

Abstract
We describe a robust method to direct the differentiation of pluripotent stem cells into retinal pigment epithelial cells (RPE). The purpose of providing a detailed and thorough protocol is to clearly demonstrate each step and to make this readily available to researchers in the field. This protocol results in a homogenous layer of RPE with minimal or no manual dissection needed. The method presented here has been shown to be effective for induced pluripotent stem cells (iPSC) and human embryonic stem cells. Additionally, we describe methods for cryopreservation of intermediate cell banks that allow long-term storage. RPE generated using this protocol might be useful for iPSC disease-in-a-dish modeling or clinical application.
Keywords: Developmental Biology, Issue 128, retinal pigment epithelial cells (RPE), retinal pigment epithelium, induced pluripotent stem cells, human embryonic stem cells, differentiation, retinal development, cell culture
Introduction
The retinal pigment epithelium is a monolayer of pigmented cells that provides crucial support for photoreceptors. Retinal pigment epithelial cells (RPE) have numerous functions in vision, including light absorption, nutrient and ion transport, retinoid cycling, photoreceptor outer segment phagocytosis, and growth factor secretion1. There are a variety of retinal dystrophies that affect the function of RPE and result in a loss of vision, including age-related macular degeneration and retinitis pigmentosa. Generation of RPE from pluripotent stem cells may facilitate research to understand these eye diseases, and can provide an unlimited source of RPE for cell therapies2. In fact, multiple clinical trials are underway using RPE derived from pluripotent stem cells3.
This differentiation protocol was originally described by Buchholz4 and was based on the previously published method from Clegg5. The procedure mimics the normal in vivo developmental process to direct undifferentiated pluripotent stem cells towards an RPE fate via manipulation of the insulin growth factors (IGF), basic fibroblast growth factor (FGF-2; FGF-basic), transforming growth factor beta (TGF-β), and WNT pathways4,5. The protocol was significantly improved by addition of a WNT pathway agonist late in the protocol, which yielded 97.77% ± 0.1% pre-melanosome protein (PMEL) positive cells, and has been adapted to xeno-free conditions6,7. The resulting RPE have been shown to express RPE markers at the transcript and protein levels, to secrete known RPE growth factors with appropriate polarity, and carry out phagocytosis of photoreceptor outer segments8. This protocol is more rapid and reliable than "spontaneous" protocols of differentiation that involve simple removal of basic fibroblast growth factor8. Furthermore, RNA sequencing data show that RPE obtained using this protocol are very similar to those obtained using the more common spontaneous approach8. The 14-day method generates RPE that fit the "5 P's" mentioned by Mazzoni9 (pigmented, polarized, phagocytic, post-mitotic, polygonal)9. While this procedure has proven to be reproducible in multiple labs, we wish to acknowledge several additional directed differentiation methods that have been published in recent years10,11,12,13.
Protocol
1. Preparation of Reagents for Day 0 to Day 14 of the Protocol
- Prepare the following medium components:
- Make 100 mL of retinal differentiation medium (RDM) by adding 1 mL of 100x N2 supplement, 2 mL of 50x B27 supplement, and 1 mL of 100x non-essential amino acid (NEAA) to 96 mL of Dulbecco's modified essential medium/nutrient mixture F12 9 (DMEM/F12).
- Make 10 mL of 1 M nicotinamide (NIC) by dissolving 1.221 g of NIC in 8 mL of sterile water, vortexing, and bringing the volume to 10 mL with sterile water. Sterile filter the solution.
- Prepare the following growth factors and small molecules:
- Reconstitute recombinant mouse noggin, human dickkopf WNT signaling pathway inhibitor 1 (DKK-1), and IGF-1 to 100 µg/mL each in 0.1% bovine serum albumin (BSA) in phosphate-buffered solution (PBS). Aliquot as needed and store at -20 °C for up to 3 months.
- Reconstitute FGF-basic to 10 µg/mL and recombinant human/mouse/rat Activin A to 100 µg/mL each in 0.1% BSA in PBS. Aliquot as needed and store at -80 °C for up to 1 year.
