

Abstract
DNA double-strand breaks (DSB) are serious DNA lesions. Analysis of the formation and repair of DSB is relevant in a broad spectrum of research areas including genome integrity, genotoxicity, radiation biology, aging, cancer, and drug development. In response to DSB, the histone H2AX is phosphorylated at Serine 139 in a region of several megabase pairs forming discrete nuclear foci detectable by immunofluorescence microscopy. In addition, 53BP1 (p53 binding protein 1) is another important DSB-responsive protein promoting repair of DSB by nonhomologous end-joining while preventing homologous recombination. According to the specific functions of γH2AX and 53BP1, the combined analysis of γH2AX and 53BP1 by immunofluorescence microscopy may be a reasonable approach for a detailed analysis of DSB. This manuscript provides a step-by-step protocol supplemented with methodical notes for performing the technique. Specifically, the influence of the cell cycle on γH2AX foci patterns is demonstrated in normal fibroblasts of the cell line NHDF. Further, the value of the γH2AX foci as a biomarker is depicted in x-ray irradiated lymphocytes of a healthy individual. Finally, genetic instability is investigated in CD34+ cells of a patient with acute myeloid leukemia by immunofluorescence microscopy of γH2AX and 53BP1.
Keywords: Cellular Biology, Issue 129, Immunofluorescence microscopy, γH2AX, 53BP1, X-rays, genetic instability, DNA double-strand breaks, DNA repair
Introduction
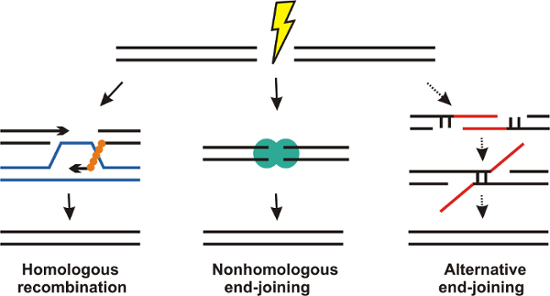
DNA is continuously damaged by endogenous (e.g., replication stress, reactive oxygen species, intrinsic instability of DNA) and exogenous (e.g., chemical radicals, irradiation) sources (Figure 1)1,2,3,4. Among DNA damage, DNA double-strand breaks (DSB) are particularly serious lesions and may induce cell death or carcinogenesis. About 50 DSB may arise per cell and cell cycle5. In mammalian cells, homologous recombination (HR) and nonhomologous end-joining (NHEJ) developed as major pathways for the repair of DSB (Figure 2). HR occurs in late S/G2 phase and uses an intact sister chromatid as a template for potentially error-free repair. In comparison, NHEJ is active throughout the cell cycle and potentially mutagenic as base pairs may be added or resected before ligation of the broken ends. In addition, alternative end-joining may be engaged as a slow mutagenic back-up repair mechanism in case of NHEJ deficiency6,7.

DSB induce the phosphorylation of the histone H2AX at Serine 139 in a region of several megabase pairs around each DSB. The forming nuclear foci are named γH2AX foci and are detectable by immunofluorescence microscopy8. γH2AX promotes the recruitment of further DSB-responsive proteins and is involved in chromatin remodeling, DNA repair, and signal transduction9. As each γH2AX focus is considered to represent a single DSB, the quantification of DSB by immunofluorescence microscopy is possible, which has been demonstrated in cancer cell lines and patient specimens10,11,12,13,14. 53BP1 (p53 binding protein 1) is another key protein in mediating DSB repair. It is involved in the recruitment of DSB-responsive proteins, checkpoint signaling, and the synapsis of DSB ends15. In addition, 53BP1 plays a critical role in the DSB repair pathway choice. It triggers the repair of DSB towards NHEJ while HR is prevented16. Considering the genuine functions of γH2AX and 53BP1 in DSB repair, the simultaneous analysis of γH2AX and 53BP1 by immunofluorescence microscopy may be a useful method for the precise analysis of the formation and repair of DSB.
