

Abstract
Advances in fluorescent microscopy and cell biology are intimately correlated, with the enhanced ability to visualize cellular events often leading to dramatic leaps in our understanding of how cells function. The development and availability of super-resolution microscopy has considerably extended the limits of optical resolution from ~250-20 nm. Biologists are no longer limited to describing molecular interactions in terms of colocalization within a diffraction limited area, rather it is now possible to visualize the dynamic interactions of individual molecules. Here, we outline a protocol for the visualization and quantification of cellular proteins by ground-state depletion microscopy for fixed cell imaging. We provide examples from two different membrane proteins, an element of the endoplasmic reticulum translocon, sec61β, and a plasma membrane-localized voltage-gated L-type Ca2+ channel (CaV1.2). Discussed are the specific microscope parameters, fixation methods, photo-switching buffer formulation, and pitfalls and challenges of image processing.
Keywords: Structural Biology, Issue 129, CaV1.2, confocal microscopy, endoplasmic reticulum (ER), super-resolution, total internal reflection fluorescence (TIRF) microscopy, voltage gated calcium channels, ground state depletion microscopy followed by individual molecule return (GSDIM)
Introduction
Cellular signaling reactions translate changing internal and external environments to initiate a cellular response. They regulate all aspects of human physiology, serving as the foundation for hormone and neurotransmitter release, the heartbeat, vision, fertilization, and cognitive function. Disruption of these signaling cascades can have severe consequences in the form of pathophysiological conditions including cancer, Parkinson's, and Alzheimer's disease. For decades, biological and medical investigators have successfully used fluorescent proteins, probes, and biosensors coupled with fluorescence microscopy as the primary tools to understand the precise spatial and temporal organization of these cellular signals.
The strengths of optical techniques such as epifluorescence, confocal, or total internal reflection fluorescence (TIRF) microscopy are their sensitivity, speed, and compatibility with live cell imaging, while the major limitation is their diffraction-limited resolution, meaning structures or protein complexes smaller than 200-250 nm cannot be resolved. With the theoretical and practical development of deterministic super-resolution (e.g., stimulated emission depletion microscopy (STED1), structured illumination microscopy (SIM2) or stochastic super-resolution (e.g., photoactivated localization microscopy (PALM3), or ground state depletion (GSD4,5)), lateral and axial resolution in fluorescence microscopy has been extended beyond the diffraction barrier, to the order of tens of nanometers. Thus, investigators now have the unparalleled ability to visualize and understand how protein dynamics and organization translates to function at the near-molecular level.
Ground state depletion microscopy followed by individual molecule return (GSDIM), or simply GSD as it is known, circumvents the diffraction limit by reducing the number of simultaneously emitting fluorophores4,5. High energy laser light is used to excite the fluorophore-labelled sample, bombarding electrons with photons and increasing the probability they will undergo a 'spin-flip' and enter the triplet or 'dark-state' from the excited state4. This effectively depletes the ground state, hence the name 'ground state depletion'. In the triplet state, fluorophores do not emit photons and the sample appears dimmer. However, these fluorophores stochastically return to the ground state and can go through several photon emitting excited-to-ground state transitions before returning to the triplet state. With less fluorophores emitting at any given time, photon bursts emitted from individual fluorophores become spatially and temporally distinct from neighboring fluorophores. The burst of photons can be fit with a gaussian function, the calculated centroid of which corresponds to the position of the fluorophore with a localization precision that is dependent on the numerical aperture (NA) of the lens, the wavelength of light used for excitation and crucially, the number of photons emitted per fluorophore. One limitation of GSD is that, since only a subset of fluorophores actively emits at any time, thousands of images must be collected over several minutes to build up a complete localization map. The long acquisition time combined with the high laser power requirement, means that GSD is better suited to fixed rather than live samples.
This article, describes the preparation of fixed samples for super-resolution microscopy imaging of membrane and endoplasmic reticulum (ER)-resident proteins (for a list of necessary consumables and reagents see the Table of Materials). Examples of how this protocol can be easily adapted to quantify the size and degree of clustering of L-type voltage-gated Ca2+ channels (Cav1.2) in the sarcolemma of cardiac myocytes, or used to visualize the cellular distribution of the ER, are demonstrated. Understanding the distribution and organization of these cellular components is critically important in understanding the initiation, translation, and ultimately the function of many Ca2+-dependent signaling cascades. For example, Cav1.2 channels are fundamentally important for excitation-contraction coupling, while receptor-mediated Ca2+ release from the ER is perhaps the most ubiquitous signaling cascade in mammalian cells.
Protocol
1. Washing Glass Coverslips
- The day before sample processing, clean #1.5 glass coverslips by sonicating in a 1 M solution of KOH for 1 h. This removes any fluorescent contaminants that may be present on the coverslips.
- Thoroughly rinse the KOH from the coverslips with de-ionized water.