- Reconstitute SU 5402 (FGF receptor-specific tyrosine kinase inhibitor) and CHIR99021 (glycogen synthase kinase 3, GSK-3β, inhibitor) to 10 mM each in dimethyl sulfoxide (DMSO). Aliquot and store at -20 °C for up to 1 year or 6 months, respectively.
Obtain the following for day 0 and/or day 14: 1x ethylenediaminetetraacetic acid (EDTA) solution (0.2 g EDTA per 1 L of PBS), 1X PBS -/- (PBS without calcium or magnesium, pH 7.4), 1x trypsin-like dissociation enzyme (TDE), DPBS (Dulbecco's PBS), RPE supporting medium (RSM), and Y-27632 dihydrochloride (use at 10 µM).
2. Day 0: Day of Pluripotent Stem Cell Passage for Differentiation
Grow stem cell colonies in feeder-free, serum-free conditions to approximately 80% confluence before passaging. NOTE: See discussion for details on optimizing this step.
Coat a 12-well plate with extracellular matrix-based hydrogel (ECMH) as per manufacturer recommendations. Allow to set for 1 h at room temperature or overnight at 4 °C.
Aliquot the volume of RDM and PBS -/- needed for day 0 and warm in a water bath to 37 °C before adding growth factors . Bring EDTA to room temperature.
Add the growth factors necessary for day 0 to the warmed RDM with 10 mM NIC, 50 ng/mL noggin, 10 ng/mL DKK-1, and 10 ng/mL IGF-1. From the stocks described in step 1.2, add 100 µL of NIC, 5 µl of noggin, 1 µL of DKK-1, and 1 µL of IGF-1 to 10 mL of RDM.
Pick to remove all differentiated colonies based on morphology from the stem cells that will be passaged for differentiation. Use a P10 pipet tip to manually remove the differentiated cells. NOTE: Fibroblastic cells between colonies as well as the opaque cells within colonies indicate differentiated cells to be removed. See discussion for details about differentiated cells.
- Passage a single well of a 6-well plate into 4 wells of a 12-well plate (1:4). NOTE: See discussion for details on passaging stem cells at this stage.
- Aspirate the stem cell medium from the stem cells and wash the wells once with 2 mL of pre-warmed PBS -/-.
- Aspirate PBS -/- and rinse each well three times with 1 mL of EDTA per well of a 6-well plate.
- Gently tilt the plate and aspirate the EDTA. Do not agitate the plate in any way to avoid prematurely lifting the cells.
- After the third wash, add 1 mL of EDTA and incubate at room temperature in the hood for 3-5 min. Do not disturb the plate during this incubation.
- Aspirate the EDTA and add 1 mL of RDM per well that will be seeded with 0.5 mL of extra medium. For example, wash 1 well of a 6-well plate with 4.5 mL of RDM to plate on 4 wells of a 12-well plate.
- Use a cell scraper to gently detach the cells. Collect all cells in a conical tube and triturate the cells in RDM by pipetting up and down 5 times. Dissociate large clumps of cells, but do not triturate to single cell suspension. Distribute the cells evenly in the pipet. Complete this step quickly to prevent reattachment to the plate.
- Seed cells on the ECM-coated 12-well plates (1 mL of cell suspension per well).
- Tilt the plate back and forth to distribute the cells evenly throughout the wells. Gently place in a cell culture incubator at 37 °C and 5% CO2 until the next medium change.
- Note the exact time. Change medium at the same time each day.
3. Day 1 to 14: Addition of Growth Factors
Day 1: Change the medium on all wells (1 mL per well) using RDM with the growth factor composition for day 0 (see step 2.4).
Day 2: Change the medium using RDM (1 mL per well) with 10 mM NIC, 5 ng/mL FGF-basic, 10 ng/mL noggin, 10 ng/mL DKK-1, and 10 ng/mL IGF-1. From the stocks described in step 1.2, add 100 µL of NIC, 5 µL of FGF-basic, 1 µL of noggin, 1 µL of DKK1, and 1 µL of IGF1 to 10 mL of RDM.