This manuscript provides a step-by-step protocol for performing immunofluorescence microscopy of γH2AX and 53BP1 in cell nuclei. Specifically, the technique is applied in normal fibroblasts of the cell line NHDF, in x-ray irradiated lymphocytes of a healthy individual and in CD34+ cells of a patient with acute myeloid leukemia. The details of the method are pointed out in the context of the presented results.
Protocol
All methods described here have been approved by the Ethics Committee II of the Medical Faculty Mannheim of the Heidelberg University. Written informed consent was obtained from all individuals.
1. Preparation of Materials
Anticoagulant stock solution: Prepare an anticoagulant stock solution of 200 I.U. heparin per mL in 0.9% sodium chloride. Fill each of the collection tubes (draw volume 9 mL) with 2 mL of the anticoagulant stock solution before withdrawal of the blood or bone marrow samples.
Lysis solution for red cells: Prepare the 10x lysis solution for red cells with 82.91 g ammonium chloride, 7.91 g ammonium bicarbonate, and 2 mL of 0.5 M ethylenediaminetetraacetic acid solution to a pH of 8.0 by dissolving the agents in double-distilled water for a total volume of 1 L.
Fixation solution: Add 8.3 µL of a 1 M potassium hydroxide solution to 360 mg paraformaldehyde (PFA) in a microtiter tube. Fill the tube with phosphate buffered saline (PBS) to the 1 mL mark of the tube and heat the tube in a heating block at 95 °C to get 1 mL of a 36% PFA solution. Transfer the 1 mL 36% PFA solution to a tube with a capacity of 15 mL. Add 8 mL PBS to the 1 mL 36% PFA solution, to get 9 mL of a 4% PFA stock solution. Aliquot the 4% PFA stock solution and store at -18 °C until use for fixation of the cells. CAUTION: 1) Potassium hydroxide solution is corrosive. Be aware of eye and skin protection. 2) PFA is carcinogenic. Avoid skin contact and inhalation. Prepare the fixation solution under a hood.
Permeabilization solution: Add 50 µL Octoxinol 9 to 50 mL PBS to get a solution of approximately 0.1% Octoxinol 9.
Blocking solution: Prepare 5% and 2% blocking solutions by mixing the protein blocking agent with PBS and store at 6 – 8 °C.
2. Preparation of the Samples
- Fibroblasts in cell culture: Cultivate human fibroblasts of the cell line NHDF in a humidified 5% CO2 atmosphere at 37 °C in RPMI 1640 medium supplemented with 10% fetal calf serum, 4 mM glutamine, and 1% penicillin/streptomycin.
- Preparation of a microscope slide with adherent fibroblasts
- Remove the medium from the flask (75 cm2 growth area) with exponentially growing NHDF fibroblasts. Add 10 mL PBS to the flask. Wash the adherent fibroblasts gently by manually shaking the flask for 1 min.
- Remove the PBS and add 1 mL trypsin into the flask. Shake the flask gently to ensure that all adherent cells are covered by trypsin. Remove the trypsin from the flask. Put the flask into the incubator for 5 min at 37 °C.
- Take the flask out of the incubator and add 10 mL RPMI 1640 medium into the flask. Pipette the medium up and down several times to disperse the fibroblasts.
- Pipette 0.3 mL of the dispersed fibroblasts onto each of the two fields of the microscope slide and put the slide into the incubator for 24 h so that the fibroblasts adhere to the slide. For immunostaining of γH2AX and 53BP1 in nuclei of fibroblasts, move on to step 3.1.
- Patient samples
- Blood samples: Use the collection tubes that were filled with 2 mL of the anticoagulant stock solution and add 7 mL blood into the tubes. Store the samples overnight at room temperature (i.e., 18 - 20 °C). The next day, isolate the G0/G1 phase resting mononuclear cells by density gradient centrifugation (step 2.2.3).