- Once cleaned in KOH, store the coverslips in 70% ethanol until ready to use.
2. Coating Glass Coverslips
NOTE: Steps in this section should be performed in a cell culture hood to prevent contamination.
Use forceps to remove an individual coverslip from the 70% ethanol solution and swiftly flame to remove the excess. Place the coverslips into a 6-well plate. If flaming in a culture hood is not an option, consider autoclaving the coverslips after rinsing with de-ionized water and/or sterilization under the UV light in the culture hood for at least 30 min.
To aid adhesion of cells, coat coverslips with sterile 0.01% poly-L-lysine for 1 h at room temperature.
Aspirate the poly-L-lysine and rinse the coverslips with sterile phosphate buffered saline (PBS). Air dry and place in the refrigerator overnight.
The next morning, remove the coverslips from the refrigerator and add 300 µL of laminin, at a concentration of 50 µg/mL, for at least 30 min at room temperature. Ensure that the laminin is carefully placed directly onto the coverslip. NOTE: This laminin step is not necessary for COS-7 cells or HEK293 cells but is required for isolated ventricular myocytes. Other primary cells such as vascular smooth muscle cells or neurons may adhere better to collagen or fibronectin coated coverslips.
3. Preparation of Cells
- Cultured cells
- Grow COS-7 cells (or preferred cell line) on a 10 cm culture dish in 10 mL Dulbecco's Modified Eagles Medium (DMEM) culture medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (PS) solution. Grow the cells in a cell culture incubator at 37 °C and 5% CO2.
- When the cells reach 90% confluency, harvest them from the 10 cm dish by aspirating the DMEM media and adding 5 mL of 0.05% trypsin-EGTA solution. Once the cells begin to round up and detach, immediately neutralize the Trypsin with supplemented DMEM.
- Use a 5 mL serological pipette to remove the cells from the dish. Pipette the medium against the base of the dish several times to remove any cells that remain adherent and to homogenously distribute the cells throughout the culture medium.
- Plate cells onto 35 mm culture dishes to achieve 70% confluency for transfection. Make the volume of medium in the dish up to 2 mL with fresh DMEM/FBS/PS solution.
- Transfect COS-7 cells with the desired plasmid DNA constructs (e.g., sec61β-mCherry plasmid), using an appropriate transfection reagent and according to the manufacturer's instructions. NOTE: It may take 24-48 h for the protein to express, depending on the plasmid and/or the transfection reagent used.
- Adult mouse ventricular myocytes
- Isolate adult mouse ventricular myocytes using the well-established Langendorff perfusion method6. Re-suspend the resulting pellet of myocytes in Medium 199 (M199) supplemented with 2.5% FBS and 1% PS.
4. Plating Cells
- Transfected COS-7 cells
- Aspirate 2 mL DMEM/FBS/PS solution from 35 mm dish and add 1 mL Ca2+-free PBS. Return the dish to the incubator for 2 min. Harvest cells from the dish by first aspirating the Ca2+-free PBS and then adding 1 mL of 0.05% trypsin-EGTA solution.
- Once the cells begin to round up and detach, immediately neutralize the Trypsin with 1 mL of supplemented DMEM. Gently pipette up and down several times to ensure the transfected cells are evenly distributed throughout the 2 mL trypsin-EGTA-DMEM suspension.
- Plate transfected COS-7 cells onto poly-L-lysine coated coverslips at an appropriate density so that single cells can be visualized, and fill the dish volume up to 2 mL with DMEM/FBS/PS (e.g., add 90 µL cell suspension to the coverslip then add 1,910 µL supplemented DMEM, swirl dish to distribute cells evenly). NOTE: Cells do not need to be counted but the volume of cells to be added to each dish will vary depending on the confluency of the cells.
- Return plated cells to the cell culture incubator overnight to allow the cells to adhere and recover.
- Adult mouse ventricular myocytes
- Aspirate laminin and plate myocytes directly onto the coated coverslips at an appropriate density so that individual cells can be visualized. This will depend on the density of viable cells obtained from the isolation.
- Balance the cell suspension in a dome on the coverslip and place in a cell culture incubator at 37 °C and 5% CO2 for ~45 min to permit adhesion.
5. Fixation of Cells
- Remove cells from the incubator and carefully aspirate the medium.
- Rinse the adherent cells once with PBS.
- Fix cells with a fixative optimized to the individual application and antibodies. NOTE: An important criterion to keep in mind when selecting a fixative is the preservation of the cellular structure of the sample, while also ensuring that there is no conformational change in the antigen of interest. For the purposes of this protocol fixation with 3% paraformaldehyde/0.1% glutaraldehyde (PFA/GA) and fixation with 100% methanol is discussed.
- PFA/GA fixation of transfected COS-7 cells expressing Sec61β-mCherry
- Mix 1.9 mL 16% PFA and 20 µL 50% GA and add PBS to make a final volume of 10 mL.