Day 4: Change the medium using RDM (1 mL per well) with 100 ng/mL activin A, 10 ng/mL DKK-1, and 10 ng/mL IGF-1. From the stocks described in step 1.2, add 10 µL of activin A, 1 µL of DKK1, and 1 µL of IGF-1 to 10 mL of RDM. NOTE: Observe that cells are confluent at this stage.
Day 6: Change the medium using RDM (1 mL per well) with 100 ng/mL activin A and 10 µM SU 5402. From the stocks described in step 1.2, add 10 µL of activin A and 10 µL of SU 5402 to 10 mL of RDM.
Days 8, 10, and 12: Change the medium using RDM (1 mL per well) with 100 ng/mL activin A, 10 µM SU 5402, and 3 µM CHIR99021.From the stocks described in step 1.2, add 10 µL of activin A, 10 µL of SU 5402, and 3 µL of CHIR99021 to 10 mL of RDM.
4. Day of Enrichment to Passage 0 of RPE
Coat a 6-well plate with growth factor reduced ECMH as per manufacturer recommendations. Allow to set for 1 h at room temperature or overnight at 4 °C.
Aliquot the volume of DPBS needed and 1 mL of RDM per well of enrichment and warm in a water bath to 37 °C. Bring the TDE to room temperature and warm necessary volume of RSM, antimicrobial reagent, and Y-27632 to 37 °C.
Add antimicrobial reagent and Y-27632 obtaining 0.5x and 10 µM compositions respectively to RSM. Use this medium for the first 4-7 days to improve attachment.
Aspirate spent medium from all wells and add 1 mL per well of pre-warmed RDM (no growth factors required).
Using a dissecting microscope, manually dissect and scrape away all non-RPE cells using a P10 pipet tip. NOTE: See the representative results section for examples.
After dissection, aspirate RDM and all cell debris. Wash twice with 1 mL of pre-warmed DPBS per well.
Add 0.5 mL of TDE per well of a 12-well plate and incubate at 37 °C for 5 min. Use a cell scraper to gently remove the cells from the plate. Use a P1000 pipet to gently triturate the cell/TDE suspension by pipetting up and down 3-4 times to create a uniform suspension.
Dilute the cell/TDE suspension 1:10 in pre-warmed RSM, without Y-27632. Centrifuge cell suspension at 173 x g for 5 min at room temperature.
Aspirate the medium from the cell pellet and resuspend the cells in RSM with 10 µM Y-27632 (1 mL per enriched well).
Strain the cells using a nylon mesh cell strainer with 40 µm pores. Count the number of cells in a specified volume using a hemocytometer and calculate the concentration of cells in the strained solution.
Seed cells on the growth factor reduced ECM-coated plates at 1 x 105 cells/cm2 in 4 mL of RSM with 10 µM Y-27632.
Replace the RSM with 10 µM Y-27632 48 h after cell seeding and continue to replace media every 3-4 days (e.g. on Mondays and Thursdays). Do not replace the 10 µM Y-27632 after 4-7 days.
Allow the cells to mature for 28 to 35 days at 37 °C and 5% CO2. Continue to replace the RSM every 3-4 days (e.g. on Mondays and Thursdays).
5. Maturation: Passage 1 and 2 of RPE
NOTE: volumes are indicated for 1 well of a 6-well plate or a T75 flask as indicated by parentheses.
Between days 28 to 35 of passage 0, coat a 6-well plate (T75 flask) with ECMH as per manufacturer recommendations.
Aliquot the volume of DPBS and RSM needed and warm in a water bath to 37 °C. Bring TDE to room temperature.
Aspirate spent medium from wells and wash each well twice with 2 mL (10 mL) of pre-warmed DPBS. NOTE: Do not use 10 µM Y-27632.