- Bone marrow samples: Use the collection tubes that were filled with 2 mL of the anticoagulant stock solution (step 1.1) and add 7 mL bone marrow into the tubes. Store the samples overnight at room temperature (i.e.,18 - 20 °C). The next day, isolate the G0/G1 phase resting mononuclear cells by density gradient centrifugation (step 2.2.3). Use CD34 microbeads and cell separation columns for the isolation of the CD34+ cells.
- Isolation of mononuclear cells by density gradient centrifugation NOTE: Mononuclear cells are isolated from blood and bone marrow samples by density gradient centrifugation17,18,19.
- Dilute the heparinized blood sample in a ratio of 1:1 with PBS and the heparinized bone marrow sample in a ratio of 1:3 with PBS. Suspend the cells gently by drawing them in and out of the pipette two times.
- Add a volume of density gradient medium to a fresh tube. Gently layer the diluted blood or bone marrow on top of the density gradient medium. Take care not to mix the two layers.
- Centrifuge the tube for 30 min at room temperature and 400 x g. Draw the upper plasma layer off with the pipette. Then carefully harvest the mononuclear cells in the layer above the density gradient medium. Transfer the mononuclear cells into a fresh tube.
- Add at least three volumes of PBS to the mononuclear cells in the tube. Suspend the cells gently by drawing them in and out of the pipette two times. Centrifuge the tube for 10 min at room temperature and 400 x g.
- After the spin remove the supernatant and add 10 mL of 4 °C, 1x lysis solution for red cells. Resuspend the cells gently by drawing them in and out of the pipette two times, and place the tube on ice for 5 min. Then add 30 mL PBS at 4 °C and centrifuge the tube for 10 min at room temperature and 400 x g.
- Preparation of the cytospins
- Prepare two cytospins with mononuclear cells of the patient samples (i.e., one cytospin for γH2AX immunofluorescence staining and one cytospin for γH2AX and 53BP1 combined immunofluorescence staining) by centrifugation (300 x g, 10 min) of 1.0 x 105 cells for each preparation (Figure 3 and 4).

3. γH2AX and 53BP1 Immunofluorescence Staining
Fixation and permeabilization: Fix the cells with 200 µL of 4% PFA for 10 min. After fixation, wash the cells gently three times with 30 mL of PBS for 5 min each on a lab shaker. Then permeabilize the cells with 200 µL of 0.1% Octoxinol 9 for 10 min. Wash the cells gently three times with 30 mL of 5% blocking solution for 5 min each on a lab shaker. Block the cells in 30 mL of fresh 5% blocking solution for 1 h.
- Incubation with antibodies
- Incubate one preparation of the cells with a mouse monoclonal anti-γH2AX antibody (1:500) and the other preparation of the cells with a mouse monoclonal anti-γH2AX antibody (1:500) and a polyclonal rabbit anti-53BP1 antibody (1:500) overnight at 4 °C.
- After incubation wash the cells gently 3 times with 30 mL of 2% blocking solution for each 5 min on a lab shaker.
- Remove the blocking solution and incubate the first preparation of the cells with an Alexa488-conjugated goat anti-mouse secondary antibody (1:500) and the second preparation of the cells with an Alexa488-conjugated goat anti-mouse secondary antibody (1:500) and an Alexa555-conjugated donkey anti-rabbit secondary antibody (1:500) for 1 h at room temperature.
Mounting medium: Put the slide with the cells in a bowl and wash the cells gently three times with 30 mL of PBS for each 5 min on a lab shaker. Remove the PBS and mount the cells with mounting medium containing 4,6-diamidino-2-phenylindole. Cautiously put a coverslip on top of the mounting medium so that no air bubbles are embedded. Wait at least 3 h for hardening of the mounting medium before analyzing the cells by fluorescence microscopy.
4. Analysis of γH2AX and 53BP1 Foci
Fluorescence microscopy: Analyse the γH2AX and 53BP1 foci in the cell nuclei with a fluorescence microscope equipped with filters for DAPI, Alexa 488, and Cy3 during imaging at a 100X objective magnification. Record images with a camera and process the images with appropriate imaging software.