- Add 1 mL freshly prepared PFA/GA solution to each dish of cells and fix for 10 min at room temperature.
- Aspirate the PFA/GA and rinse cells 3 times with PBS.
- Wash the adhered cells with PBS, 5 times for 5 min each. Perform all washing steps on a rotator set to a moderate speed (e.g., 50 cycles per minute).
- Next, reduce the free aldehyde groups with 1 mL freshly prepared 0.1% mass/volume sodium borohydride in de-ionized water.
- Rinse cells twice then wash 3 times for 5 min each in PBS to remove all traces of sodium borohydride and the alcohol it produces when it reacts with free aldehydes.
- Methanol fixation of adult mouse ventricular myocytes
- Carefully add 1 mL of ice-cold 100% methanol to the cells. Tilt the 6-well plate and pipette the methanol against the side wall rather than directly onto the coverslip. Then tilt the 6-well plate back to horizontal to allow the methanol to evenly wash over the cells. Fix for 5 min at -20 °C.
- Aspirate the fixative and rinse cells 3 times with PBS.
- Wash the adhered cells with PBS, 5 times for 5 min each on a rotator.
6. Blocking Non-specific Binding
Block for 1 h at room temperature in the blocking solution (see the Table of Materials). NOTE: Various solutions may be used to block non-specific antibody binding including 3% bovine serum albumin (BSA) or non-immune serum from the same species as the primary antibody.
7. Detection
- Primary antibody incubation
- Aspirate the blocking solution. Do not wash or rinse.
- Prepare the primary antibody solution as follows (see step 7.1.5): dilute the primary antibody to a concentration of 10 µg/mL (or manufacturer's suggested concentration) in the antibody incubation solution (see the Table of Materials). Balance this small volume on the coverslip and carefully set a square of plastic paraffin film on top. This helps to spread the small volume of antibody over the coverslip and guards against evaporation.
- Place the 6-well plate in a humidity chamber and incubate overnight in the refrigerator.
- In the morning, remove the cells from the humidity chamber, carefully remove the paraffin film square from the surface of the coverslip and aspirate the primary antibody solution.
- Rinse 3 times with PBS, then wash 5 times for 5 min with washing block solution (see Table of Materials) on the rotator. NOTE: This protocol uses the following antibodies, anti-RFP mouse monoclonal antibody for images displayed in Figure 2 and rabbit polyclonal FP17 anti-CaV1.2 antibody (a gift from Prof. Johannes Hell, UC Davis) for images displayed in Figure 3. With regards to the humidity chamber, a plastic lunch box with a tight-fitting lid, lined with wet paper towels works. PBS may be used for washing steps, however this protocol improves labeling specificity and reduces background by using a diluted blocking solution for washing steps.
- Secondary antibody incubation
- Aspirate the washing solution and add 200 µL conjugated secondary antibody solution diluted 1:1,000 in antibody incubation solution. Place a square of paraffin film on top of the coverslip (see step 7.1.2).
- Incubate for 1 h at room temperature in the dark.
- Aspirate secondary antibody and rinse 3 times with PBS.
- Wash cells with diluted blocking solution 5 times for 5 min on the rocker.
- Wash 3 times for 5 min with PBS. NOTE: This protocol describes the use of anti-mouse Alexa 647 conjugated secondary antibody and anti-rabbit Alexa 647 conjugated secondary antibody for Figure 2 and Figure 3, respectively. For single protein labeling, Alexa 647 is the preferred fluorophore owing to its high photon count (improves localization precision) and low duty cycle (improves temporal resolution)8. Many other fluorophore-conjugated secondary antibody options, such as Atto or Cy-dyes, are available. Refer to Dempsey et al.8 and Chozinski et al.9 for comprehensive evaluations of the suitability of several fluorophores/dyes for super-resolution imaging.

8. Post-fixation (Optional Step)
Dilute 50 µL 50 % GA in 10 mL PBS to obtain a 0.25% GA solution. Post-fix cells for 10 min in this solution at room temperature.
Wash with PBS 3 times for 5 min on a rotator.
9. Storage of Samples
Store samples in the refrigerator (4 °C) in an aluminum foil lined container to protect from light until imaging.
For long term storage of samples (up to several months), store in PBS containing 3 mM sodium azide to prevent bacterial growth.
10. GSD Super-resolution Imaging Photoswitching Buffer Preparation
- Prepare the oxygen scavenging 'GLOX' solution as follows:
- In a 1.5 mL microcentrifuge tube, add 12.5 µL catalase (stock 34 mg/mL), 3.5 mg glucose oxidase, and 0.5 µl 1 M Tris (pH = 8) to 49.5 µL PBS.
