PBS solution. Dissolve in 37 °C water bath for at least 15 min. Filter to sterilize solution with a 2 µm filter and store aliquots at -20 °C.
- Prepare the Fluorescence Activated Cell Sorting (FACS) buffer as a solution of 5% FBS in IMDM immediately prior to use.
- Prepare OP9 media as a solution of α-MEM, 20% FBS, 2 mM L-glutamine, and 1x antibiotics. Filter to sterilize with a 2 µm filter and store at 4 °C.
- Obtain methylcellulose-based media (e.g., MethoCult, referred to as MeC hereon) as pre-aliquoted 3 mL quantities or as a 100 mL bottle. If using the 100 mL MeC, upon arrival, divide into 2.5 mL aliquots in 14 mL conical tubes. NOTE: All aliquots should be stored at -20 °C. MeC medium aliquots should be thawed immediately prior to use.
2. Mesoderm Differentiation of hPSCs
Grow the hPSC cell lines on MEFs to 70 - 80% confluency, in a 37 °C incubator with 5% CO2. NOTE: The hPSCs should have sharp, undifferentiated borders.
- Prepare MAT-coated plasticware 24 h before passaging the hPSCs. NOTE: The total number of MAT-coated 6-well dishes varies across hPSC lines. Normally, three 6-well dishes are sufficient per 6-well dish of confluent hPSCs on MEFs.
- Perform a trial feeder-depletion with different ratios of confluent hPSCs:MAT wells (1:4, 1:3, 1:2, etc.) before the first differentiation to determine how many MAT-coated 6-well dishes should be used for subsequent differentiations. NOTE: 24 h after plating onto MAT, the hPSCs should be 80 - 90% confluent, with cell clumps of approximately 10 - 20 cells in size.
- Aspirate the hPSC media and add 1 mL 37 °C 0.25% Trypsin-EDTA to each well for 1 min. Aspirate and add 1 mL STOP media to each well. Using a cell scraper, scrape the cells off the plate. Add 1 mL WASH buffer to each well.
- With a 2 mL serological pipet, triturate 3 - 5 times to break up large clumps of cells and transfer the cells to a 50 mL conical tube. Centrifuge the hPSCs at 330 x g for 5 min.
Resuspend the cells in a total volume of hPSC media equivalent to 2 mL per well of the MAT-coated plasticware to be used. Add 2 mL/well of hPSCs to the plasticware and incubate overnight in a 37 °C incubator with 5% CO2.
24 h later, aspirate the media and replace with 37 °C 0.05% Trypsin-EDTA for 1 min. Aspirate the Trypsin-EDTA and add 1 mL STOP media. Scrape the cells vigorously with a cell scraper. Add 1 mL WASH media to each well. With a 2 mL serological pipet, triturate the cells 1 - 2 times and transfer to a 50 mL conical tube. Centrifuge cells at 220 x g for 5 min. NOTE: The length of time of Trypsin-EDTA treatment should be determined by the investigator for each hPSC line. The Trypsin treatment should be applied for as long as possible without causing single-cell dissociation. This step should detach all the hPSCs from the MAT-coated plates, but not result in single-cell dissociation. Resultant EBs should be 6 - 10 cells, on average.
Aspirate the media. Using a 5 mL serological pipet, gently resuspend the cells in 20 mL WASH media. Centrifuge at 220 x g for 5 min.
Gently resuspend the EBs in Day 0 media (Table 1). Dispense 2 mL of differentiation media containing EBs into each well of a 6-well low-adherent cell culture dish using a 10 mL serological pipet. Place into a 37 °C 5% CO2 5% O2 (multi-gas) incubator. NOTE: Day 0 media: SFD media supplemented with 10 ng/mL BMP4 (Table 1). For every two plates of hPSCs, use one 6-well low-adherent cell culture dish.
24 h later, add 2 mL Day 1 media (Table 1). Incubate the cells overnight in a multi-gas incubator. NOTE: Day 1 media: SFD media supplemented with 10 ng/mL BMP4 and 10 ng/mL bFGF to each well (Table 1).
18 h later, gently harvest the EBs with a 5 mL serological pipet and spin at 100 x g for 5 min. Wash and combine the entire culture with 10 mL IMDM. Spin at 100 x g for 5 min. Aspirate the media. NOTE: Debris will exist in the cultures. This centrifugation step at a low speed (100 x g) will remove the debris.
- Gently resuspend the EBs in SFD media supplemented with Day 2 media (Table 1), and then dispense 2 mL per well into 6-well low-adherent cell culture plates. Incubate in a 37 °C multi-gas incubator for 30 h. NOTE: Day 2 media: 10 ng/mL BMP4, and 5 ng/mL bFGF, and the appropriate WNT agonist or antagonist (Table 1).
- Add 3 µM CHIR99021 to specify definitive hematopoietic progenitors. Alternatively, add 3 µM IWP2 and 1 ng/mL Activin A to specify primitive hematopoietic progenitors.
Continue to Section 3 for hematopoietic progenitor specification.
