

Abstract
Background
Long terminal repeat (LTR) retrotransposons are highly abundant in plant genomes and require transcriptional activity for their proliferative mode of replication. These sequences exist in plant genomes as diverse sublineages within the main element superfamilies (i.e., gypsy and copia). While transcriptional activity of these elements is increasingly recognized as a regular attribute of plant transcriptomes, it is currently unknown the extent to which different sublineages of these elements are transcriptionally active both within and across species. In the current report, we utilize next generation sequencing methods to examine genomic copy number abundance of diverse LTR retrotransposon sublineages and their corresponding levels of transcriptional activity in three diploid wild sunflower species, Helianthus agrestis, H. carnosus and H. porteri.
Results
The diploid sunflower species under investigation differ in genome size 2.75-fold, with 2C values of 22.93 for H. agrestis, 12.31 for H. carnosus and 8.33 for H. porteri. The same diverse gypsy and copia sublineages of LTR retrotransposons were identified across species, but with gypsy sequences consistently more abundant than copia and with global gypsy sequence abundance positively correlated with nuclear genome size. Transcriptional activity was detected for multiple copia and gypsy sequences, with significantly higher activity levels detected for copia versus gypsy. Interestingly, of 11 elements identified as transcriptionally active, 5 exhibited detectable expression in all three species and 3 exhibited detectable expression in two species.
Conclusions
Combined analyses of LTR retrotransposon genomic abundance and transcriptional activity across three sunflower species provides novel insights into genome size evolution and transposable element dynamics in this group. Despite considerable variation in nuclear genome size among species, relatively conserved patterns of LTR retrotransposon transcriptional activity were observed, with a highly overlapping set of copia and gypsy sequences observed to be transcriptionally active across species. A higher proportion of copia versus gypsy elements were found to be transcriptionally active and these sequences also were expressed at higher levels.
Electronic supplementary material
The online version of this article (10.1186/s12870-017-1223-z) contains supplementary material, which is available to authorized users.
Keywords: Retrotransposons, Gypsy, Copia, Genomic abundance, Transcriptional activity, Sunflower
Background
Long terminal repeat (LTR) retrotransposons are Class 1 transposable elements found in the nuclear genome of diverse forms of life [1]. These elements have been particularly successful at replicating in plants genomes [2, 3], where they often comprise a majority of nuclear DNA. Gypsy and copia elements represent the main autonomous superfamilies found in plants though additional nonautonomous forms (e.g., TRIMs, LARDs), also are recognized [4].
The ‘copy-and-paste’ mode of transposition of LTR retrotransposons requires transcriptional activation, with resulting RNA molecules serving as templates both for translation and reverse transcription [5]. Products of translation (e.g., gag, RT, INT) function in the multistep, autonomous life cycle whereas products of reverse transcription serve as the physical daughter copies that insert at new locations in the genome. This transcriptionally based, replicative mode of transposition enables LTR retrotransposons to achieve the exceptionally high copy numbers found within plant genomes.
It long has been suggested that LTR retrotransposons are transcriptionally inactive under normal conditions due to epigenetic suppression by the host genome. Natural selection should favor element suppression given potentially negative mutagenic effects of unchecked element activity. It is increasingly recognized, however, that transcriptional activity of LTR retrotransposons is more common than previously supposed, and may actually be a regular attribute of plant transcriptomes [6–12].
Though highly repetitive, LTR retrotransposons exist in plant genomes as diverse populations of sequences represented by different sublineages within the larger classification groups (i.e., superfamilies gypsy and copia). Genomic copy number abundance of elements within different sublineages can be highly variable, with elements from a small number of sublineages often comprising a disproportionately large fraction of the total genomic abundance [13–17]. The extent to which transcriptionally active LTR retrotransposons are representative of the total element diversity present in plant genomes is not well understood, nor is the relationship between expression level and genomic copy number abundance of specific sublineages. In the current report, we use NGS sequencing to investigate these relationships in three diploid wild Helianthus sunflower species by comparing genomic abundance and transcriptional activity levels for multiple sublineages of gypsy and copia LTR retrotransposons. We focus specifically on genomic abundance and transcriptional activity for a diverse panel of 40 full-length gypsy and 12 full-length copia elements previously shown to represent much of the LTR retrotransposon diversity in sunflowers [16–19]. Insertion age estimates of these elements in the common sunflower Helianthus annuus indicate that most have been active recently, with a mean insertion age of 0.7 million years in the H. annuus genome [19].
Helianthus species have been the focus of a range of investigations examining LTR retrotransposons and their contribution to genome evolution (reviewed in Giordani, et al. [20]) and transcriptional activity of these sequences has been reported for multiple species within the genus [7, 12, 17, 21–23]. Species under investigation in the current study include H. agrestis, the sunflower species with the largest estimated diploid genome size [17, 24], plus additional species H. carnosus and H. porteri. All three sunflower species exhibit relatively restricted geographic ranges in the SE United States, with populations of H. agrestis and H. carnosus largely restricted to the state of Florida [25, 26] and populations of H. porteri confined to granite outcrops in the southern Piedmont region of eastern Alabama, Georgia and South Carolina [25–27]. Despite similarly restricted ranges and geographic occurrences, these species occupy disparate locations in the Helianthus phylogeny, with H. carnosus belonging to a Southeastern perennial clade, H. agrestis, an annual, subtending a monophyletic group consisting of all perennial Helianthus, and with H. porteri, also an annual, positioned basal to all of Helianthus [26]. The phylogenetic placement of these species enable LTR retrotransposon genomic abundance and transcriptional activity patterns to be interpreted broadly across the sunflower genus.