- Vortex briefly to dissolve components into solution then centrifuge at 20,800 x g for 3 min at 4 °C. NOTE: oxygen-scavenging solutions such as GLOX, have been found to oppose photobleaching. GLOX should be kept in the refrigerator for up to one week. Repeat the centrifuge step each time it is used.
- Prepare the photoswitching-inducing thiol solution as follows:
- Prepare 100 mM β-mercaptoethylamine (MEA) solution in PBS and modify the pH to pH 8 or alternatively use β-mercaptoethanol (βME). Store aliquoted MEA solution in the freezer (-20 °C) for up to 1 week.
- Immediately before imaging, prepare the final switching buffer on ice by adding the thiol and oxygen-scavenging components together. For 500 µL GLOX-MEA, add 5 µL GLOX to 50 µL MEA (pH = 8) and 445 µL saline buffer (2.5 mL 1 M Tris pH = 8, 29.22 mg NaCl (10 mM final concentration), 5 g glucose and 47.5 mL water). Alternatively, for GLOX-βME solution add 5 µL GLOX to 5 µL βME and 490 µL saline buffer.
- Keep the solutions on ice and use as needed to mount samples on glass depression slides. NOTE: Thiol containing solutions such as βME or MEA induce photoswitching of cyanine-based dyes such as Alexa 64710 or xanthene-based dyes such as Alexa 568. βME produces better results with Alexa 647 whereas MEA is preferred for Alexa 568 or when 2 proteins are to be imaged with a double labeling approach utilizing both Alexa 568 and 647. The buffer will deteriorate over a period of hours due to acidification. This will lead to a reduction in the average photon count of the sample. To prevent this, it is advisable to make fresh photoswitching buffer after 2-3 h or if either a drop in the average photon count or a notable extension of time taken to induced photoswitching is observed. A recent article described a new imaging buffer Ox-EA which we have not yet tested but reportedly exhibits stable pH levels for several days and performs better than GLOX for multi-color imaging applications11.
11. Mounting Samples
Mount coverslips with labeled cells (see sections 1-9) on depression slides, using 100 µL imaging buffer.
Seal the edges of the coverslip with a silicone glue to prevent rapid oxidation of the imaging buffer. Wait several minutes until the silicone glue has fully cured otherwise the coverslip will drift when imaging. NOTE: Silicone glue will not harden if it comes into contact with βME. Be sure to dry the edges of the coverslip to prevent the silicone glue from contacting the imaging buffer.
12. Image Acquisition
- See Table 1 for a list of imaging parameters used in Figure 2 and Figure 3.
- To begin, select the appropriate laser to illuminate the sample depending on the fluorophore selected. The SR-GSD system used in this protocol is equipped with high-power lasers (488 nm, 1.4 KW/cm2; 532 nm, 2.1 KW/cm2; 561 nm, 2.1 KW/cm2 642 nm, 2.1 KW/cm2). Figure 2 used the 642 nm laser.
Use an oil-immersion objective lens with a high NA. The SR-GSD system used here is equipped with an HC PL APO 160X/1.43 NA objective.
Chose the appropriate dichroic imaging cube. The SR-GSD system in this protocol is equipped with 488 HP-T, 532 HP-T, 568 HP-T, 642 HP-T with emission band-pass filters of 505-605 nm, 550-650 nm, and 660-760 nm.
Select the 2D acquisition mode. Set the camera to frame-transfer mode and exposure time to 10 ms (100 Hz). Set the EM gain to 300. Select the detection threshold. NOTE: Low thresholds produce images with high background. High thresholds may filter out the signal of interest. Determine the threshold based on negative controls such as imaging coverslips of non-transfected cells or cells incubated with secondary antibody only.
Turn on auto event control and set events per images to 8.
Enter the pixel size in the GSD-acquisition tab (start with 10 nm or 20 nm pixel size). Ensure that "Histogram Mode" is selected in the GSD tools tab under the High resolution image calculation options before image acquisition.
To image proteins at the plasma membrane, select the TIRF mode and modify the incidence angle to determine the penetration depth.
To send electrons to the dark state (called pumping), set laser intensity to 100%.
Select single molecule detection while pumping and set to acquire automatically when the frame correlation is 0.20.
For acquisition, set laser intensity to 50%.
Set the number of acquisition frames to 60,000. NOTE: The investigator may find that a sample requires more or less frames than this. Modify the frame count based on observations of fluorophore photobleaching and stop acquisition when no further events are detectable.
Start acquisition. NOTE: At the beginning of acquisition, all dye molecules are in the fluorescent state and then switch to the dark state. When the frame correlation reaches 0.20, images will start to be acquired. When less than 8 events are detected per frame, the 405 laser will switch on, encouraging fluorophores to transition from the dark state to the ground state. When acquiring images for a dataset of n = x cells from n = y animals, parameters such as TIRF penetration depth, detection threshold, and number of frames acquired should be kept constant.