3. Specification of CD34+ Hematopoietic Progenitors
After 30 h, isolate the EBs with a 2 mL serological pipet at Day 3 of differentiation. Transfer to a 50 mL conical tube and centrifuge at 330 x g for 5 min. Gently resuspend and wash with 10 mL of IMDM. Centrifuge the cells again at 330 x g for 5 min.
Resuspend the EBs in Day 3 media (Table 2) and dispense 2 mL into each well of low-adherent 6-well plates. Incubate cells in a 37 °C multi-gas incubator for 72 h. NOTE: Day 3 media: SP34, supplemented with 15 ng/mL VEGF and 5 ng/mL bFGF (Table 2). Add more media per well if the pH of the media harvested on Day 3 of differentiation has significantly changed (i.e., yellow-orange media). Alternatively, use fewer EBs per well on Day 0.
After 72 h of incubation, add 2 mL of Day 6 media (Table 2) to each well. Incubate in a 37 °C multi-gas incubator for an additional 48 h. NOTE: Day 6 media: SP34 media supplemented with 15 ng/mL VEGF, 5 ng/mL bFGF, 200 ng/mL SCF, 4 IU EPO, 20 ng/mL IL-6, 10 ng/mL IL-11, and 50 ng/mL IGF-1 (Table 2). If more media was added in step 3.1, then add the equivalent amount of media in step 3.2 so that the final cytokine concentrations remain the same.
Isolate the CHIR99021-treated EBs on Day 8 of differentiation. Proceed to Section 4.
IWP2-treated EBs are harvested on Day 8 of differentiation into a 50 mL conical tube. Centrifuge at 330 x g for 5 min. Gently resuspend in Day 8 media (Table 2). Incubate for 24 h in a 37 °C 5% CO2 incubator in low adherent dishes. Proceed to Section 4. NOTE: Day 8 media: SP34 media supplemented with 100 ng/mL SCF, 2 IU EPO, 10 ng/mL IL-6, 5 ng/mL IL-11, 25 ng/mL IGF-1, 30 ng/mL TPO, 10 ng/mL Flt3-L, and 30 ng/mL IL-3 (Table 2).
4. Enzymatic Dissociation of EBs and Hematopoietic Progenitor Isolation
Isolate the EBs with a serological pipet into a 50 mL conical tube. Centrifuge at 330 x g for 5 min. Aspirate off the supernatant.
Add 2 mL of 0.25% Trypsin-EDTA to the EBs. Vortex for 5 s. Incubate in a 37 °C water bath for 8 min. Add 5 mL of STOP solution. Centrifuge at 330 x g for 5 min. Aspirate off the supernatant.
Resuspend the EBs in 5 mL of Collagenase II. Vortex for 5 s and incubate in a 37 °C water bath. After 60 min, vortex for 5 s and add 5 mL of STOP solution. Spin at 330 x g for 5 min. Aspirate off the supernatant.
In 1 mL of FACS buffer, resuspend the cells. Pass the cells through a 40 µm cell strainer. This removes any undissociated cell clumps.
- Count the cells. Aliquot 1 x 106 cells into a 14 mL conical tube. Spin the cells at 330 x g for 5 min. Resuspend the cells at a concentration of 5 x 106 cells/mL in FACS buffer. Aliquot and pool 10% of the cells in each condition for flow cytometric color controls. Incubate the cells in antibodies for 30 min on ice, in the dark. NOTE: See suggested fluorophore combinations in Table of Materials.
- For IWP2-treated cells, use anti-CD34 and anti-CD43 antibodies.
- For CHIR99021-treated cells, use anti-CD34, anti-CD43, anti-CD73, and anti-CD184 antibodies.
Rinse the cells twice using FACS buffer. Spin at 330 x g for 5 min. Resuspend the cells at a final concentration of 5 x 106 cells/mL.
Analyze or isolate cells by flow cytometry. For primitive hematopoietic progenitors, analyze the CD34 and CD43 expression (Figure 2A). For definitive hematopoietic progenitors, isolate the HE (CD34+CD43−CD73−CD184−) by FACS (Figure 2B). Collect the cells in tubes containing chilled FACS buffer.
To assess the definitive hematopoietic potential from isolated HE, continue to either Section 5 for the endothelial-to-hematopoietic transition, or Section 7 for T-lymphoid assay. For primitive hematopoietic CFC assays, continue to Section 6.

5. Endothelial-to-hematopoietic Transition
Spin the isolated CD34+CD43−CD73−CD184− cells at 330 x g for 5 min. Resuspend cells in HE media at 200,000 cells/mL (Table 2). Distribute the cells in 50 µL aliquots in a 96-well low-adherent cell culture plate. Incubate the cells overnight in a 37 °C 5% CO2 5% O2 multi-gas incubator. NOTE: HE media: SP34 media supplemented with 5 ng/mL VEGF, 5 ng/mL bFGF, 100 ng/mL SCF, 2 IU EPO, 10 ng/mL IL-6, 5 ng/mL IL-11, 25 ng/mL IGF-1, 30 ng/mL TPO, 10 ng/mL FLT3-L, 30 ng/mL IL-3, 10 ng/mL BMP4, 20 ng/mL SHH, 10 µg/mL Angiotensin II, and 100 µM Losartan potassium at 2 x 105 cells/mL (Table 2).