Aspirate DPBS and add 1 mL (5 mL) of TDE. Place in incubator at 37 °C and 5% CO2 for 5 min. After incubation, view cells on an inverted microscope to confirm the cells are contracting and detaching.
Using an appropriately sized cell scraper, gently remove the cells from the bottom of the well or flask.
Use a P1000 tip (10 mL serological pipet) to gently triturate the cell/TDE suspension up and down 3-4 times to create a uniform suspension.
Dilute cell suspension 1:10 in RSM. Reserve 2 mL (5 mL) of RSM to rinse the well/flask and add to the diluted cell suspension. NOTE: Do not allow enzyme exposure time to exceed 25 min.
Centrifuge the cell suspension at 173 x g for 5 min at room temperature.
Aspirate the medium from the cell pellet and resuspend the cells in 1 mL (5 mL) of RSM
Strain the cells using a nylon mesh cell strainer with 40 µm pores. Count the number of cells in a specified volume using a hemocytometer and calculate the concentration of cells in the strained solution.
Seed cells on the ECMH-coated plates at 1 x 105 cells/cm2 in 4 mL (15 mL) of RSM.
Allow the cells to mature for 30 days. Continue to change the RSM every 3-4 days.
Repeat the above procedure (step 5.2-5.11) at day 30 to passage the cells from passage 1 to 2.
6. Creating an Intermediate Cell Bank: Cryopreservation of Passage 2 day 3-5 RPE
NOTE: Cryopreserve cells while they are subconfluent (~50%) and have not regained pigment.
Based on the number of cells, calculate the volume of cryopreservation medium with 10% DMSO needed to resuspend the cells at a concentration of 3x106 cells/mL.
Follow steps 5.2 to 5.8. Resuspend the cell pellet in the cryopreservation medium with 10% DMSO to 3 x 106 cells/mL and transfer 1 mL of the cell suspension to 1.2 mL cryogenic vials.
Place cryogenic vials in a freezing container designed to cool at -1 °C/min and place at -80 °C overnight. Transfer to liquid nitrogen for long-term storage. NOTE: These cells will be passage 3 upon thawing. Culture the cells for 30 more days before characterization. Seed passage 3 RPE at 1.5 x 105 per cm2 upon thawing.2,4,6,7
Representative Results
This method results in the production of a homogeneous, pigmented, and cuboidal monolayer of RPE. The timeline in Figure 1 corresponds to the images depicted in Figure 2. As shown in Figure 2A, the stem cell colonies are tightly packed with defined edges and no fibroblastic cells between colonies or opaque cells within colonies. Figure 2B provides a representation of immature RPE that are subconfluent. If the cells are already confluent at this stage, they cannot extend projections that are critical to the differentiation process. Cells that are severely subconfluent will not be able to establish a monolayer and form tight junctions, characteristic of epithelium. Details on how to optimize this confluence are outlined in the discussion section. Figure 2C shows the morphology of the two most common types of non-RPE that may arise during this differentiation process: neural or fibroblastic patches. It is important to note that these neural patches appear especially opaque on a dissecting microscope, whereas the defined, fibroblastic-like patches are nearly translucent on a dissecting microscope. It can be helpful to mark these areas on a tissue culture plate with an ethanol-proof lab pen to more easily identify them on both a compound light microscope and dissecting microscope. Figures 2D-F show the characteristic bright borders, cobblestone morphology, and pigmentation that indicate a healthy, maturing culture of RPE. Figure 3 is a higher magnification image to show the different appearance of fully mature RPE depicted by phase contrast and bright field microscopy. At passage 3 day 30, the cells are ready for the characterization that has been described in previous publications, including RNA expression, protein expression, growth factor secretion, and phagocytosis2,4,6,7. These characterizations show that the cells represented in these images are not only pigmented and cuboidal, but also phagocytic, post-mitotic, and polarized.