13. Image Analysis
- To quantify protein cluster size, use the 'analyze particles' feature of ImageJ. NOTE: Further analysis may be performed using stochastic optical reconstruction microscopy-based relative localization analysis (STORM-RLA) or similar13.
- Perform cluster analysis (Figure 3) using ImageJ as follows:
- Open the image file to be analyzed. This should be a 10 nm histogram single molecule localization map as generated in the imaging software described above (section 12).
- Adjust the brightness and contrast to visualize the image: Click on the image menu, select adjust, then brightness and contrast. Click the auto option.
- Change the image type to 8-bit: Select the image menu, then select type, and choose 8-bit.
- Set the scale of the image: Open the analyze menu, click set scale and enter the following parameters: distance = 1, known distance = 10, 1 pixel = 10 nm.
- To select the measurements to be obtained, open the analyze menu, select set measurements, and check the box beside the area option.
- Adjust the threshold of the image. Open the image menu, select adjust, and then select threshold, select over/under. Move the maximum value to 255, and move the minimum value to 1 and click apply.
- To analyze the particles, open the analyze menu, select analyze particles. Place a checkmark to select pixel units, display results, and summarize. Set the size to 4-infinity (since resolution is ~20 nm), select the bare outlines option and click ok. A summary file will be created which will contain the analysis details such as the average cluster size and the number of clusters in the image. NOTE: Caution should be exercised when making conclusions about the number of proteins within a particular cluster, as the number of localized points is not linearly correlated with the number of proteins. GSD localizes dyes that are conjugated to antibodies, resulting in a linkage-error that displaces the fluorescent label from the epitope by >10 nm in any direction. Additionally, if polyclonal antibodies are used, more than one antibody can bind to a single antigen and to confound the issue even further, commercial secondary antibodies are conjugated to, on average, 3-6 molecules of dye so one fluorophore does not always equal one protein. Linkage-error can be reduced by conjugating a dye directly to the primary antibody (eliminating the secondary) or by using a nanobody-based labeling scheme14.
Representative Results
As documented in the introduction, there are many different super-resolution microscopy imaging modalities. This protocol, focuses on GSD super-resolution imaging. Representative images and localization maps are shown in Figure 2 and Figure 3.
Figure 2 shows a COS-7 cell transfected with the ER protein, mCherry-Sec61β, and processed using the method described above. Figure 2A-2B allow the comparison of images of the ER taken using a super-resolution microscope in TIRF mode (A) and using GSD acquisition mode (B). The images show an improvement in the axial resolution when acquired in GSD mode. This is further demonstrated in the accompanying plot profile, that can be generated using ImageJ, which shows the difference in the distribution of the normalized intensity curves at the areas of interest (yellow line). These curves represent the diameter of the ER tubules which appear to be much narrower when examined in GSD mode. GSD microscopy has a lateral localization precision of approximately 20 nm and thus represents approximately a ten-fold increase in resolution beyond the diffraction limit. This improvement in resolution results in a more accurate representation of the structure of the ER tubules.
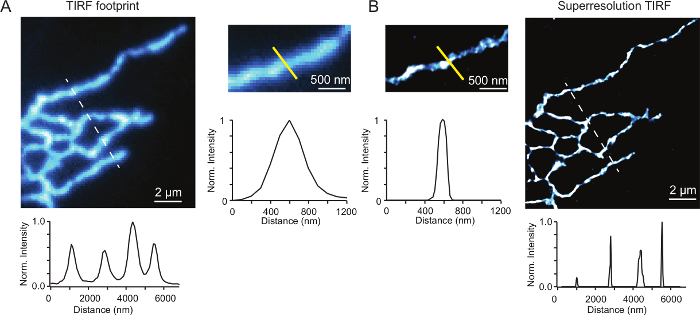
The improvement in resolution offered by super-resolution GSD imaging is further demonstrated in Figure 3, showing a labeled cardiac myocyte with an anti-Cav1.2 antibody, processed as described above. The image in Figure 3A was taken using a GSD super-resolution microscope in TIRF setting. Clusters of CaV1.2 channels can be seen to organize along the t-tubule network. Figure 3B shows the same cell, however this image was acquired using GSD mode. The improvement in the axial resolution is more prominent in the panels which focus on single clusters of channels (Figure 3B), when a comparison is made between the same ROI imaged using TIRF and GSD, separate clusters of channels are easier to identify as a result of the improvement in resolution offered by GSD imaging. These panels also compare the difference in the detection threshold defined as the number of photons per pixel. This parameter must be chosen carefully and adapted to the requirements of the experiment. Panel 3D shows cluster outlines when the GSD image was processed using ImageJ software, from this the cluster area for each individual cluster in the image can be determined (see note 13.1.8 above and the Discussion section for important considerations when performing such a quantification). This information can then be used to generate the frequency histogram in panel 3C. This analysis can be used to examine changes in cluster size, or the number of clusters between samples under control versus test conditions (e.g., WT vs mutant channels or untreated vs drug-treated cells). Using the approaches outlined in this protocol alongside complementary step-wise photobleaching experiments, investigators have determined that CaV1.2 channels are distributed in clusters of, on average, 8 channels in cardiac myocytes15.
