

Abstract
Mutants are invaluable genetic resources for gene function studies. To generate mutant collections, three types of mutagens can be utilized, including biological such as T-DNA or transposon, chemical such as ethyl methanesulfonate (EMS), or physical such as ionization radiation. The type of mutation observed varies depending on the mutagen used. For ionization radiation induced mutants, mutations include deletion, duplication, or rearrangement. While T-DNA or transposon-based mutagenesis is limited to species that are susceptible to transformation, chemical or physical mutagenesis can be applied to a broad range of species. However, the characterization of mutations derived from chemical or physical mutagenesis traditionally relies on a map-based cloning approach, which is labor intensive and time consuming. Here, we show that a high-density genome array-based comparative genomic hybridization (aCGH) platform can be applied to efficiently detect and characterize copy number variations (CNVs) in mutants derived from fast neutron bombardment (FNB) mutagenesis in Medicago truncatula, a legume species. Whole genome sequence analysis shows that there are more than 50,000 genes or gene models in M. truncatula. At present, FNB-induced mutants in M. truncatula are derived from more than 150,000 M1 lines, representing invaluable genetic resources for functional studies of genes in the genome. The aCGH platform described here is an efficient tool for characterizing FNB-induced mutants in M. truncatula.
Keywords: Genetics, Issue 129, Medicago truncatula, fast neutron bombardment mutants, array-based comparative genomic hybridization, copy number variation, mutation detection, mutation
Introduction
Legumes (Fabaceae) are the third largest family of flowering plants, with many economically important species such as soybean (Glycine max) and alfalfa (Medicago sativa). Legume plants can interact with nitrogen-fixing soil bacteria, generally called Rhizobia to develop root nodules in which the atmospheric dinitrogen is reduced to ammonia for use by the host plant. As such, cultivation of legume crops requires little input of nitrogen fertilizers and thus contributes to sustainable agriculture. Legume crops produce leaves and seeds with high protein content, serving as excellent forage and grain crops. However, cultivated legume species generally have complex genome structures, making functional studies of genes that play key roles in legume-specific processes cumbersome. Medicago truncatula has been widely adopted as a model species for legume studies primarily because (1) it has a diploid genome with a relatively small haploid genome size (~550 Mbp); (2) plants can be stably transformed for gene functional studies; and (3) it is closely related to alfalfa (M. sativa), the queen of forages, and many other economically important crops for translational studies. Recently, the genome sequence of M. truncatula cv Jemalong A17 has been released1,2. Annotation of the genome shows that there are more than 50,000 predicted genes or gene models in the genome. To determine the function of most of the genes in the M. truncatula genome is a challenging task. To facilitate functional studies of genes, a comprehensive collection of mutants in the range of over 150,000 M1 lines has been generated using fast neutron bombardment (FNB) mutagenesis in M. truncatula cv Jemalong A173,4. Fast neutron, a high energy ionization mutagen, has been used in generating mutants in many plant species including Arabidopsis5,6, rice (Oryza sativa)7, tomato (Solanum lycopersicum), soybean (Glycine soja; G. max)8,9, barley (Hordeum vulgare), and Lotus japonicus10. A large portion of mutations derived from FNB mutagenesis are due to DNA deletions that range in size from a few base pairs to mega base pairs9,11. Many phenotype-associated genes have been successfully identified and characterized4,12,13,14,15,16,17,18,19. Previously, molecular cloning of the underlying genes from FNB mutants relied on a map-based approach, which is time consuming and limits the number of mutants to be characterized at the molecular level. Recently, several complimentary approaches including transcript-based methods, genome tiling array-based comparative genomic hybridization (CGH) for DNA copy number variation detection, and whole genome sequencing, have been employed to facilitate the characterization of deletion mutants in diverse organisms including animals and plants20,21,22,23,24,25,26,27,28,29,30,31.
To facilitate the characterization of FNB mutants in M. truncatula, a whole-genome array-based comparative genomic hybridization (CGH) platform has been developed and validated. As reported in animal systems, the array-based CGH platform allows detection of copy number variations (CNVs) at the whole genome level in M. truncatula FNB mutants. Furthermore, lesions can be confirmed by PCR and deletion borders can be identified by sequencing. Overall, the array CGH platform is an efficient and effective tool in identifying lesions in M. truncatula FNB mutants. Here, the array CGH procedure and PCR characterization of deletion borders in an M. truncatula FNB mutant are illustrated.