Representative Results
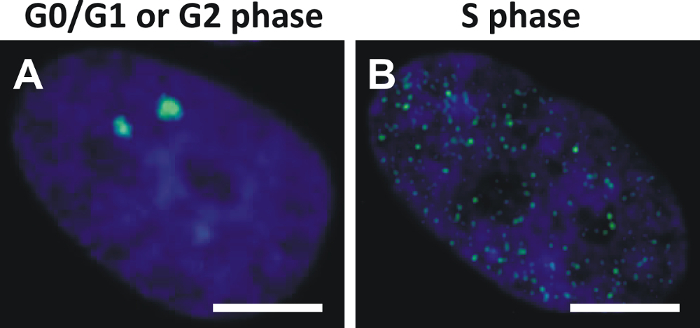
Analysis of γH2AX foci in cells is most accurate in the G0/G1 phase and the G2 phase when γH2AX foci appear as distinct fluorescent dots (Figure 5A). In contrast, analysis of γH2AX foci in cells during the S phase is complicated by dispersed pan-nuclear γH2AX speckles caused by the replication process (Figure 5B).
Fixation of the cells was performed with 4% PFA (10 min) and permeabilization with 0.1% Octoxinol 9 (10 min). Methanol may be used for fixation and permeabilization, too; however, methanol may break up the cell nuclei quite severely and smear out the γH2AX foci so the γH2AX foci may not appear distinct when compared to the γH2AX foci obtained by treatment with 4% PFA and 0.1% Octoxinol 9.
The choice of efficient antibodies is critical for effective immunofluorescence microscopy. Experiments were performed with a primary mouse monoclonal anti-γH2AX antibody and a primary rabbit polyclonal anti-53BP1 antibody for immunofluorescence staining of γH2AX and 53BP1, respectively.
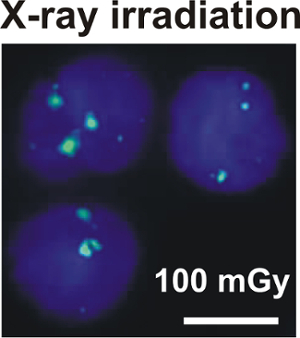
As each γH2AX focus is considered to represent a single DSB, immunofluorescence staining of γH2AX can be used for the quantification of DSB in irradiated tissues. Levels of γH2AX foci in irradiated lymphocytes (Figure 6) are proportional to the irradiation dose20,21. γH2AX foci may achieve a maximum already 5 min of irradiation20,22. The decay of γH2AX foci at later points in time can be monitored and reflects the repair of DSB20,21,22.
Genetic instability can be analyzed by combined immunofluorescence staining of γH2AX and 53BP1 in cell lines and tumor specimens10,11,12,13,14. An exemplary analysis was performed in the CD34+ cells of a patient with acute myeloid leukemia (Figure 7). Co-localization of γH2AX and 53BP1 indicated promotion of 53BP1-mediated NHEJ repair mechanisms at sites of DSB while HR was prevented. However, the percentage of co-localizing γH2AX and 53BP1 foci in blasts of acute myeloid leukemia was variable. The γH2AX foci without an additional 53BP1 signal might have engaged HR for the repair of DSB in late S/G2 phase. The role of singular 53BP1 foci remained elusive.
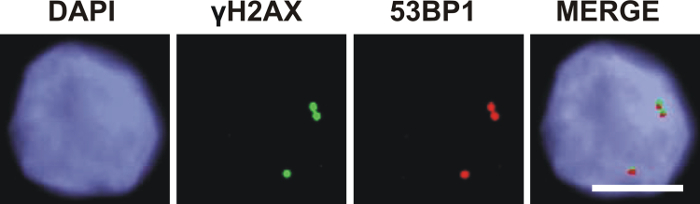
Figure 1: Sources and consequences of endogenous and exogenous DNA damage1,2,3,4. DSB, DNA double-strand breaks; NHEJ, nonhomologous end-joining.