Figure 1: Schematic representation of the timeline of events for super-resolution imaging of membrane proteins. Day 0 refers to the first day of processing for antibody labeling. The protocol section refers to the section in the protocol where detailed information is found on each step. Please click here to view a larger version of this figure.
Figure 2: Super-resolution microscopy offers improved signal to noise and increased spatial resolution. (A) Left COS-7 cells expressing mCherry-Sec61β, fixed and labelled as described in the protocol and imaged using conventional TIRF microscopy. Right, upper panel: single ER tubule. Right, lower panel: plot profile taken across the width of the ER tubule (yellow line). (B) Same cell as (A) except the localization map was generated using GSD super-resolution. Please click here to view a larger version of this figure.
Figure 3: Super-resolution imaging of Cav1.2 channels in cardiac myocytes. (A) TIRF footprint from an isolated cardiac myocyte, fixed, and stained with anti-CaV1.2 antibody (FP1). (B) Left: Super-resolution TIRF footprint from the same cardiac myocyte as in (A). Right: enlarged portions of the localization map that have been subjected to different detection thresholds. Note that as one increases the detection threshold, the apparent size of CaV1.2 channel clusters decrease. (C) Frequency histogram of cluster sizes obtained from the localization map in (B). (D) Left: Outlines of the CaV1.2 clusters from (B). Right: enlarged portions of the image have been subjected to different detection thresholds. Note, similar to (B), as the detection threshold increases, the area occupied by the CaV1.2 channel clusters decreases. Please click here to view a larger version of this figure.
| Fluorophore used | Alexa-647 |
| Laser | 642 nm |
| Emission filters | 623 HP-T |
| Lens | 160X 1.43 NA |
| Exposure time | 10 ms |
| Detection threshold | 65 |
| Incidence Angle (penetration depth) | 65.04° (150 nm) |
| EM gain | 300 |
| #frames acquired | 30,000–60,000 |
| Laser intensity for pumping | 100% |
| Laser intensity for acquisition | 50% |
Table 1: List of imaging parameters.
Discussion
The recent explosion of technologies that allow imaging beyond the diffraction limit have offered new windows into the complexities of mammalian cell signaling in space and time. These technologies include STORM, STED, PALM, GSD, SIM, and their variants (e.g., dSTORM, FPALM). The ingenuity of the scientists behind these techniques has allowed us to circumvent the limitations imposed by laws of physics governing the diffraction of light. In spite of this huge accomplishment, each of these techniques makes some sort of compromise to achieve super-resolution and, as such, has its limitations. The ideal situation that cell biologists and biophysicists strive for is optimum sensitivity, high resolution, and fast acquisition, with no photobleaching. In addition, one should remain cognizant that by removing cells from animals, we inherently change them and potentially change molecular dynamics. Therefore, the quest to develop a super-resolution technology that permits dynamic, live cell, single molecule imaging in a living, breathing animal goes on. For now, we have tools at our disposal that can generate images with 10-fold improvements on diffraction limited resolution. In this article, we discuss sample preparation, image acquisition, and analysis for GSD that enable resolutions down to 20 and 50 nm in the lateral and axial dimensions, respectively.
Although the focus of this protocol is on Sec61β and CaV1.2 L-type Ca2+ channels, the protocol described above can serve as a template for researchers who wish to image different proteins in their own labs on a GSD microscope. The positive attributes of this type of protocol are that it is relatively straightforward, can be modified and applied to a number of different cell types, and can be easily adapted to multi-color imaging. The obvious limitations are that super-resolution systems, equipped with a sensitive EM-CCD camera capable of single-molecule detection, still tend to be prohibitively expensive for many laboratories.
As the number of laboratories that use super-resolution microscopy as their preferred imaging technique grow, more uniform understanding and agreement is needed about image acquisition, processing, and interpretation. How, for example, does one quantify the level of proximity of two proteins that appear colocalized in a diffraction-limited pixel but transpire to actually display little overlap at all in a super-resolution image/map? Indeed, this scenario has already arisen for several previously assumed colocalized proteins, such as the adhesion complex proteins paxillin and zyxin. As the resolution that microscopes achieve invariably increases, perhaps proteins will no longer be reported to 'colocalize' but rather to have non-randomly distributed patterns of preferred-localization around one another. After all, it is impossible for two proteins to physically occupy exactly the same 3-dimensional space. Some solutions to this analysis conundrum are starting to emerge in the literature, including stochastic optical reconstruction microscopy-based relative localization analysis (STORM-RLA), which can reportedly quantify the frequency and degree of overlap between two co-labeled proteins and also provide quantitative measurements of the distance between non-overlapping proteins13. When comparing one channel to another in super-resolution, it is of course critical to ensure you have correct registry between the two channels. This can be evaluated using multi-color microsphere fluorescent beads and imaging in each individual channel then overlaying the images. Additionally, since the SR-GSD 3D microscope can also function as a TIRF microscope and generate super-resolution localization maps in TIRF, it is important to match the penetration depth between two-color channels, as TIRF angles change depending on the wavelength of light used to excite the sample.