Methods
Plant materials and growing conditions
Seeds of the plant species utilized in this study were acquired from the United States Department of Agriculture (USDA) National Plant Germplasm System (Table 1). Seeds were germinated on moist filter paper in Petri dishes and then transferred to 10 cm plastic pots containing a mixture (2:1) of Metro-mix 350: all purpose sand. Plants were grown in the Kansas State University greenhouse facility under a daily light cycle of 16 h light: 8 h dark, with supplemental lighting. Young, fully-expanded leaves were harvested from mature plants on the same date from three biological replicates per species, flash-frozen in liquid nitrogen, and stored at −70 °C prior to processing as described below.
Table 1.
Genome size estimates for three Helianthus sunflower species
| Species | Accession | Genome size [2C (SE)] accession mean | Genome size [2C (SE)] species mean |
|---|---|---|---|
| H. agrestis | 673199 | 23.19 (0.30) | 22.93 (0.32) |
| 468414 | 22.29 (0.64) | ||
| 468416 | 23.31 (0.05) | ||
| H. carnosus | 649956 | 12.07 (0.64) | 12.31 (0.24) |
| 664671 | 12.55 (0.01)) | ||
| H. porteri | 649911 | 8.16 (0.18) | 8.33 (0.12) |
| 649912 | 8.56 (0.06) | ||
| 649918 | 8.27 (0.07) |
Genome size estimation
Nuclear genome size was determined by flow cytometry using a Guava PCA-96 micro capillary system (Guava Technologies, Hayward, CA.). Sample preparation consisted of co-chopping with razor blades approximately 30 mg fresh leaf tissue each for the sample and an internal standard (Secale cereal cv. Dankovské) in a nucleus isolation buffer modified after Bino et al. [28] and consisting of 15 mM HEPES, 1 mM EDTA, 80 mM KCl, 20 mM NaCl, 300 mM sucrose, 0.5 mM spermine, 0.25 mM PVP-40, 15 mM β-mercaptoethanol and 0.2% Triton-X. Samples were then filtered through 30-μm nylon mesh, centrifuged to collect nuclei, and stained with propidium iodide solution (BioSure). Nuclear genome size estimates were obtained for 3 different populations of H. agrestis and H. porteri and for 2 different populations of H. carnosus (Table 1). Three biological replicates were assayed for each population, resulting in a total of 24 samples.
DNA and RNA extraction, library construction and sequencing
DNA and RNA were extracted from leaf tissue using a DNeasy Plant Mini Kit (Qiagen, Valencia, CA) and TRIzol reagent (Invitrogen, Carlsbad, CA), respectively, following manufacturer instructions. Total RNA was purified to avoid any genomic DNA contamination using DNAse I and a RNeasy Mini Kit (Qiagen, Valencia, CA). One microgram of total DNA and RNA per sample was utilized for library preparation and sequencing on an Illumina HiSeq2500 platform, generating 2 × 100 bp paired-end reads for both datasets. Library preparation was performed following the Illumina TruSeq DNA PCR-free Library Prep Kit and the Illumina TruSeq RNA Sample Prep Kit v2 (Illumina Inc., San Diego, CA, USA). Average insert sizes were 470 bp and 260 bp for DNA and RNA libraries, respectively. Library construction and sequencing were performed at the University of Kansas Genome Sequencing Core Facility, Lawrence, KS, USA (http://gsc.drupal.ku.edu/).
Raw reads from DNA- and RNA-seq were trimmed using CLC Genomics Workbench v8.5.1 (Qiagen, Valencia, CA) to remove low quality sequences (PHRED scores <30) and sequences <80 bp and <40 bp, respectively. More stringent filtering of DNA-seq data was performed on account of the program RepeatExplorer [29] requiring input sequences ≥ 80 bp in length for analysis. After quality filtering, chloroplast- and mitochondrial-derived sequences were identified and removed by mapping reads against the complete chloroplast genome (GenBank accession number DQ383815) and complete mitochondrial genome (KF815390) of Helianthus annuus.
Genomic repetitive fraction
The genomic repetitive fraction of each species was determined using a graph-based clustering approach developed by Novak et al. [29] and implemented in RepeatExplorer [30] on the Galaxy Server (http://www.repeatexplorer.org/). Briefly, approximately 3 M single end DNA-seq reads were randomly sampled from each biological replicate per species and clustered based on an all-by-all comparison of sequence similarity (≥ 90%) and sequence overlap (≥ 55%). Individual clusters were identified and counted toward the genomic repetitive fraction if they contained ≥0.01% of the starting number of sampled sequences (e.g., for 3 M sequences, minimum cluster size = 300 sequences). These parameter values represent default setting of RepeatExplorer.