Figure 2: Schematic view of the major DNA double-strand break (DSB) repair mechanisms in mammalian cells.Homologous recombination uses an intact sister chromatid in late S/G2 phase for potentially error-free repair of DSB. Nonhomologous end-joining (NHEJ) is active throughout the cell cycle and potentially error-prone as few base pairs may be added or resected before ligation of the DSB ends. Alternative end-joining is a slow mutagenic back-up repair mechanism which might be engaged in the case of NHEJ deficiency. Please click here to view a larger version of this figure.
Figure 3: Device for preparing the cytospins. Please click here to view a larger version of this figure.
Figure 4: Preparation of the cytospins.Two cytospins of each sample are prepared by centrifugation of 1.0 x 105 cells at 300 x g for 10 min. One cytospin is used for γH2AX immunofluorescence staining and another cytospin is utilized for combined γH2AX and 53BP1 immunofluorescence staining. Please click here to view a larger version of this figure.

Figure 5: Cell cycle dependent patterns of γH2AX foci in normal fibroblasts of the cell line NHDF. (A) γH2AX foci (green, Alexa 488) in nuclei (blue, DAPI) of NHDF fibroblasts appear as distinct dots in G0/G1 or G2 phase. (B) γH2AX foci in nuclei of fibroblasts in S phase appear as dispersed pan-nuclear speckles (scale bar = 5 µm). Please click here to view a larger version of this figure.
Figure 6: γH2AX foci in x-ray irradiated lymphocytes of a healthy individual. The images of γH2AX foci (green, Alexa 488) in nuclei (blue, DAPI) of lymphocytes are recorded at 5 min after irradiation with 100 mGy (scale bar = 5 µm). Please click here to view a larger version of this figure.
Figure 7: Co-localization of γH2AX and 53BP1 foci in CD34+ cells of a patient with acute myeloid leukemia. Co-localization of γH2AX foci (green, Alexa 488) and 53BP1 foci (red, Cy3) indicates promotion of 53BP1-mediated nonhomologous end-joining repair mechanisms at sites of DNA double-strand breaks (scale bar = 5 µm). Please click here to view a larger version of this figure.
Discussion
Immunofluorescence microscopy of γH2AX and 53BP1 is a useful method for analyzing formation and repair of DSB in a broad spectrum of research areas. Critical parameters that influence the outcome of the experiments are the phase of the cell cycle, the agents used for the fixation and permeabilization of the cells, the choice of the antibodies, and the hardware and software of the fluorescence microscope.
The influence of the cell cycle on γH2AX foci patterns was demonstrated by exponentially growing normal fibroblasts of the cell line NHDF. The γH2AX foci appeared as distinct fluorescent dots in the G0/G1and G2 phases as compared to the dispersed pan-nuclear γH2AX speckles in S phase (Figure 5). The distinct γH2AX foci in the G0/G1 phase and in G2 phase indicated transient and/or constitutive DNA damage that might have been acquired during repetitive cell cycle passages; conversely, the dispersed speckles in S phase occurred only transiently and were presumably associated with the replication process. As the pan-nuclear γH2AX speckles may disturb the analysis of genuine γH2AX foci, the exclusion of S phase cells from analysis is recommended, which is readily done by the S phase specific γH2AX pattern. Alternatively, cells in the S phase can be excluded by measurement of the intensity of the DAPI signal or by application of S phase specific markers (e.g., Cyclin A). Moreover, the induction of growth arrest with synchronization of cells in G0/G1 phase may be useful before preparing the cytospins by storing the samples at room temperature (i.e., 18 - 20 °C) overnight.