Further, as depicted in Figure 3B and Figure 3D, a localization map can appear vastly different depending on the degree of 'thresholding'. If the detection threshold is set too low, structures may appear to be larger than they truly are as the probability that one includes spurious non-specific labeling of 'noise' increases. Conversely, if the detection threshold is set too high, then structures may appear smaller than they truly are as 'real' events are excluded. Thus, thresholding should be applied with reason and caution. So how can a reasonable detection threshold for a given sample be determined? Controls are key. As with any immuno-labeling approach, positive and negative controls should be performed to demonstrate the specificity of antibodies. With appropriate controls, it is possible to derive a detection threshold above which only specific labeling should be observed. Appropriate controls include samples in which the primary antibody incubation has been omitted so a sample exposed only to secondary antibody can be observed. From such a control, the non-specific binding of a secondary antibody can be discerned. In a well-prepared sample, this should result in a much smaller event count per image, and a lower mean number of photons per pixel when compared to the primary and secondary incubated sample. The detection threshold of the image can then be set so that non-specific labeling can be eliminated. The same detection threshold should then be used when imaging the primary and secondary incubated sample to enhance confidence that 'noise' is eliminated. Further controls include those testing the specificity of the primary antibody. In transfected cells, if the labeled protein is not natively expressed in the cell line, then a simple test of the primary antibody specificity is to perform the labeling procedure on untransfected cells. To demonstrate primary antibody specificity in primary cells, a genetic knockout of the labeled protein is the gold standard. Detection thresholds may also be set using these primary antibody control experiments. In the absence of the target antigen, any fluorescence emission is non-specific. As with the secondary only controls, with a good primary antibody, less events per image should be observed and therefore, over the same number of frames, a lower mean number of photons per pixel should be evident. The detection threshold can thus be set to a level that will eliminate non-specific background staining due to off-target primary antibody binding. For complete transparency, it is suggested that authors should present images with their thresholding parameters clearly stated and perhaps with a link to the raw data in an online depository.
The investigator must also remain cognizant that multiple secondary antibodies can bind to a single primary antibody so a 1:1 primary:secondary binding ratio should not be assumed. Furthermore, commercial secondary antibodies are commonly conjugated to multiple fluorophores, for example, the secondary antibodies employed in this protocol are noted by the manufacturer to be conjugated to 2-8 fluorophores. As a work-around to this, a fluorophore can be directly conjugated to a primary antibody. Spectrophotometry can then be used to determine the average number of fluorophores per primary antibody. Several commercially available kits are available to perform this conjugation process. However, even if a 1:1 stoichiometry is achieved, the fluorophore molecule itself can create further overcounting problems due to the blinking and reversible switching of fluorophores. In practice, this means that a fluorophore may cycle several times between the ground and excited state emitting clusters of photons as it does so. These photons may be detected in several neighboring pixels and lead to overestimation of cluster size. Other investigators have addressed this issue by choosing to combine fluorescent emissions clustered over short periods of time and space16,17. This is a somewhat empirical approach that still does not eliminate the possibility that the same fluorophore could cycle back to the dark state for an extended period, then reverse to a cycling/emitting state once more and be judged as a second molecule. Therefore, the number of molecules in a cluster cannot be calculated based solely on the cluster size. At the moment, there is no perfect solution to these issues and, therefore, the use of super-resolution imaging in conjunction with another technique such as step-wise photobleaching can give an approximate estimate of the number of molecules per cluster. Relevant controls to increase confidence in cluster area measurements include comparing cluster sizes of known monomeric proteins (e.g., CD86) and known dimeric proteins (e.g., CTLA-4) to those of the protein of interest as suggested by Fricke et al.18.
In summary, in the present article, a straightforward immunolabeling protocol is set forth describing the preparation of fixed samples for super-resolution imaging. In addition, some common pitfalls in image acquisition and analysis are discussed. As super-resolution imaging becomes more commonplace, it may become necessary for journals to set forth a new set of guidelines to avert inappropriate manipulation of these complex images/localization maps. Super-resolution microscopy has added a powerful new tool to the toolbox of cell biologists and biophysicists and has already made an extraordinary impact on our understanding of cellular architecture and molecular organization.
Disclosures
The authors have no competing interests to disclose.