The following protocol provides experimental steps and information about reagents, equipment and analysis tools for researchers who are interested in carrying out whole genome array-based comparative genomic hybridization (CGH) analysis of copy number variations in plants. As an example, Medicago truncatula FN6191 mutant was used to identify deletion regions and candidate genes associated with mutant phenotypes. M. truncatula FN6191 mutant, originally isolated from a fast neutron bombardment-induced deletion mutant collection32 (see Table of Materials), exhibited a hyper-nodulation phenotype after inoculation with the soil bacterium, Sihorhizobium meliloti Sm1021, in contrast to wild type plants.
Protocol
NOTE: Figure 1 shows the five steps for the array CGH protocol. They are: 1) Preparation of plant materials; 2) Isolation of high quality DNA samples; 3) Labeling and purification of DNA samples; 4) Hybridization, washing, and scanning of whole genome arrays; and 5) CGH data analysis. M. truncatula whole genome tiling arrays contain a total of 971,041 unique oligo probes targeting more than 50,000 genes or gene models in the genome (See Table of Materials). The unique probes are spaced approximately every 150 base pairs (bp) in exonic regions and 261 bp in intronic regions of the M. truncatula genome.
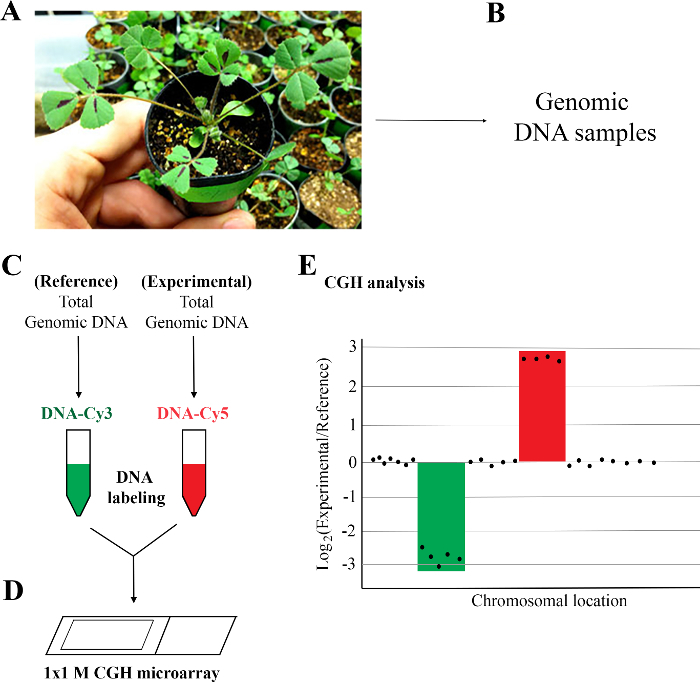
1. Preparation of Plant Materials
Scarify wild type (WT; M. truncatula cv Jemalong A17) and FN6191 mutant seeds with concentrated sulfuric acid for 8 min (see Table of Materials). Remove and discard sulfuric acid to a liquid waste container.
Rinse seeds with autoclaved deionized water three times.
Surface sterilize seeds with 20% bleach solution for 10 min.
Rinse seeds with autoclaved deionized water three times.
Store seeds at 4 °C for 3 days. After vernalization, transfer and germinate seeds in soil for one week in a growth chamber with 16 h/8 h light/dark cycle and 150 µE/m2/s light intensity.
Transplant seedlings to 1 gallon pots and grow them in a greenhouse with 16 h/8 h light/dark cycle, 150 µE/m2/s light intensity for three to four weeks. Collect young leaf samples from the plants for DNA isolation.
2. Isolation of High Quality DNA Samples
Take 1 g of young leaf tissue from a single plant and freeze it in liquid nitrogen immediately.
Grind frozen leaf tissue in liquid nitrogen with a mortar and pestle to fine powder.
Isolate genomic DNA with a DNA isolation kit (see Table of Materials).
Resuspend purified DNA samples in 50 µL of autoclaved double deionized H2O (ddH2O) or 1x TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 7.0).
Measure DNA concentrations with a spectrophotometer (see Table of Materials).
Evaluate 260 nm/280 nm and 260 nm/230 nm ratios of DNA samples. NOTE: High quality DNA samples should have a 260 nm/280 nm ratio ≥1.8 and a 260 nm/230 nm ratio ≥2.0.