LTR-RT identification and global expression
Sequence clusters obtained from DNA-seq reads were identified as gypsy and copia LTR retrotransposons based on a similarity search, implemented in RepeatExplorer, of clustering output against a custom database consisting of 40 full-length gypsy and 12 full-length copia LTR retrotransposons derived from the H. annuus genome [18, 19]. These sequences represent a panel of diverse and variably abundant elements found in the genome of H. annuus [19], several additional Helianthus species [17], as well as in additional species in Asteraceae outside Helianthus [16]. These sequences are available as supplementary File S1 in Tetreault and Ungerer [17]. We followed the classification of sublineages in [17] for our LTR-RT identification and expression analysis.
Transcriptional activity was estimated by mapping the filtered RNA-seq reads to this same custom database using the CLC Genomics Workbench v8.5.1 (Qiagen, Valencia, CA) with sequence similarity ≥80%. Each mapped paired-end read was counted as one mapped fragment, as was each broken mapped read. The total mapped fragments of each sublineage were standardized by the average length of full-length elements within the focal gypsy and copia sublineages and by the total number of filtered reads (fragments per kilobase per million sequences, FPKM) to quantify expression level. Transcriptionally active elements were defined as those with FPKM values ≥1. Only uniquely mapped reads were utilized in transcriptional activity assays, though results were qualitatively similar when analyses were performed with multiply mapped reads.
Correlation between genomic repetitive fraction and expression level
Pearson’s r was calculated to evaluate correlation between genomic repetitive fraction and expression level for sublineages within gypsy and copia superfamilies. To account for the evolutionary history of different sublineages, Phylogenetic Independent Contrasts (PICs) were conducted with CONTRAST of PHYLIP v3.695 [31, 32]. Maximum likelihood trees of gypsy and copia elements were constructed using MEGA v6.0 [33] based on amino acid sequences of the reverse transcriptase domain. The best evolutionary model of protein sequences was estimated by ProtTest v2.0 [34].
Results
Nuclear genome size, sequencing data and genomic repetitive fraction
Within species, estimates of nuclear genome size varied 22.29–23.31 for H. agrestis, 12.07–12.55 for H. carnosus and 8.16–8.56 for H. porteri. Differences in 2C values among populations of the same species were not significant based on the sampling performed (H. agrestis, F = 1.8792, P = 0.23; H. carnosus, t = 0.7403, P = 0.50; H. poteri, F = 3.1982, P = 0.11). Nuclear genome size varies 2.75-fold across species with mean estimates of 22.93 for H. agrestis, 12.31 for H. carnosus and 8.33 for H. porteri (Table 1).
The Illumina Hi-Seq platform generated from 34.0 M to 63.5 M, and from 35.8 M to 56.9 M raw reads for DNA- and RNA-seq, respectively, for the species under investigation (Table 1 and Additional file 1: Table S1). After trimming and quality filtering, the numbers of reads in the final datasets ranged, respectively, from 24.6 M to 48.0 M and from 32.6 M to 51.7 M. Read lengths were reduced from their original size of 101 bases to mean lengths of 99.6 bases for DNA-seq and 93.8 bases for RNA-seq (Additional file 1: Table S1).
Graph-based clustering of ~3 M DNA-seq reads per species implemented in RepeatExplorer [30] yielded genomic repetitive fraction estimates of 83.33 ± 0.02% for H. agrestis, 74.48 ± 0.04% for H. carnosus, and 74.78 ± 0.20% for H. porteri (Table 1). The repetitive fraction estimate for H. agrestis is similar to a previous report for that species (82.12 ± 0.15%) [17] and newly generated estimates for H. carnosus and H. porteri are at the upper end, but within the range, reported for other diploid Helianthus species (68.17% - 75.26%) [17].
LTR-RT identification and abundance
To identify and quantify DNA-seq reads belonging to a diverse panel of annotated gypsy and copia LTR retrotransposons previously identified in the common sunflower (H. annuus) genome, sequence similarity searches of clustering output data from the graph-based clustering analysis were performed against a panel of 40 gypsy and 12 copia full length LTR-RT sequences reported originally in Buti et al. [18] and Staton et al. [19]. Between 22.92% ± 0.07 (H. porteri) and 35.85% ± 0.12 (H. agrestis) of sampled DNA-seq reads were identified as gypsy, and between 4.86% ± 0.01 (H. carnosus) and 5.41% ± 0.04 (H. porteri) were identified as copia (Fig. 1a), with gypsy sequences 4.2 to 7.1-fold more abundant than copia elements in these species genomes. While significant differences in abundance were observed among species for both read types (gypsy, F = 5.246, P < 10−9; copia, F = 118, P < 10−4, Fig. 1a), only gypsy sequence abundance exhibited a positive correlation with nuclear genome size (r = 0.998, P = 0.040 for gypsy; r = −0.494, P = 0.671 for copia, Additional file 1: Figure S1).
Fig. 1.