Prepare the 24-well MAT-coated plates (see step 1.2.7.1), as needed.
The cells will aggregate into clumps of 6 - 10 cells overnight. For each 50 µL volume of reaggregated cells, gently transfer the cells into the center of a 24-well MAT-coated plate on the next morning. Gently place plate in 37 °C multi-gas incubator for 4 - 6 h, until the cells are attached.
Add 1 mL of fresh HE media to each well, after the cells are attached. Incubate the cells in a 37 °C 5% CO2 5% O2 multi-gas incubator until the hematopoietic cells are visible (typically 5-7 days; Figure 2D). Visualize under 100X magnification using a light microscope.
- Harvest the definitive hematopoietic progenitors by gently removing the HE media from cells. Pass this media through a 40 µm cell strainer and collect. To the adherent cells in the well, add 0.5 mL 0.25% Trypsin-EDTA to cells and incubate at 37 °C for 5 min. Add 0.5 mL STOP solution to each well. Pass through cells through the same 40 µm cell strainer to pool all adherent and non-adherent cells together. Spin at 330 x g for 5 min.
- To assess the CFC potential of this bulk culture, proceed to Section 6.
Wash the cells twice in FACS buffer. Spin at 330 x g for 5 min.
Resuspend the cells in FACS buffer at 5 x 106 cells/mL. Stain the cells with anti-CD45 and anti-CD34 antibodies for 30 min on ice in the dark (suggested fluorophores are in the Table of Materials).
Wash the cells twice in FACS buffer. Spin at 330 x g for 5 min.
Isolate the CD34+CD45+ cells by FACS (Figure 2E). Sort the cells into chilled FACS buffer.
Assess the definitive CD34+CD45+ hematopoietic progenitors as needed (CFC assay in Section 6, lymphoid potential in Section 7, or another investigator-initiated assay).
6. CFC Assay
Thaw aliquots of MeC for each sample used for the CFC assay.
Add the cells to be assayed (steps 4.5, 4.7, 5.5, or 5.9) in MeC. Vortex thoroughly and let the MeC cultures stand for 5 - 10 min until air bubbles dissipate, as per the manufacturer's instructions. Using a 16 ½ gauge needle on a 3 mL syringe, carefully remove 2 mL of the MeC. NOTE: Typical plating densities range from 1,000 - 20,000 cells/mL, depending on the anticipated progenitor frequency.
Carefully, dispense 1 mL MeC cell mixture in each of two 35 mm tissue culture dishes. Evenly distribute the MeC in the 35 mm dishes by gently tapping and rotating the dish. Place each of the above dishes into a 15 cm tissue culture dish filled with water. Incubate in a 37 °C 5% CO2 incubator.
Visualize the MeC cultures using a light microscope using 40X magnification. Quantify the various CFCs obtained. Analyze the Primitive CFC assays 8 - 10 days after plating (Figure 2C). Analyze the Definitive CFC assays 14 days after plating. Representative CFC assay colony morphologies can be seen in Figure 2F.
7. T Cell Assay to Establish Definitive Hematopoietic Potential
- Grow the OP9-DL4 cells until confluent in a 100 mm tissue culture dish30 31 32.
- Thaw the OP9-DL4 cells 72 - 96 h prior to plating the T cell assays. Resuspend the OP9-DL4 cells in 10 mL OP9 media and dispense into a 10 cm plate. Grow in a 37 °C 5% CO2 incubator until 70 - 90% confluent.
- To harvest the OP9-DL4 cells from a 100 mm dish, remove the media and add 5 mL 0.25% Trypsin-EDTA for 5 min. Stop the trypsin activity with 5 mL OP9 media. Spin the cells at 330 x g for 5 min. Resuspend the cells in 1 mL OP9 media and count the cells. NOTE: One 10 cm plate of confluent OP9-DL4 cells will yield approximately 10 x 106 OP9-DL4 cells.
- 48 h prior to the T cell assay, plate 2 x 106 cells in 12 mL OP9 media in a 24-well dish (0.5 mL/well). Grow in a 37 °C 5% CO2 incubator.
Add 2,000 - 10,000 cells to be assayed (as obtained in steps 4.5, 4.7, 5.5, or 5.9) to 1 well of OP9-DL4 cells in OP9 media supplemented with: 30 ng/mL SCF (first 6 days only), 5 ng/mL IL-7, and 5 ng/mL FLT3-L. Incubate in a 37 °C 5% CO2 incubator. No media changes occur at this step.