Further evaluate DNA samples by running them in 1% agarose gels. NOTE: High quality DNA samples should have high molecular weight and should not be contaminated by RNA. Ideally, the final concentration of DNA samples should be ~150 ng/µL.
3. DNA Labeling and Purification
Dilute 1 µg of genomic DNA samples with ddH2O to a final volume of 20 µL in a 500 µL centrifuge tube.
Add 5 µL of a random primer (see Table of Materials) to the tube.
Vortex the tube briefly and put it into a thermocycler to incubate and denature DNA at 98 °C for 10 min.
Take the tube out of the thermocycler and put it immediately on ice water to chill for 5 min.
Prepare two labeling mixes as shown in Table 1.
Add 25 µL of labeling mixes 1 and 2 to reference DNA and sample DNA tubes, respectively.
Mix the labeling mix and DNA by pipetting three times and spin the tubes briefly.
Put the tubes into a thermocycler to incubate at 37 °C for 2 h and then at 65 °C for 20 min to inactivate the exo-Klenow enzyme (see Table of Materials).
Add 430 µL of 1x TE buffer to each tube.
Mix the contents and spin the tubes briefly.
Transfer the solution in the tube into a purification column with a 2 mL collection tube (see Table of Materials).
Centrifuge the purification column at 14,000 x g for 10 min.
Discard the pass through solution.
Add 480 µL 1x TE buffer to each column.
Centrifuge the column again at 14,000 x g for 10 min.
Discard the pass-through solution.
Transfer the labeled DNA sample that remains at the bottom portion of the column to a new centrifuge tube.
Measure the volume of the labeled DNA sample using a pipette and bring the final volume to 80 µL with 1x TE buffer.
Measure the concentration of the labeled DNA sample using a spectrophotometer (see Table of Materials).
Mix an equal amount of labeled sample DNA and reference DNA and bring the final volume to 160 µL with 1x TE buffer.
Add 256 µL of 2x hybridization buffer, 50 µL of human Cot-1 DNA and 50 µL of 10× aCGH blocking agent (see Table of Materials) and mix them well by pipetting for three times.
Spin the tubes briefly and put them into a thermocycler to incubate at 98 °C for 10 min.
Incubate the tubes at 37 °C for 20 min.
4. Hybridization, Washing, and Scanning of the Genome Arrays
Pre-warm the hybridization oven to 67 °C at least 4 h before hybridization starts.
Place a gasket slide onto the base of a hybridization chamber.
Load 490 µL of hybridization solution from Step 3.23 onto the gasket slide.
Cover the gasket slide with a Medicago truncatula genome microarray chip to form a hybridization chamber.
Put on the hybridization chamber covers and tighten the chamber firmly with clamps.
Put the assembled hybridization chamber into the hybridization oven and incubate at 67 °C for 40 - 48 h.
Prepare three slide washing dishes: two with 250 mL washing buffer I kept at room temperature, and one with washing buffer II kept at 37 °C.
Prepare two slide washing jars: one with 70 mL acetonitrile and one with 70 mL stabilization and drying solution; keep both in a fume hood at room temperature (see Table of Materials).
Remove the hybridization chamber from the oven and remove the chamber clamps and cover.
Move the microarray chip and gasket slide to a slide washing dish with wash buffer I and separate the chip from the cover slide.
Move the microarray chip to the second slide washing dish with wash buffer I and gently circulate the solution with a stir bar on a magnetic stir plate at room temperature for 5 min.
Transfer the microarray chip to the third slide washing dish with pre-warmed washing buffer II and wash it gently with a stir bar on a magnetic stir plate at 37 °C for 1 min.
Transfer the microarray chip to a slide washing jar with acetonitrile in a fume hood and wash it for 30 s.
Transfer the microarray chip to a slide washing jar with stabilization and drying solution and wash it for 30 s.
Take out the chip slowly and dry it for 1 min.
Scan the microarray chip using a scanner (see Table of Materials) under 2 µm resolution. Set parameters to save multiple images and whole slide scans. Set channels 1 and 2 gains to 100% and cancel the auto gain.
5. CGH Data Analysis
Extract the Cy3 and Cy5 fluorescence intensities for each probe on the array using scanning software (see Table of Materials).