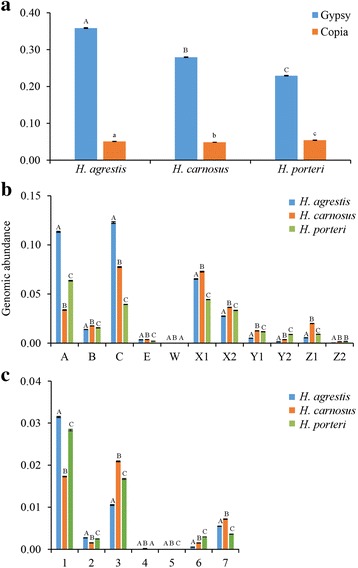
a Genomic abundance of gypsy and copia elements in three sunflower species based on similarity searches of graph-based clustering output to a reference panel of 52 full-length LTR retrotransposons. b and c Same information as in (a), but for different sublineages of gypsy (b) and copia (c). Error bars represent ±1 SE based on three biological replicates. Within a given element superfamily (panel a) or sublineage (panels b and c), histogram bars are labelled with different letter (A, B, C or a, b, c) when significant difference was determined (post-hoc Tukey test, P < 0.05)
Sequence diversity is considerable within the LTR-RT reference panel utilized in this study, with the 40 full-length gypsy and 12 full-length copia sequences comprising multiple, well-supported sublineages based on phylogenetic analyses of the reverse transcriptase amino acid domain (Additional file 1: Figure S2). We thus examined genomic abundance of these various gypsy and copia sublineages in the context of this diversity. Considerable variation was observed within all three sunflower species in density of DNAseq reads assigned to different sublineages, with density of gypsy reads ranging from 3.97 × 10−6 to 0.12 for H. agrestis, from 8.56 × 10−6 to 0.08 for H. carnosus and from 3.56 × 10−6 to 0.06 for H. porteri and density of copia reads ranging from 5.67 × 10−7 to 0.03 for H. agrestis, from 2.78 × 10−6 to 0.02 for H. carnosus and from 1.16 × 10−5 to 0.03 for H. porteri (Fig. 1b and c). Within individual sublineages of gypsy and copia, significant differences in DNA-seq read abundance were observed among species in all instances, with the largest interspecific differences characterized by elevated abundance in H. agrestis for two sublineages of gypsy (sublineages A and C; Fig. 1b).
Expression characteristics
Transcriptional activity of copia and gypsy sequences was determined by mapping RNAseq data to the reference panel of full-length LTR retrotransposons. Expression was detected for a total of 11 individual elements, consisting of 6 copia and 5 gypsy sequences (Fig. 2). Proportionally more copia (50%) versus gypsy (12.5%) elements were found to be expressed given the 1 FPKM threshold used for detection, and pooled across species, expression levels of copia elements were significantly higher than those for gypsy (t = 2.77; P = 0.011). Interestingly, expression of the same elements often was detected across multiple species, with 5 of 11 elements expressed in all three species, 3 of 11 expressed in 2 species and only the remaining 3 elements expressed in a single species. For elements determined to be active transcriptionally, regression analyses of FPKM and genomic abundance yielded nonsignificant results (Fig. 3a-c), though a negative trend was evident for H. agrestis and H. carnosus. Elements with the highest genome abundance estimates (e.g., those in gypsy sublineages C and X1) lacked transcriptional activity and the single copia element for which highest expression was detected in all three Helianthus species (i.e, copia sublineage 6) exhibited among the lowest genomic abundance estimates (Fig. 2).
Fig. 2.
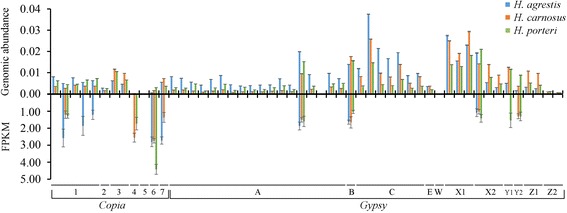
Genome abundance and expression level (FPKM) of 40 individual gypsy and 12 individual copia elements in three sunflower species. FPKM ≥1 was used as a cutoff for detectable expression. Error bars represent ±1 SE based on three biological replicates. Numerical and letter designations refer to different sublineages depicted in Additional file 1: Figure S2
Fig. 3.
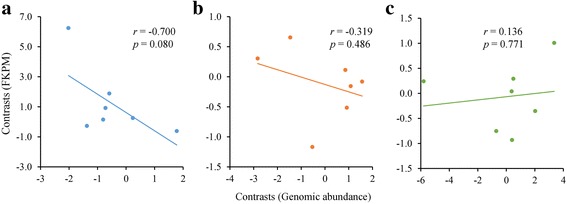
Phylogenetic independent contrasts between expression level (FPKM) and genomic abundance for gypsy and copia elements exhibiting FPKM ≥1 (see Fig. 2). Each data point represents an individual contrast, with Pearson’s r and P values calculated based on PIC analyses performed for H. agrestis (a), H. carnosus (b) and H. porteri (c)
Discussion
Our understanding of LTR retrotransposon diversity and abundance in plant genomes has increased substantially in recent years on account of advances in genomics technology and the rapidly increased rate at which sequence data can be generated for both model and nonmodel species [13–17, 35]. For a majority of plant species examined thus far, both gypsy and copia superfamilies exist as diverse sublineages, with a small number of these sublineages often representing a disproportionately large fraction of the LTR retrotransposon and thus genomic component ([4], but see [35]). Growing evidence supported by unbiased RNAseq studies also indicates that LTR retrotransposons are expressed more commonly than previously supposed, despite longstanding belief that these sequences are transcriptionally inactive. It remains unknown, however, the extent to which diverse elements in plant genomes are expressed at detectable levels, and the precise relationship between genomic abundance of individual elements and likelihood, or level, of transcriptional activity. In the present work, we combined DNAseq and RNAseq datasets to investigate both genomic abundance and transcriptional activity levels of a diverse panel of LTR retrotransposons in three diploid wild sunflower species.