Every 4 - 5 days, passage the T cell progenitors onto fresh OP9-DL4 stromal cells in a 6-well dish. Triturate the cells with a 1 mL pipet and pass through a 40 µm filter to remove clumps. Spin at 330 x g for 5 min. Aspirate off the media. Resuspend the cells in 2 mL of fresh OP9 media supplemented with 5 ng/mL IL-7 and 5 ng/mL Flt3-L, and place on fresh OP9-DL4 cells. NOTE: 48 h prior to passaging, plate 2 x 106 fresh OP9-DL4 cells in 12 mL OP9 media, and distribute 2 mL/well into 6-well dishes.
After at least 21 days of co-culture, triturate the cells vigorously and pass through a 40 µm filter to remove cell debris. Spin at 330 x g for 5 min and aspirate the supernatant.
Wash the cells twice in cold FACS buffer. Spin at 330 x g for 5 min.
Resuspend the cells in FACS buffer at 5 x 106 cells/mL. Stain cells with anti-CD45, anti-CD56, anti-CD4, and anti-CD8 antibodies for 30 min on ice in the dark (suggested fluorophore combinations are in Table of Materials). Apply a live/dead cell stain (DAPI, etc.) to remove debris from the analysis.
Wash the cells twice in FACS buffer. Spin at 330 x g for 5 min.
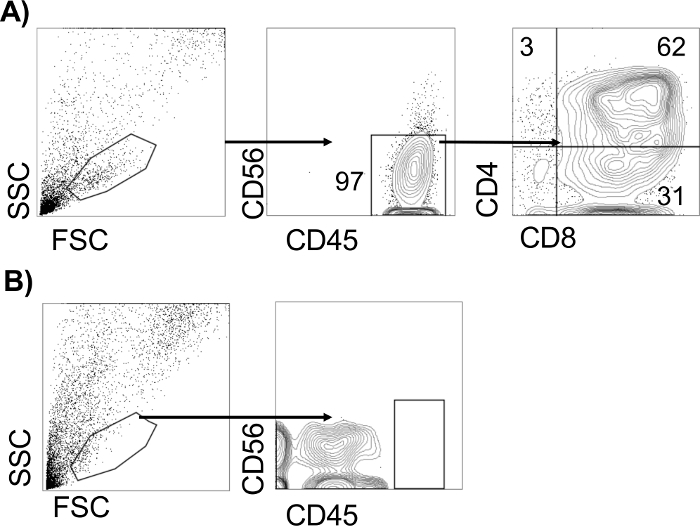
Analyze the cells using a flow cytometer to assess the presence of T-lymphocyte progenitors (Figure 3).
Representative Results
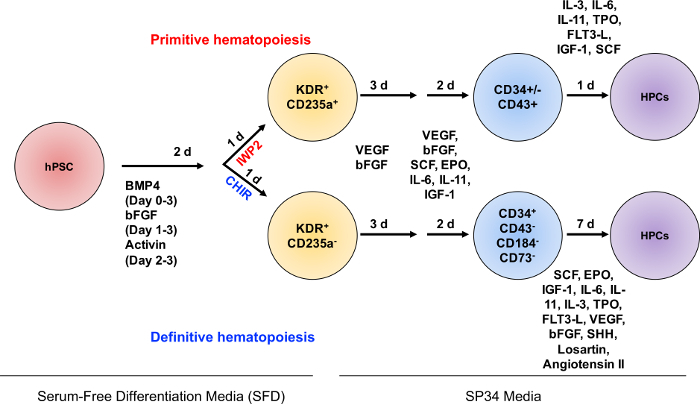
A schematic depicting the induction of primitive and definitive hematopoietic progenitors from hPSCs is illustrated in Figure 1. Mesoderm patterning by canonical WNT signaling occurs during days 2 - 3 of differentiation, followed by hematopoietic progenitor specification.
Representative flow cytometric analysis and colony forming methylcellulose assays of hPSC-derived hematopoietic differentiation cultures are shown in Figure 2. IWP2-treated differentiation cultures will yield a distinct CD34+CD43− and CD34low/−CD43+ population, while CHIR-treated differentiation cultures will have <1% CD43+ cells on Day 8 of differentiation (Figure 2A,B)26. Primitive hematopoietic progenitors derived under IWP2 conditions isolated on Day 9 will give rise to predominantly primitive erythroid (EryP-CFC) and myeloid progenitors in the methylcellulose assays, while CHIR-treated cultures will be largely devoid of CFC potential at this stage (Figure 2C,F). Instead, the CD34+CD43- population derived from the CHIR99021 treatment is a heterogeneous population, containing endothelial progenitors, as well as CD34+CD43-CD73-CD184- HE (Figure 2B), which when isolated by FACS, will initially form a monolayer of endothelial-like cells (Figure 2D, left panel), that then undergoes an endothelial-to-hematopoietic transition, resulting in round, refractile non-adherent hematopoietic cells (Figure 2D, right panel). These hematopoietic cells can be assessed by flow cytometry for the expression of CD34 and CD45 (Figure 2E). Hematopoietic progenitors isolated from the EHT assays can be used in methylcellulose assays and give rise to large burst-like erythroid colony-forming units (BFU-E), small erythroid colony-forming units (CFU-E), and myeloid progenitors (CFU-M; Figure 2F).