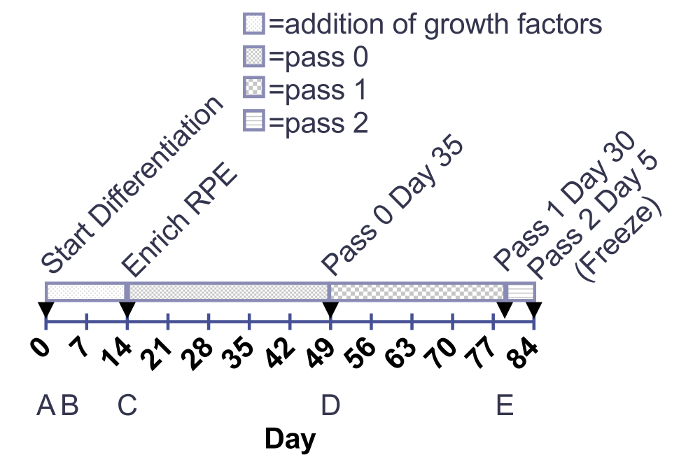

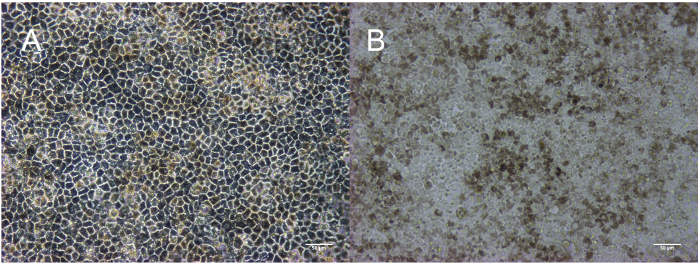
Figure 1:. Timeline for the addition of growth factors and maturation of RPE. Growth factors are added to 12-well plates from day 0-14. Maturing RPE are cultured in 6-well plates or T75 flasks from day of enrichment to 30 days post-thaw (passage 3 day 30). Arrows indicate enzymatic cell passaging. (A-E) below the timelines correspond to the images in Figure 2. Please click here to view a larger version of this figure.
Figure 2: Representative morphology and confluence of maturing RPE. Induced pluripotent stem cells immediately before passaging for differentiation (A). Immature RPE cells subconfluent at day 2 (B) and before pick-to-remove enrichment on day 14; non-RPE patches (indicated by white arrows) appear as patches or opaque "ribbons" (C). RPE at passage 0, 1, and 3 on day 30 (D, E, and F). Scale bar = 200 µm. Please click here to view a larger version of this figure.
Figure 3: Mature RPE at passage 3: Day 30 Cuboidal morphology depicted in phase contrast (A) and pigmentation depicted in bright-field (B). Scale bar = 50 µm. Please click here to view a larger version of this figure.
Discussion
This protocol describes how to produce retinal pigment epithelial cells from pluripotent stem cells. The method was optimized using both human embryonic and induced pluripotent stem cells from a feeder-free, serum-free culture method. Since the initial isolation of human embryonic stem cells in 1998 and the derivation of induced pluripotent stem cells (iPSC) in 2007, a multitude of stem cell culture methods have been developed14,15,16,17. These methods should be sufficient for producing stem cell colonies that are susceptible to this differentiation. There are no known limitations to the applicability of this method to properly derived and maintained pluripotent stem cells.
The most critical steps are the passaging of stem cells to day 0 of differentiation (step 2.5) and the potential need for manual dissection at day 14 of the process (step 4.5). When picking to remove differentiated cells from the stem cell colonies, refer to the images in Kent18. As indicated, the fibroblastic cells between colonies and the opaque cells within colonies indicate differentiated cells that need to be removed before beginning this protocol18. Only undifferentiated, tightly packed colonies with defined edges should be passaged for differentiation.