The procedures of fixation and permeabilization are critical steps in immunostaining of γH2AX and 53BP1. Fixation is necessary to preserve the morphology and antigenicity of the cells23. Permeabilization enables access to the intracellular and nuclear antigens24. PFA is a relatively gentle agent for fixation and stabilizes cells while preserving protein structures by reacting with basic amino acids to form methylene crosslinks. After fixation with PFA, cytospins can be stored for days and weeks until further processing. For detection of γH2AX and 53BP1, the cells have to be permeabilized. Octoxinol 9 was used as a non-ionic detergent containing uncharged hydrophilic head groups consisting of polyoxyethylene moieties. Octoxinol 9 breaks up the cell nuclei gently and preserves γH2AX foci quite distinctly. Alternatively, methanol can be used for the fixation and permeabilization of the cells, too. Methanol precipitates proteins and dissolves lipids from the cell membrane, making them permeable to antibodies. However, treatment with methanol is presumably more aggressive and γH2AX foci may not appear as distinctly when compared to the γH2AX foci obtained by treatment with 4% PFA and 0.1% Octoxinol 9. Notwithstanding, efficient antibodies play a key role for the immunostaining of γH2AX and 53BP1. Therefore, the use of proven antibodies is recommended (see Table of Materials/Equipment). A serial dilution of the antibodies may be useful for determination of the optimal antibody concentrations.
γH2AX foci are used as a marker in radiation biodosimetry at low doses > 1 mGy25. γH2AX foci were analyzed in lymphocytes at 5 min after x-ray irradiation with a dose of 100 mGy in vitro (Figure 6). As H2AX is phosphorylated rapidly at sites of DSB after irradiation, the number of γH2AX foci may already reach a maximum at 5 min after irradiation20,22. However, the repair process starts instantly after irradiation and γH2AX foci are removed at a distinct decay rate. Hence, for monitoring the number of γH2AX foci in a chronological sequence, it is advisable to interrupt the repair process by putting the sample on ice at the specified point in time after irradiation.
Immunofluorescence microscopy of γH2AX and 53BP1 is a useful method for analyzing genetic instability in cancer cells. CD34+ cells were enriched from the bone marrow of a patient with acute myeloid leukemia using CD34 microbeads. Images of γH2AX foci and 53BP1 foci were obtained using specific fluorescence filters. A bright illumination of the cytospin is important to induce adequate fluorescence of the γH2AX and 53BP1 foci. After recording the image, adaption of the intensity of the foci and the background signal is necessary using the imaging software. For analyzing the co-localization of γH2AX and 53BP1 foci, the images are merged (Figure 7). A basic epifluorescence microscope equipped with filters for DAPI, Alexa 488, and Cy3 in combination with a camera system and imaging software is sufficient for the analysis of γH2AX and 53BP1 foci in DAPI stained cell nuclei. However, certain applications (e.g., analysis of DNA damage along single particle tracks) may require higher resolution and reduction of the background fluorescence (as provided e.g., by confocal laser scanning microscopy). Moreover, microscope technology is a rapidly developing field and a resolution below 200 nm is feasible with state-of-the-art microscopes.
In summary, this manuscript highlights critical issues of DSB formation and repair and provides a step-by-step protocol for a detailed analysis of DSB by immunofluorescence microscopy of γH2AX and 53BP1. The research on DSB will clearly benefit from recent and future developments in microscope technology.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The project was supported by the German José Carreras Leukemia Foundation (DJCLS 14 R/2017).
References
- Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411(6835):366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362(6422):709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol. 2014;16(1):2–9. doi: 10.1038/ncb2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IAEA. Biological effects of low doses of ionizing radiation: A fuller picture. 1994. https://www.iaea.org/sites/default/files/36405843745.pdf.