Figure 3 depicts representative flow cytometric analyses of the T-lymphocyte assays potential from CHIR99021-derived CD34+CD43−CD73−CD184− hematopoietic progenitors (Figure 3A) and IWP2-derived (Figure 3B) CD34+CD43− hematopoietic progenitors, following 21 days of OP9-DL4 co-culture. The presence of a CD45+CD56−CD4+CD8+ population in CHIR-derived progenitors is indicative of definitive hematopoietic potential within the input CD34+ population. CD34+ and CD43+ progenitors isolated from IWP2-derived differentiations do not give rise to T-lymphocytes in this assay, under these differentiation conditions18,26.
Figure 1: Schematic diagram of protocol for the specification of definitive and primitive hematopoietic progenitors. Depiction of the major experimental steps and timelines of the differentiation from EBs to definitive and primitive hematopoietic progenitors. EBs are generated on Day 0 from hPSCs on MAT-coated plasticware. EBs are grown in SFD media containing BMP4 in a multi-gas incubator. On Day 1 of differentiation, cultures are fed with media supplemented with BMP4 and bFGF. On Day 2, a media change occurs with WNT manipulation through the addition of CHIR99021 (CHIR) for definitive hematopoietic progenitors and IWP2 for primitive hematopoietic progenitors. On Day 3, media is changed to SP34, supplemented with VEGF and bFGF. At Day 6, cultures are fed with media supplemented with hematopoietic cytokines. On Day 8 of the primitive hematopoietic specification, the media is changed and cells are moved to a 5% CO2 incubator for 24 h, followed by the hematopoietic progenitor cell (HPC) assay. On Day 8 of definitive hematopoietic specification, CD34+CD43-CD73-CD184- cells are isolated by FACS and assayed for definitive hemogenic endothelial potential, or T-lymphoid potential (not depicted). Please click here to view a larger version of this figure.
Figure 2: Analysis of differentiation cultures and generation of primitive hematopoietic progenitors. (A) Representative flow cytometric analyses of primitive hematopoietic progenitors from IWP2-treated EBs on Day 8. (B) Representative flow cytometric analyses from CHIR99021-treated EBs on Day 8. Hemogenic endothelium (HE) can be identified and isolated as a CD34+CD43-CD73-CD184- population. (C) Colony-forming potential of day 9 hematopoietic progenitors from IWP2- and CHIR99021- (CHIR) treated differentiation cultures. n = 3, error bars represent SD of the mean, asterisks indicate statistical significance, using student's t-test *** p <0.0001. (D) Representative photographs of isolated HE cells after 4 days (left) or 7+ days (right) of growth on MAT-coated plasticware. Original magnification, 100X. Scale bars = 100 µm. (E) Representative flow cytometry of CD34 and CD45 expression from definitive hematopoietic progenitors after 9 days of culture. (F) Representative photograph showing morphology of EryP-CFC (left), CFU-M (middle), BFU-E and CFU-E (right). Original magnification, 40X. Scale bars = 100 µm. Arrows indicate named colonies. Please click here to view a larger version of this figure.
Figure 3: Analysis of definitive hematopoietic potential. Representative flow cytometric analyses of T lymphocyte potential of CHIR-derived CD34+CD43−CD73−CD184− hematopoietic progenitors (A) or IWP2-derived (B) CD34+CD43− hematopoietic progenitors, 28 days after co-culture with OP9-DL4 cells. The T-lymphocyte potential from a sample is considered positive if there are more than 100 CD45+ events detected. Please click here to view a larger version of this figure.
| SFD Media | Day 0 | Day 1 | Day 2 Definitive | Day 2 Primitive |
| BMP4 | 10 ng/mL | 10 ng/mL | 10 ng/mL | 10 ng/mL |
| bFGF | – | 10 ng/mL | 5 ng/mL | 5 ng/mL |
| Activin A | – | – | – | 1 ng/mL |
| CHIR99021 | – | – | 3 µM | – |
| IWP2 | – | – | – | 3 µM |
Table 1: SFD-based media for Days 0 - 2.
| SP34 Media | Day 3 | Day 6 | Day 8 | HE |
| VEGF | 15 ng/mL | 15 ng/mL | – | 5 ng/mL |
| bFGF | 5 ng/mL | 5 ng/mL | – | 5 ng/mL |
| SCF | – | 200 ng/mL | 100 ng/mL | 100 ng/mL |
| EPO | – | 4 IU | 2 IU | 2 IU |
| IL-6 | – | 20 ng/mL | 10 ng/mL | 10 ng/mL |
| IL-11 | – | 10 ng/mL | 5 ng/mL | 5 ng/mL |
| IGF-1 | – | 50 ng/mL | 25 ng/mL | 25 ng/mL |
| TPO | – | – | 30 ng/mL | 30 ng/mL |
| Flt-3L | – | – | 10 ng/mL | 10 ng/mL |
| IL-3 | – | – | 30 ng/mL | 30 ng/mL |
| BMP4 | – | – | – | 10 ng/mL |
| SHH | – | – | – | 20 ng/mL |
| Angiotensin II | – | – | – | 10 µg/mL |
| Losartan potassium | – | – | – | 100 µM |
Table 2: SP34-based media for Days 3 - 8 and HE.