Acknowledgments
This work was supported by a grant from the AHA to R.E.D. (15SDG25560035). Authors would like to acknowledge Dr. Fernando Santana for use of his Leica SR GSD 3D microscope, and Dr. Johannes Hell for kindly providing the FP1 antibody.
References
- Willig KI, Rizzoli SO, Westphal V, Jahn R, Hell SW. STED microscopy reveals that synaptotagmin remains clustered after synaptic vesicle exocytosis. Nature. 2006;440(7086):935–939. doi: 10.1038/nature04592. [DOI] [PubMed] [Google Scholar]
- Gustafsson MG. Nonlinear structured-illumination microscopy: wide-field fluorescence imaging with theoretically unlimited resolution. Proc Natl Acad Sci U S A. 2005;102(37):13081–13086. doi: 10.1073/pnas.0406877102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betzig E, et al. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313(5793):1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- Hell SW, Kroug M. Ground-state-depletion fluorscence microscopy: A concept for breaking the diffraction resolution limit. JAppl Phys B. 1995;60(5):495–497. [Google Scholar]
- Bretschneider S, Eggeling C, Hell SW. Breaking the diffraction barrier in fluorescence microscopy by optical shelving. Phys Rev Lett. 2007;98(21):218103. doi: 10.1103/PhysRevLett.98.218103. [DOI] [PubMed] [Google Scholar]
- Flynn JM, Santana LF, Melov S. Single cell transcriptional profiling of adult mouse cardiomyocytes. J Vis Exp. 2011. p. e3302. [DOI] [PMC free article] [PubMed]
- Davare MA, Horne MC, Hell JW. Protein phosphatase 2A is associated with class C L-type calcium channels (Cav1.2) and antagonizes channel phosphorylation by cAMP-dependent protein kinase. J Biol Chem. 2000;275(50):39710–39717. doi: 10.1074/jbc.M005462200. [DOI] [PubMed] [Google Scholar]
- Dempsey GT, Vaughan JC, Chen KH, Bates M, Zhuang X. Evaluation of fluorophores for optimal performance in localization-based super-resolution imaging. Nat Methods. 2011;8(12):1027–1036. doi: 10.1038/nmeth.1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chozinski TJ, Gagnon LA, Vaughan JC. Twinkle, twinkle little star: photoswitchable fluorophores for super-resolution imaging. FEBS Lett. 2014;588(19):3603–3612. doi: 10.1016/j.febslet.2014.06.043. [DOI] [PubMed] [Google Scholar]
- Dempsey GT, et al. Photoswitching mechanism of cyanine dyes. J Am Chem Soc. 2009;131(51):18192–18193. doi: 10.1021/ja904588g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahidiazar L, Agronskaia AV, Broertjes J, van den Broek B, Jalink K. Optimizing Imaging Conditions for Demanding Multi-Color Super Resolution Localization Microscopy. PLoS One. 2016;11(7):e0158884. doi: 10.1371/journal.pone.0158884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovesny M, Krizek P, Borkovec J, Svindrych Z, Hagen GM. ThunderSTORM: a comprehensive ImageJ plug-in for PALM and STORM data analysis and super-resolution imaging. Bioinformatics. 2014;30(16):2389–2390. doi: 10.1093/bioinformatics/btu202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeraraghavan R, Gourdie RG. Stochastic optical reconstruction microscopy-based relative localization analysis (STORM-RLA) for quantitative nanoscale assessment of spatial protein organization. Mol Biol Cell. 2016;27(22):3583–3590. doi: 10.1091/mbc.E16-02-0125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ries J, Kaplan C, Platonova E, Eghlidi H, Ewers H. A simple, versatile method for GFP-based super-resolution microscopy via nanobodies. Nat Methods. 2012;9(6):582–584. doi: 10.1038/nmeth.1991. [DOI] [PubMed] [Google Scholar]
- Dixon RE, et al. Graded Ca2+/calmodulin-dependent coupling of voltage-gated CaV1.2 channels. Elife. 2015;4 doi: 10.7554/eLife.05608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annibale P, Vanni S, Scarselli M, Rothlisberger U, Radenovic A. Identification of clustering artifacts in photoactivated localization microscopy. Nat Methods. 2011;8(7):527–528. doi: 10.1038/nmeth.1627. [DOI] [PubMed] [Google Scholar]
- Nan X, et al. Single-molecule superresolution imaging allows quantitative analysis of RAF multimer formation and signaling. Proc Natl Acad Sci U S A. 2013;110(46):18519–18524. doi: 10.1073/pnas.1318188110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricke F, Beaudouin J, Eils R, Heilemann M. One, two or three? Probing the stoichiometry of membrane proteins by single-molecule localization microscopy. Sci Rep. 2015;5:14072. doi: 10.1038/srep14072. [DOI] [PMC free article] [PubMed] [Google Scholar]