Analyses of DNAseq datasets indicate considerably higher genomic abundance of gypsy versus copia LTR retrotransposons in all three species, a result consistent with previous analyses of other sunflowers [16–19, 36], and other plant taxa [37–39], though biases of elevated gypsy abundances in plant genomes are not universal [40–43]. The three sunflower species under investigation here generally displayed similar patterns regarding which sublineages of gypsy and copia were most versus least abundant in the genome, though significant differences were detected among species in densities of reads within most sublineages. Highest read densities were observed in H. agrestis, with sequences represented by two gypsy sublineages comprising approximately 23% of all reads for that species. These observations are consistent with this species having the largest estimated nuclear genome size (Table 1), the highest genomic repetitive fraction (Table 2), and point to specific gypsy sublineages that likely contributed to genome expansion.
Table 2.
Summary statistics of genomic data
| Species | Accession used for sequencing | Raw paired-end reads (DNA-, RNA-seq) (M) | Trimmed paired-end reads (DNA-, RNA-seq) (M) | Repetitive fraction (%) (SE) |
|---|---|---|---|---|
| H. agrestis | 673199 | 38.3–40.4, 40.0–44.0 |
28.6–29.5, 36.3–39.9 |
83.33 (0.02) |
| H. carnosus | 649956 | 34.0–38.6, 48.5–56.9 |
24.6–28.3, 44.0–51.7 |
74.48 (0.04) |
| H. porteri | 649918 | 42.4–63.5, 35.8–49.7 |
31.1–48.0, 32.6–45.2 |
74.78 (0.20) |
M million, SE standard error
Transcriptional activity of both gypsy and copia LTR retrotransposons has been documented previously in both cultivated and wild sunflowers using targeted [12, 23, 44, 45] and unbiased [7, 21, 22] approaches. Low but detectable transcriptional activity was observed for multiple gypsy and copia elements in the current study, but with a higher proportion of copia elements found to be active transcriptionally and with these copia elements expressed at higher levels. These observations are consistent with the notion that elements that are more abundant in the genome (i.e., gypsy) are more likely to be targeted for silencing by the host genome and those at lower abundances more likely to escape such targeting. Expression variation of LTR retrotransposons has been observed in other plant species as well [46–48]. For example, in maize, retroelements identified as low-copy were found to be expressed whereas retroelements identified as abundant were not [49]. Such results are not universally observed, however, as positive (though non-significant) correlations also have been observed between retrotransposon copy numbers and transcription level across different elements [48].
An unanticipated finding of this study was the highly overlapping patterns across species of specific elements found to be transcriptionally active, with 5 of 11 expressed elements transcriptionally active in all three species and 3 of 11 found active in 2 species. This is an interesting result given the disparate locations of these species in the Helianthus phylogeny and estimated age of the genus at 1.7–8.2 Mya [50]. These results indicate that identical or highly similar elements retain the ability to be transcriptionally active and/or avoid host-specific targeting across the genus. It currently is unknown whether transcriptional activity of these LTR retrotransposons results in associated insertional activity, as posttranscriptional mechanisms of TE repression are not well understood in sunflowers. Posttranscriptional repression perhaps is suggested by patterns whereby individual elements exhibiting some of the highest expression levels also exhibit some of the lowest genomic abundances (e.g., copia 4 and copia 6). Such patterns should be interpreted with caution, however, as expression levels of all elements were generally low.
It is unlikely that the transcriptional activity patterns of LTR retrotransposons observed in this study have arisen from DNA contamination of RNA samples. While contamination issues are of clear concern when transcriptional activity of highly repetitive sequences are assayed, our results are counter to expectations if contamination was an issue. First, contamination would be expected to lead to positive correlations between element abundance levels and transcriptional activity levels. Individual elements exhibiting the highest abundance levels typically were found to be transcriptionally inactive. Second, for elements that were found to be active transcriptionally, positive relationships between FPKM and abundance were not observed, and in fact negative trends were apparent for two of the three species studied.
Epigenetic silencing mechanisms are known to function in host genomes to control retrotransposon proliferation and prevent potentially negative mutagenic effects of element transpositional activity [51]. These mechanisms can be achieved through both pre-transcriptional (such as RNA-directed DNA methylation; [51]) and post-transcriptional mechanism (such as RNA interference; [52, 53]). Studies investigating these silencing mechanisms mostly have focused on model plant systems such as Arabidopsis thaliana, rice and maize ([51]; and references therein). Genomic analyses of methylation and small RNA targeting in a comparative manner in sunflower may lead to a better understanding of the dynamics of host-TE interactions in the wild species under investigation here.
Conclusions
While the three sunflower species under investigation in the current study differ markedly in genome size, patterns of LTR retrotransposon transcriptional activity were relatively conserved across species, with highly overlapping sets of copia and gypsy elements found to be transcriptionally active. For elements classified as transcriptionally active, negative but nonsignificant trend was observed between expression level (FPKM) and genomic abundance for two of three species. Transcriptional activity was not detected for individual elements exhibiting highest abundance. Additional experiments will be required to elucidate the mechanisms of transcriptional control and repression.
Acknowledgements
We thank Hannah Tetreault and Anastasia Weston for technical assistance.