- Vilenchik MM, Knudson AG. Endogenous DNA double-strand breaks: production, fidelity of repair, and induction of cancer. Proc Natl Acad Sci U S A. 2003;100(22):12871–12876. doi: 10.1073/pnas.2135498100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simsek D, Jasin M. Alternative end-joining is suppressed by the canonical NHEJ component Xrcc4-ligase IV during chromosomal translocation formation. Nat Struct Mol Biol. 2010;17(4):410–416. doi: 10.1038/nsmb.1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstock DM, Brunet E, Jasin M. Formation of NHEJ-derived reciprocal chromosomal translocations does not require Ku70. Nat Cell Biol. 2007;9(8):978–981. doi: 10.1038/ncb1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273(10):5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- Scully R, Xie A. Double strand break repair functions of histone H2AX. Mutat Res. 2013;750(1-2):5–14. doi: 10.1016/j.mrfmmm.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters DK, et al. Evidence for ongoing DNA damage in multiple myeloma cells as revealed by constitutive phosphorylation of H2AX. Leukemia. 2011;25(8):1344–1353. doi: 10.1038/leu.2011.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu T, MacPhail SH, Banath JP, Klokov D, Olive PL. Endogenous expression of phosphorylated histone H2AX in tumors in relation to DNA double-strand breaks and genomic instability. DNA Repair (Amst) 2006;5(8):935–946. doi: 10.1016/j.dnarep.2006.05.040. [DOI] [PubMed] [Google Scholar]
- Sedelnikova OA, Bonner WM. GammaH2AX in cancer cells: a potential biomarker for cancer diagnostics, prediction and recurrence. Cell Cycle. 2006;5(24):2909–2913. doi: 10.4161/cc.5.24.3569. [DOI] [PubMed] [Google Scholar]
- Chen E, et al. JAK2V617F promotes replication fork stalling with disease-restricted impairment of the intra-S checkpoint response. Proc Natl Acad Sci U S A. 2014;111(42):15190–15195. doi: 10.1073/pnas.1401873111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp HD, et al. Increase of DNA damage and alteration of the DNA damage response in myelodysplastic syndromes and acute myeloid leukemias. Leuk Res. 2017;57:112–118. doi: 10.1016/j.leukres.2017.03.011. [DOI] [PubMed] [Google Scholar]
- Panier S, Boulton SJ. Double-strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol. 2014;15(1):7–18. doi: 10.1038/nrm3719. [DOI] [PubMed] [Google Scholar]
- Escribano-Diaz C, et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol Cell. 2013;49(5):872–883. doi: 10.1016/j.molcel.2013.01.001. [DOI] [PubMed] [Google Scholar]
- Boyum A. Separation of White Blood Cells. Nature. 1964;204:793–794. doi: 10.1038/204793a0. [DOI] [PubMed] [Google Scholar]
- Boyum A. Isolation of mononuclear cells and granulocytes from human blood. Isolation of monuclear cells by one centrifugation, and of granulocytes by combining centrifugation and sedimentation at 1 g. Scand J Clin Lab Invest Suppl. 1968;97:77–89. [PubMed] [Google Scholar]
- Bain B, Pshyk K. Enhanced reactivity in mixed leukocyte cultures after separation of mononuclear cells on Ficoll-Hypaque. Transplant Proc. 1972;4(2):163–164. [PubMed] [Google Scholar]
- Popp HD, et al. Leukocyte DNA damage after reduced and conventional absorbed radiation doses using 3rd generation dual-source CT technology. Eur J Radiol Open. 2016;3:134–137. doi: 10.1016/j.ejro.2016.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothkamm K, Balroop S, Shekhdar J, Fernie P, Goh V. Leukocyte DNA damage after multi-detector row CT: a quantitative biomarker of low-level radiation exposure. Radiology. 2007;242(1):244–251. doi: 10.1148/radiol.2421060171. [DOI] [PubMed] [Google Scholar]
- Lobrich M, et al. In vivo formation and repair of DNA double-strand breaks after computed tomography examinations. Proc Natl Acad Sci U S A. 2005;102(25):8984–8989. doi: 10.1073/pnas.0501895102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamur MC, Oliver C. Cell fixatives for immunostaining. Methods Mol Biol. 2010;588:55–61. doi: 10.1007/978-1-59745-324-0_8. [DOI] [PubMed] [Google Scholar]
- Jamur MC, Oliver C. Permeabilization of cell membranes. Methods Mol Biol. 2010;588:63–66. doi: 10.1007/978-1-59745-324-0_9. [DOI] [PubMed] [Google Scholar]
- Rothkamm K, Lobrich M. Evidence for a lack of DNA double-strand break repair in human cells exposed to very low x-ray doses. Proc Natl Acad Sci U S A. 2003;100(9):5057–5062. doi: 10.1073/pnas.0830918100. [DOI] [PMC free article] [PubMed] [Google Scholar]