The number of stem cells seeded per well (step 2.6.7) is complicated by the fact that the stem cells cannot be triturated into a single cell suspension upon passage and cannot be accurately counted using a hemocytometer. The approximation of 80% confluent stem cells is indicated for passaging 1 well of a 6-well plate into 4 wells of a 12-well plate. Differences between stem cell lines, such as growth rate, can affect how quickly the immature RPE reach confluence between days 0 to 4. The stem cells will produce RPE regardless of precise confluence, but the cell yield will be negatively affected if the cells are too sparse at this stage. The immature RPE cells should be approximately 40-50% confluent on day 1 and nearly 100% confluent by day 4. If the cells are not producing a confluent monolayer by day 4 or 6, the protocol should be repeated at a higher seeding density at day 0. For example, if 1 well of a 6-well plate was passaged to 4 wells of a 12-well plate at day 0 and the immature RPE are not 100% confluent at day 4, reduce the seeding to a 1:3 or 1:2 passage on day 0 or allow the stem cells to become more confluent before passaging. It is critical to establish a consistent seeding density when comparing multiple cell lines.
The manual dissection step at day 14 is only necessary when non-RPE cells are present in culture (Figure 2C). Since the addition of CHIR99021 to the protocol, many pluripotent stem cell lines require little to no manual dissection. Some preparations have a higher incidence of neural patches and it is critical to remove those cells. If the RPE are not viable at passage 0 through passage 3, it is possible to repeat the differentiation protocol taking sufficient time to remove all non-RPE cells. This does not happen often, but it is mentioned here to note that the dissection step on day 14 can be optimized when needed.
There are a variety of RPE differentiation protocols that vary in cost as well as culture methods, efficiency, quantification, and functional assessment, the latter of which has been reviewed thoroughly2. We prefer the 14-day method detailed here because of its efficacy, adaptability, and applicability to a wide range of cell lines4,7,8. The cryopreservation step in this protocol also provides a major advantage in creating an intermediate cell bank for future use, avoiding lot-to-lot variability in experiments. Starting with only 4 wells of a 12-well plate, it is possible to expand into 6-well plates at passage 0 and T75 flasks at passage 1 and 2. At passage 2 day 3-5, when the cells are still subconfluent and have not regained pigment, it is possible to cryopreserve tens of millions of cells and then thaw the mature RPE, designated passage 3 day 30, to check RNA expression, protein expression, growth factor secretion, phagocytosis, etc. We have also established protocols to expand RPE for up to 13 passages 19.
Looking forward, this method will be useful for iPSC modeling of ocular disease and for generation of RPE for cellular therapy. With regards to iPSC disease modeling, this protocol is currently being used in the lab to produce RPE from CRISPR-corrected lines with non-corrected controls from the same patient. Furthermore, this protocol is adaptable to synthetic substrates and xeno-free conditions that are useful for adhering to the good manufacturing practices required for a cellular therapy.
Disclosures
Dr. Clegg is a co-founder of Regenerative Patch Technologies LLC.
Acknowledgments
This work was supported by the Garland Initiative for Vision, the California Institute for Regenerative Medicine (CIRM; grants DR1-01444, CL1-00521, TB1-01177, FA1-00616 and TG2-01151), The Vermont Community Foundation, The Breaux Foundation, and the Foundation Fighting Blindness Wynn-Gund Translational Research Acceleration Program.
References
- Strauss O. The retinal pigment epithelium in visual function. Physiol. Rev. 2005;85(3):845–881. doi: 10.1152/physrev.00021.2004. [DOI] [PubMed] [Google Scholar]
- Leach LL, Clegg DO. Concise Review: Making Stem Cells Retinal: Methods for Deriving Retinal Pigment Epithelium and Implications for Patients With Ocular Disease. Stem Cells. 2015;33(8):2363–2373. doi: 10.1002/stem.2010. [DOI] [PubMed] [Google Scholar]
- Pennington BO, Clegg DO. Pluripotent Stem Cell-Based Therapies in Combination with Substrate for the Treatment of Age-Related Macular Degeneration. J. Ocul. Pharmacol. Ther. 2016;32(5):261–271. doi: 10.1089/jop.2015.0153. [DOI] [PubMed] [Google Scholar]
- Buchholz DE, Pennington BO, Croze RH, Hinman CR, Coffey PJ, Clegg DO. Rapid and efficient directed differentiation of human pluripotent stem cells into retinal pigmented epithelium. Stem Cells Transl. Med. 2013;2(5):384–393. doi: 10.5966/sctm.2012-0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clegg DO, Buchholz D, Hikita S, Rowland T, Hu Q, Johnson LV. Retinal Pigment Epithelial Cells: Development In Vivo and Derivation from Human Embryonic Stem Cells In Vitro for Treatment of Age-Related Macular Degeneration. Stem Cell Res. Ther. 2008. pp. 1–24.