Funding
This work was supported by Kansas State University. Contribution no. 17-217-J from the Kansas Agricultural Experiment Station. Publication of this article was funded in part by the Kansas State University Open Access Publishing Fund.
Availability of data and materials
Raw sequence reads are available from the NCBI Short Read Archive (SRA) with project #PRJNA418245.
Abbreviations
- FPKM
Fragments per kilobase per million sequences
- INT
Integrase
- LARDs
Large retrotransposon derivatives
- LTR
Long terminal repeat
- NGS
Next generation sequencing
- PICs
Phylogenetic independent contrasts
- RT
Reverse transcriptase
- TRIM
Terminal-repeat retrotransposons in miniature
Additional file
Pearson’s correlation between density of reads and nuclear genome size for gypsy and copia elements. Figure S2. Phylogenetic relationship among different sublineages of (A) gypsy and (B) copia based on amino acid sequences of the reverse transcriptase domain (gypsy, 173 a.a.; copia, 250 a.a.) using maximum likelihood. Bootstrap scores are presented for internal branches with >50% support. Table S1. Summary statistics of DNA-seq and RNA-seq data. (PDF 488 kb)
Authors’ contributions
FQ and MCU conceived of and designed the study. FQ and MCU conducted the experiments, FQ performed all analyses. FQ and MCU wrote the manuscript. Both authors have read and approved this manuscript.
Ethics approval and consent to participate
Seeds of the plant species utilized in this study were acquired from the United States Department of Agriculture (USDA) National Plant Germplasm System. The experimental research on plants performed in this study complies with institutional, national and international guidelines.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12870-017-1223-z) contains supplementary material, which is available to authorized users.
References
- 1.Eickbush TH, Malik HS. Origins and evolution of retrotransposons. In: Craig NL, Craigie R, Gellert M, Lambowitz AM, editors. Mobile DNA II. Washington, D.C.: ASM press; 2002. p. 1111–44.
- 2.Grover CE, Wendel JF. Recent insights into mechanisms of genome size change in plants. J Bot. 2010;2010:382732. [Google Scholar]
- 3.Kumar A, Bennetzen JL. Plant retrotransposons. Annu Rev Genet. 1999;33:479–532. doi: 10.1146/annurev.genet.33.1.479. [DOI] [PubMed] [Google Scholar]
- 4.Sabot F, Schulman AH. Parasitism and the retrotransposon life cycle in plants: a hitchhiker's guide to the genome. Heredity. 2006;97(6):381–388. doi: 10.1038/sj.hdy.6800903. [DOI] [PubMed] [Google Scholar]
- 5.Schulman AH. Retrotransposon replication in plants. Curr Opin Virol. 2013;3(6):604–614. doi: 10.1016/j.coviro.2013.08.009. [DOI] [PubMed] [Google Scholar]
- 6.Grandbastien MA. LTR retrotransposons, handy hitchhikers of plant regulation and stress response. Biochim Biophys Acta. 2015;1849(4):403–416. doi: 10.1016/j.bbagrm.2014.07.017. [DOI] [PubMed] [Google Scholar]
- 7.Kawakami T, Darby BJ, Ungerer MC. Transcriptome resources for the perennial sunflower Helianthus maximiliani obtained from ecologically divergent populations. Mol Ecol Resour. 2014;14(4):812–819. doi: 10.1111/1755-0998.12227. [DOI] [PubMed] [Google Scholar]
- 8.Neumann P, Pozarkova D, Macas J. Highly abundant pea LTR retrotransposon ogre is constitutively transcribed and partially spliced. Plant Mol Biol. 2003;53(3):399–410. doi: 10.1023/B:PLAN.0000006945.77043.ce. [DOI] [PubMed] [Google Scholar]
- 9.Neumann P, Yan H, Jiang J. The centromeric retrotransposons of rice are transcribed and differentially processed by RNA interference. Genetics. 2007;176(2):749–761. doi: 10.1534/genetics.107.071902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parchman TL, Geist KS, Grahnen JA, Benkman CW, Buerkle CA. Transcriptome sequencing in an ecologically important tree species: assembly, annotation, and marker discovery. BMC Genomics. 2010;11:180. doi: 10.1186/1471-2164-11-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vicient CM, Jaaskelainen MJ, Kalendar R, Schulman AH. Active retrotransposons are a common feature of grass genomes. Plant Physiol. 2001;125(3):1283–1292. doi: 10.1104/pp.125.3.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vukich M, Giordani T, Natali L, Cavallini A. Copia and gypsy retrotransposons activity in sunflower (Helianthus annuus L.) BMC Plant Biol. 2009;9:150. doi: 10.1186/1471-2229-9-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baucom RS, Estill JC, Chaparro C, Upshaw N, Jogi A, Deragon JM, Westerman RP, SanMiguel PJ, Bennetzen JL. Exceptional diversity, non-random distribution, and rapid evolution of retroelements in the B73 maize genome. PLoS Genet. 