Discussion
This protocol describes a rapid, serum-free, stroma-free method for the differentiation of either primitive or definitive hematopoietic progenitors. Mesodermal specification of either primitive or definitive hematopoietic progenitors can be reliably achieved using our protocol, which uniquely exploits small molecule inhibitors of canonical WNT signaling. Stage-specific WNT activation by the GSK3β inhibitor CHIR9902133 gives rise to definitive hematopoietic mesoderm, whereas WNT inhibition by the PORCN inhibitor IWP234 specifies primitive hematopoietic mesoderm26. Of note, this method does not robustly give rise to an engraftable HSC-like population (not shown). However, the definitive hematopoietic progenitors generated with this approach are amenable to transgene-induced engraftment potential35, highlighting their potential in future translational applications.
A crucial determinant to successful hPSC differentiation is the initial size of the EBs that are formed from hPSCs. Ideally, the EBs should be around 6 - 10 cells in size. The differentiation cultures can aberrantly specify a mixture of both programs if the EBs are too large, possibly due to improper signal activation within the EB. Differentiation cultures should be visually inspected for optimal EB size within the first 24 h of differentiation. Alternatively, if the hPSCs are completely dissociated to single cells, the hPSCs instead undergo anoikis cell death36, resulting in poor hematopoietic differentiation.
A successful differentiation will give rise to exclusively primitive hematopoietic progenitors when IWP2 is used during mesodermal specification on Days 2 and 3 of this differentiation protocol. On Day 8 of differentiation, the IWP2-derived culture should be comprised of distinct CD34+CD43−, CD34mid/lowCD43+, and CD34−CD43+ populations (Figure 2A). The CD34mid/lowCD43+ population gives rise to primitive erythroid and myeloid progenitors; while the CD34−CD43+ population primarily gives rise to erythroid progenitors with limited myeloid potential18. The primitive hematopoietic potential can be confirmed by the methylcellulose assay for the presence of EryP-CFC (Section 6), which will predominately express the embryonic globin HBE in comparison to fetal HBG26,28,37. Similarly, the presence of CD43+ hematopoietic progenitors at this stage can be reliably used to indicate the presence of primitive hematopoietic progenitors18,23,26. It should be noted that CD43 expression is not exclusive to progenitors of the primitive hematopoietic program18,23,26, and cannot be used as the sole metric to assess primitive hematopoietic potential, as the immunophenotype and NOTCH-dependence of the human EMP remains uncharacterized (reviewed in3).
When CHIR99021 is used during mesodermal patterning during days 2 and 3 of differentiation, a population of exclusively definitive hematopoietic progenitors will be specified. On Day 8 of the differentiation protocol, CHIR99021-derived cultures should be comprised of a distinctive CD34+CD43− population, with little to no CD43 expression26. The CD34+CD43− population is a heterogeneous population of endothelial cells with hematopoietic, venous, and arterial potential that can be demarcated based on CD73 and CD184 expression, with HE cells lacking the expression of both27,28. When definitive HE is isolated after undergoing a NOTCH-dependent endothelial-to-hematopoietic transition (Section 5)28 it will yield CD34+CD45+ hematopoietic progenitors that will give rise to BFU-E and myeloid cells in methylcellulose, but not EryP-CFC26,28 (Figure 2). In addition, BFU-E colonies can be isolated and assessed for globin analysis, which will predominately express the fetal globin HBG in comparison to embryonic HBE (not shown)26,28,37. In contrast, the lymphoid potential can be directly assessed from isolated HE, as the NOTCH-dependent endothelial-to-hematopoietic transition occurs on the OP9-DL4 stroma18,26,28. The definitive HE specified with this method contains erythro-myelo-lymphoid progenitors at a clonal level28, which functionally distinguishes it from our current understanding of EMP or LMPP progenitors3. As such, the presence of T-lymphoid and HBG-expressing erythroid potential, coupled with an absence of EryP-CFC potential, can be used to reliably indicate the definitive hematopoietic specification from hPSCs. Of note, solely assessing for progenitors that can give rise to HBG-expressing erythroblasts may not accurately determine the definitive potential, as, similar to that described above, the human EMP, and its signal requirements, has not been characterized to-date (reviewed in reference3).
This simple differentiation strategy is very powerful as it allows for the generation of exclusively primitive or exclusively definitive hematopoietic progenitors, enabling disease modeling comparative studies of the two programs from patient-derived iPSCs or gene-modified hPSCs. Similarly, human developmental processes can be interrogated, such as discovering what additional signal pathway(s) are required to confer self-renewal capacity, and hence HSC-like potential, from the definitive hematopoietic progenitors.