- Leach LL, Buchholz DE, Nadar VP, Lowenstein SE, Clegg DO. Canonical/β-catenin Wnt pathway activation improves retinal pigmented epithelium derivation from human embryonic stem cells. Invest. Ophthalmol. Vis. Sci. 2015;56(2):1002–1013. doi: 10.1167/iovs.14-15835. [DOI] [PubMed] [Google Scholar]
- Pennington BO, Clegg DO, Melkoumian ZK, Hikita ST. Defined culture of human embryonic stem cells and xeno-free derivation of retinal pigmented epithelial cells on a novel, synthetic substrate. Stem Cells Transl. Med. 2015;4(2):165–177. doi: 10.5966/sctm.2014-0179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach LL, et al. Induced Pluripotent Stem Cell-Derived Retinal Pigmented Epithelium: A Comparative Study Between Cell Lines and Differentiation Methods. J. Ocul. Pharmacol. Ther. 2016;32(5):317–330. doi: 10.1089/jop.2016.0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzoni F, Safa H, Finnemann SC. Understanding photoreceptor outer segment phagocytosis: use and utility of RPE cells in culture. Exp. Eye Res. 2014;126:51–60. doi: 10.1016/j.exer.2014.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoda S, Spee C, Barron E, Ryan SJ, Kannan R, Hinton DR. A protocol for the culture and differentiation of highly polarized human retinal pigment epithelial cells. Nat. Protoc. 2009;4(5):662–673. doi: 10.1038/nprot.2009.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary P, et al. Directing Differentiation of Pluripotent Stem Cells Toward Retinal Pigment Epithelium Lineage. Stem Cells Transl. Med. 2017;6(2):490–501. doi: 10.5966/sctm.2016-0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruotti J, et al. Small-molecule-directed, efficient generation of retinal pigment epithelium from human pluripotent stem cells. Proc. Natl. Acad. Sci. U.S.A. 2015;112(35):10950–10955. doi: 10.1073/pnas.1422818112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane A, et al. Engineering efficient retinal pigment epithelium differentiation from human pluripotent stem cells. Stem Cells Transl. Med. 2014;3(11):1295–1304. doi: 10.5966/sctm.2014-0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson JA, et al. Science. 5391. Vol. 282. New York, N.Y: 1998. Embryonic stem cell lines derived from human blastocysts; pp. 1145–1147. [DOI] [PubMed] [Google Scholar]
- Yu J, et al. Science. 5858. Vol. 318. New York, N.Y: 2007. Induced pluripotent stem cell lines derived from human somatic cells; pp. 1917–1920. [DOI] [PubMed] [Google Scholar]
- Takahashi K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Amit M, Itskovitz-Eldor J. Derivation and spontaneous differentiation of human embryonic stem cells. J. Anat. 2002;200(Pt 3):225–232. doi: 10.1046/j.1469-7580.2002.00032.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent L. Culture and Maintenance of Human Embryonic Stem Cells. J. Vis. Exp. 2009. pp. e1427–e1427. [DOI] [PMC free article] [PubMed]
- Croze RH, et al. ROCK Inhibition Extends Passage of Pluripotent Stem Cell-Derived Retinal Pigmented Epithelium. Stem Cells Transl. Med. 2014;3(9):1066–1078. doi: 10.5966/sctm.2014-0079. [DOI] [PMC free article] [PubMed] [Google Scholar]