2009;5(11):e1000732. doi: 10.1371/journal.pgen.1000732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.El Baidouri M, Panaud O. Comparative genomic paleontology across plant kingdom reveals the dynamics of TE-driven genome evolution. Genome Biol Evol. 2013;5(5):954–965. doi: 10.1093/gbe/evt025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hawkins JS, Kim H, Nason JD, Wing RA, Wendel JF. Differential lineage-specific amplification of transposable elements is responsible for genome size variation in Gossypium. Genome Res. 2006;16(10):1252–1261. doi: 10.1101/gr.5282906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Staton SE, Burke JM. Evolutionary transitions in the Asteraceae coincide with marked shifts in transposable element abundance. BMC Genomics. 2015;16:623. doi: 10.1186/s12864-015-1830-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tetreault HM, Ungerer MC. Long terminal repeat retrotransposon content in eight diploid sunflower species inferred from next-generation sequence data. G3 (Bethesda) 2016;6(8):2299–2308. doi: 10.1534/g3.116.029082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buti M, Giordani T, Cattonaro F, Cossu RM, Pistelli L, Vukich M, Morgante M, Cavallini A, Natali L. Temporal dynamics in the evolution of the sunflower genome as revealed by sequencing and annotation of three large genomic regions. Theor Appl Genet. 2011;123(5):779–791. doi: 10.1007/s00122-011-1626-4. [DOI] [PubMed] [Google Scholar]
- 19.Staton SE, Bakken BH, Blackman BK, Chapman MA, Kane NC, Tang S, Ungerer MC, Knapp SJ, Rieseberg LH, Burke JM. The sunflower (Helianthus annuus L.) genome reflects a recent history of biased accumulation of transposable elements. Plant J. 2012;72(1):142–153. doi: 10.1111/j.1365-313X.2012.05072.x. [DOI] [PubMed] [Google Scholar]
- 20.Giordani T, Cavallini A, Natali L. The repetitive component of the sunflower genome. Curr Plant Biol. 2014;1:45–54. doi: 10.1016/j.cpb.2014.05.001. [DOI] [Google Scholar]
- 21.Gill N, Buti M, Kane N, Bellec A, Helmstetter N, Berges H, Rieseberg LH. Sequence-based analysis of structural organization and composition of the cultivated sunflower (Helianthus annuus L.) genome. Biology. 2014;3(2):295–319. doi: 10.3390/biology3020295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Renaut S, Rowe HC, Ungerer MC, Rieseberg LH. Genomics of homoploid hybrid speciation: diversity and transcriptional activity of long terminal repeat retrotransposons in hybrid sunflowers. Philos T R Soc B. 2014;369(1648):20130345. [DOI] [PMC free article] [PubMed]
- 23.Ungerer MC, Kawakami T. Transcriptional dynamics of LTR retrotransposons in early generation and ancient sunflower hybrids. Genome Biol Evol. 2013;5(2):329–337. doi: 10.1093/gbe/evt006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sims LE, Price HJ. Nuclear DNA content variation in Helianthus (Asteraceae) Am J Bot. 1985;72(8):1213–1219. doi: 10.2307/2443401. [DOI] [Google Scholar]
- 25.Heiser CB, Smith DM, Clevenger SB, Martin WC. The North American sunflowers (Helianthus) Mem Torrey Bot Club. 1969;22:1–218. [Google Scholar]
- 26.Stephens JD, Rogers WL, Mason CM, Donovan LA, Malmberg RL. Species tree estimation of diploid Helianthus (Asteraceae) using target enrichment. Am J Bot. 2015;102(6):910–920. doi: 10.3732/ajb.1500031. [DOI] [PubMed] [Google Scholar]
- 27.Gevaert SD, Mandel JR, Burke JM, Donovan LA. High genetic diversity and low population structure in porter's sunflower (Helianthus porteri) J Hered. 2013;104(3):407–415. doi: 10.1093/jhered/est009. [DOI] [PubMed] [Google Scholar]
- 28.Bino RJ, Lanteri S, Verhoeven HA, Kraak HL. Flow cytometric determination of nuclear replication stages in seed tissues. Ann Bot-London. 1993;72(2):181–187. doi: 10.1006/anbo.1993.1097. [DOI] [Google Scholar]
- 29.Novak P, Neumann P, Macas J. Graph-based clustering and characterization of repetitive sequences in next-generation sequencing data. BMC Bioinformatics. 2010;11:378. doi: 10.1186/1471-2105-11-378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Novak P, Neumann P, Pech J, Steinhaisl J, Macas J. RepeatExplorer: a galaxy-based web server for genome-wide characterization of eukaryotic repetitive elements from next-generation sequence reads. Bioinformatics. 2013;29(6):792–793. doi: 10.1093/bioinformatics/btt054. [DOI] [PubMed] [Google Scholar]
- 31.Felsenstein J. Phylogenies and the comparative method. Am Nat. 1985;125(1):1–15. doi: 10.1086/284325. [DOI] [PubMed] [Google Scholar]
- 32.Felsenstein J. PHYLIP - phylogeny inference package (version 3.2) Cladistics. 1989;5:164–166. [Google Scholar]
- 33.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abascal F, Zardoya R, Posada D. ProtTest: selection of best-fit models of protein evolution. Bioinformatics. 2005;21(9):2104–2105. doi: 10.1093/bioinformatics/bti263. [DOI] [PubMed] [Google Scholar]