In summary, WNT manipulation during mesodermal patterning in hPSCs specifies primitive CD43+ or definitive CD34+CD45+ hematopoietic progenitors, which can be isolated and used to study hPSC-derived hematopoiesis.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
This work has been supported by the Department of Internal Medicine, Division of Hematology, Washington University School of Medicine. CD was supported by T32HL007088 from the National Heart, Lung, and Blood Institute. CMS was supported by an American Society of Hematology Scholar Award.
References
- Thomson JA, et al. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282(5391):1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- Murry CE, Keller G. Differentiation of embryonic stem cells to clinically relevant populations: lessons from embryonic development. Cell. 2008;132(4):661–680. doi: 10.1016/j.cell.2008.02.008. [DOI] [PubMed] [Google Scholar]
- Ditadi A, Sturgeon CM, Keller G. A view of human haematopoietic development from the Petri dish. Nat Rev Mol Cell Biol. 2017;18(1):56–67. doi: 10.1038/nrm.2016.127. [DOI] [PubMed] [Google Scholar]
- Chen MJ, et al. Erythroid/myeloid progenitors and hematopoietic stem cells originate from distinct populations of endothelial cells. Cell Stem Cell. 2011;9(6):541–552. doi: 10.1016/j.stem.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath KE, et al. Distinct Sources of Hematopoietic Progenitors Emerge before HSCs and Provide Functional Blood Cells in the Mammalian Embryo. Cell Rep. 2015;11(12):1892–1904. doi: 10.1016/j.celrep.2015.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath KE, et al. A transient definitive erythroid lineage with unique regulation of the beta-globin locus in the mammalian embryo. Blood. 2011;117(17):4600–4608. doi: 10.1182/blood-2010-12-325357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palis J, et al. Spatial and temporal emergence of high proliferative potential hematopoietic precursors during murine embryogenesis. Proc Natl Acad Sci U S A. 2001;98(8):4528–4533. doi: 10.1073/pnas.071002398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palis J, Robertson S, Kennedy M, Wall C, Keller G. Development of erythroid and myeloid progenitors in the yolk sac and embryo proper of the mouse. Development. 1999;126(22):5073–5084. doi: 10.1242/dev.126.22.5073. [DOI] [PubMed] [Google Scholar]
- Boiers C, et al. Lymphomyeloid contribution of an immune-restricted progenitor emerging prior to definitive hematopoietic stem cells. Cell Stem Cell. 2013;13(5):535–548. doi: 10.1016/j.stem.2013.08.012. [DOI] [PubMed] [Google Scholar]
- Bertrand JY, et al. Haematopoietic stem cells derive directly from aortic endothelium during development. Nature. 2010;464(7285):108–111. doi: 10.1038/nature08738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadland BK, et al. A requirement for Notch1 distinguishes 2 phases of definitive hematopoiesis during development. Blood. 2004;104(10):3097–3105. doi: 10.1182/blood-2004-03-1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumano K, et al. Notch1 but not Notch2 is essential for generating hematopoietic stem cells from endothelial cells. Immunity. 2003;18(5):699–711. doi: 10.1016/s1074-7613(03)00117-1. [DOI] [PubMed] [Google Scholar]
- Medvinsky A, Rybtsov S, Taoudi S. Embryonic origin of the adult hematopoietic system: advances and questions. Development. 2011;138(6):1017–1031. doi: 10.1242/dev.040998. [DOI] [PubMed] [Google Scholar]
- Robert-Moreno A, Espinosa L, de la Pompa JL, Bigas A. RBPjkappa-dependent Notch function regulates Gata2 and is essential for the formation of intra-embryonic hematopoietic cells. Development. 2005;132(5):1117–1126. doi: 10.1242/dev.01660. [DOI] [PubMed] [Google Scholar]
- Chadwick K, et al. Cytokines and BMP-4 promote hematopoietic differentiation of human embryonic stem cells. Blood. 2003;102(3):906–915. doi: 10.1182/blood-2003-03-0832. [DOI] [PubMed] [Google Scholar]
- Davis RP, et al. Targeting a GFP reporter gene to the MIXL1 locus of human embryonic stem cells identifies human primitive streak-like cells and enables isolation of primitive hematopoietic precursors. Blood. 2008;111(4):1876–1884. doi: 10.1182/blood-2007-06-093609. [DOI] [PubMed] [Google Scholar]
- Kaufman DS, Hanson ET, Lewis RL, Auerbach R, Thomson JA. Hematopoietic colony-forming cells derived from human embryonic stem cells. Proc Natl Acad Sci U S A. 2001;98(19):10716–10721. doi: 10.1073/pnas.191362598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy M, et al. T lymphocyte potential marks the emergence of definitive hematopoietic progenitors in human pluripotent stem cell differentiation cultures. Cell Rep. 2012;2(6):1722–1735. doi: 10.1016/j.celrep.2012.11.003. [DOI] [PubMed] [Google Scholar]