- 35.Kelly LJ, Renny-Byfield S, Pellicer J, Macas J, Novak P, Neumann P, Lysak MA, Day PD, Berger M, Fay MF, et al. Analysis of the giant genomes of Fritillaria (Liliaceae) indicates that a lack of DNA removal characterizes extreme expansions in genome size. New Phytol. 2015;208:596–607. doi: 10.1111/nph.13471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Natali L, Cossu RM, Barghini E, Giordani T, Buti M, Mascagni F, Morgante M, Gill N, Kane NC, Rieseberg L, et al. The repetitive component of the sunflower genome as shown by different procedures for assembling next generation sequencing reads. BMC Genomics. 2013;14:686. doi: 10.1186/1471-2164-14-686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.International Rice Genome Sequencing P The map-based sequence of the rice genome. Nature. 2005;436(7052):793–800. doi: 10.1038/nature03895. [DOI] [PubMed] [Google Scholar]
- 38.Ming R, Hou S, Feng Y, Yu Q, Dionne-Laporte A, Saw JH, Senin P, Wang W, Ly BV, Lewis KL, et al. The draft genome of the transgenic tropical fruit tree papaya (Carica papaya Linnaeus) Nature. 2008;452(7190):991–996. doi: 10.1038/nature06856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Paterson AH, Bowers JE, Bruggmann R, Dubchak I, Grimwood J, Gundlach H, Haberer G, Hellsten U, Mitros T, Poliakov A, et al. The Sorghum bicolor genome and the diversification of grasses. Nature. 2009;457(7229):551–556. doi: 10.1038/nature07723. [DOI] [PubMed] [Google Scholar]
- 40.Argout X, Salse J, Aury JM, Guiltinan MJ, Droc G, Gouzy J, Allegre M, Chaparro C, Legavre T, Maximova SN, et al. The genome of Theobroma Cacao. Nat Genet. 2011;43(2):101–108. doi: 10.1038/ng.736. [DOI] [PubMed] [Google Scholar]
- 41.Gonzalez LG, Deyholos MK. Identification, characterization and distribution of transposable elements in the flax (Linum usitatissimum L.) genome. BMC Genomics. 2012;13:644. doi: 10.1186/1471-2164-13-644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang SW, Li RQ, Zhang ZH, Li L, Gu XF, Fan W, Lucas WJ, Wang XW, Xie BY, Ni PX, et al. The genome of the cucumber, Cucumis sativus L. Nat Genet. 2009;41(12):1275–1281. doi: 10.1038/ng.475. [DOI] [PubMed] [Google Scholar]
- 43.Jaillon O, Aury JM, Noel B, Policriti A, Clepet C, Casagrande A, Choisne N, Aubourg S, Vitulo N, Jubin C, et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature. 2007;449(7161):463–467. doi: 10.1038/nature06148. [DOI] [PubMed] [Google Scholar]
- 44.Buti M, Giordani T, Vukich M, Gentzbittel L, Pistelli L, Cattonaro F, Morgante M, Cavallini A, Natali L. HACRE1, a recently inserted copia-like retrotransposon of sunflower (Helianthus annuus L.) Genome. 2009;52:904–911. doi: 10.1139/G09-064. [DOI] [PubMed] [Google Scholar]
- 45.Kawakami T, Dhakal P, Katterhenry AN, Heatherington CA, Ungerer MC. Transposable element proliferation and genome expansion are rare in contemporary sunflower hybrid populations despite widespread transcriptional activity of LTR retrotransposons. Genome Biol Evol. 2011;3:156–167. doi: 10.1093/gbe/evr005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Domingues DS, Cruz GMQ, Metcalfe CJ, Nogueira FTS, Vicentini R, Alves CD, Van Sluys MA. Analysis of plant LTR-retrotransposons at the fine-scale family level reveals individual molecular patterns. BMC Genomics. 2012;13:137. [DOI] [PMC free article] [PubMed]
- 47.Ming R, VanBuren R, Wai CM, Tang HB, Schatz MC, Bowers JE, Lyons E, Wang ML, Chen J, Biggers E, et al. The pineapple genome and the evolution of CAM photosynthesis. Nat Genet. 2015;47(12):1435–1442. doi: 10.1038/ng.3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vicient CM. Transcriptional activity of transposable elements in maize. BMC Genomics. 2010;11:601. doi: 10.1186/1471-2164-11-601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meyers BC, Tingley SV, Morgante M. Abundance, distribution, and transcriptional activity of repetitive elements in the maize genome. Genome Res. 2001;11(10):1660–1676. doi: 10.1101/gr.188201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schilling EE. Phylogenetic analysis of Helianthus (Asteraceae) based on chloroplast DNA restriction site data. Theor Appl Genet. 1997;94(6–7):925–933. doi: 10.1007/s001220050497. [DOI] [Google Scholar]
- 51.Bousios A, Gaut BS. Mechanistic and evolutionary questions about epigenetic conflicts between transposable elements and their plant hosts. Curr Opin Plant Biol. 2016;30:123–133. doi: 10.1016/j.pbi.2016.02.009. [DOI] [PubMed] [Google Scholar]
- 52.Slotkin RK, Martienssen R. Transposable elements and the epigenetic regulation of the genome. Nat Rev Genet. 2007;8(4):272–285. doi: 10.1038/nrg2072. [DOI] [PubMed] [Google Scholar]
- 53.Lisch D. Epigenetic regulation of transposable elements in plants. Annu Rev Plant Biol. 2009;60:43–66. doi: 10.1146/annurev.arplant.59.032607.092744. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Raw sequence reads are available from the NCBI Short Read Archive (SRA) with project #PRJNA418245.