- Ledran MH, et al. Efficient hematopoietic differentiation of human embryonic stem cells on stromal cells derived from hematopoietic niches. Cell Stem Cell. 2008;3(1):85–98. doi: 10.1016/j.stem.2008.06.001. [DOI] [PubMed] [Google Scholar]
- Ng ES, et al. The primitive streak gene Mixl1 is required for efficient haematopoiesis and BMP4-induced ventral mesoderm patterning in differentiating ES cells. Development. 2005;132(5):873–884. doi: 10.1242/dev.01657. [DOI] [PubMed] [Google Scholar]
- Pick M, Azzola L, Mossman A, Stanley EG, Elefanty AG. Differentiation of human embryonic stem cells in serum-free medium reveals distinct roles for bone morphogenetic protein 4, vascular endothelial growth factor, stem cell factor, and fibroblast growth factor 2 in hematopoiesis. Stem Cells. 2007;25(9):2206–2214. doi: 10.1634/stemcells.2006-0713. [DOI] [PubMed] [Google Scholar]
- Vodyanik MA, Bork JA, Thomson JA, Slukvin II. Human embryonic stem cell-derived CD34+ cells: efficient production in the coculture with OP9 stromal cells and analysis of lymphohematopoietic potential. Blood. 2005;105(2):617–626. doi: 10.1182/blood-2004-04-1649. [DOI] [PubMed] [Google Scholar]
- Vodyanik MA, Thomson JA, Slukvin II. Leukosialin (CD43) defines hematopoietic progenitors in human embryonic stem cell differentiation cultures. Blood. 2006;108(6):2095–2105. doi: 10.1182/blood-2006-02-003327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C, et al. Retinoic acid enhances the generation of hematopoietic progenitors from human embryonic stem cell-derived hemato-vascular precursors. Blood. 2010;116(23):4786–4794. doi: 10.1182/blood-2010-01-263335. [DOI] [PubMed] [Google Scholar]
- Zambidis ET, Peault B, Park TS, Bunz F, Civin CI. Hematopoietic differentiation of human embryonic stem cells progresses through sequential hematoendothelial, primitive, and definitive stages resembling human yolk sac development. Blood. 2005;106(3):860–870. doi: 10.1182/blood-2004-11-4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturgeon CM, Ditadi A, Awong G, Kennedy M, Keller G. Wnt Signaling Controls the Specification of Definitive and Primitive Hematopoiesis From Human Pluripotent Stem Cells. Nat Biotechnol. 2014;32(6):554–561. doi: 10.1038/nbt.2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi KD, et al. Identification of the hemogenic endothelial progenitor and its direct precursor in human pluripotent stem cell differentiation cultures. Cell Rep. 2012;2(3):553–567. doi: 10.1016/j.celrep.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ditadi A, et al. Human Definitive Haemogenic Endothelium and Arterial Vascular Endothelium Represent Distinct Lineages. Nat Cell Biol. 2015;17(5):580–591. doi: 10.1038/ncb3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conner DA. Mouse embryo fibroblast (MEF) feeder cell preparation. Curr Protoc Mol Biol. 2001;Chapter 23 doi: 10.1002/0471142727.mb2302s51. Unit 23.2. [DOI] [PubMed] [Google Scholar]
- La Motte-Mohs RN, Herer E, Zuniga-Pflucker JC. Induction of T-cell development from human cord blood hematopoietic stem cells by Delta-like 1 in vitro. Blood. 2005;105(4):1431–1439. doi: 10.1182/blood-2004-04-1293. [DOI] [PubMed] [Google Scholar]
- Schmitt TM, et al. Induction of T cell development and establishment of T cell competence from embryonic stem cells differentiated in vitro. Nat Immunol. 2004;5(4):410–417. doi: 10.1038/ni1055. [DOI] [PubMed] [Google Scholar]
- Holmes R, Zuniga-Pflucker JC. The OP9-DL1 system: generation of T-lymphocytes from embryonic or hematopoietic stem cells in vitro. Cold Spring Harb Protoc. 2009;2009(2) doi: 10.1101/pdb.prot5156. [DOI] [PubMed] [Google Scholar]
- Polychronopoulos P, et al. Structural basis for the synthesis of indirubins as potent and selective inhibitors of glycogen synthase kinase-3 and cyclin-dependent kinases. J Med Chem. 2004;47(4):935–946. doi: 10.1021/jm031016d. [DOI] [PubMed] [Google Scholar]
- Chen B, et al. Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat Chem Biol. 2009;5(2):100–107. doi: 10.1038/nchembio.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimura R, et al. Haematopoietic stem and progenitor cells from human pluripotent stem cells. Nature. 2017. [DOI] [PMC free article] [PubMed]
- Ohgushi M, et al. Molecular pathway and cell state responsible for dissociation-induced apoptosis in human pluripotent stem cells. Cell Stem Cell. 2010;7(2):225–239. doi: 10.1016/j.stem.2010.06.018. [DOI] [PubMed] [Google Scholar]
- Peschle C, et al. Embryonic----Fetal Hb switch in humans: studies on erythroid bursts generated by embryonic progenitors from yolk sac and liver. Proc Natl Acad Sci U S A. 1984;81(8):2416–2420. doi: 10.1073/pnas.81.8.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]