Use segmentation analysis of the software to detect copy number variations (CNVs) between the sample DNA and reference DNA. Use a threshold of the segment mean log2 sample/reference ratio ± 2.5 standard deviations for determining CNVs.
Inspect the distribution of log2 sample/reference ratios across the whole genome using a signal mapping software (see Table of Materials).
Representative Results
Figure 2 shows the distribution of normalized log2 ratios of mutant versus WT signals across the whole genome. Analysis of CGH data revealed an approximate 22 kb deletion on chromosome 4 that encompasses the entire SUNN gene33 and several other annotated genes in FN6191 mutant (Figure 2, Figure 3). The candidate deleted region was covered by 73 consecutive probes on the array with the mean normalized log2 ratios less than -2.5 (Supplemental Table 1). An inspection of deletion borders suggested that the putative deletion in this mutant is flanked by probes located between coordinates 22,313,168 and 22,334,934 on chromosome 4 (Figure 3). To confirm the deletion borders, PCR primers were designed to be approximately 1,500 bp apart from the predicted deletion borders (FN6191-DB-F: GCTAGCAAGGGTCTGCGCAAGTT; FN6191-DB-R: GTATCGAGAAGGTCTTATAGCAGC) and PCR amplification was performed with these primers and the following parameters: 94 °C, 5 min; 94 °C, 45 s, 55 °C, 30 s, 72 °C, 1.5 min; for 35 cycles; 72 °C, 10 min and then 10 °C indefinitely. As expected, a single PCR product of approximately 1.5 kb was successfully amplified from the FN6191 mutant, but not WT (Figure 4A). Based on the M. truncatula genome release v3.5, the size of the deletion in FN 6191 was estimated to be 26.67 kb (Figure 4B). The CGH results showed that no other abnormal segments are present in the FN6191 mutant genome (Figure 2).
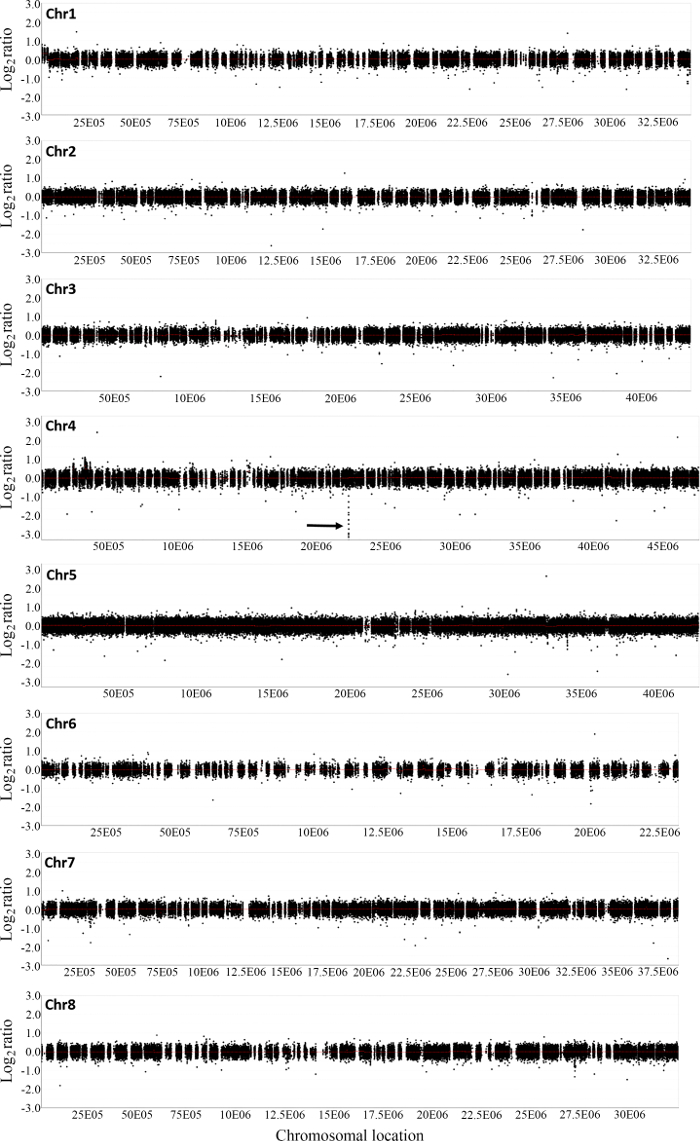
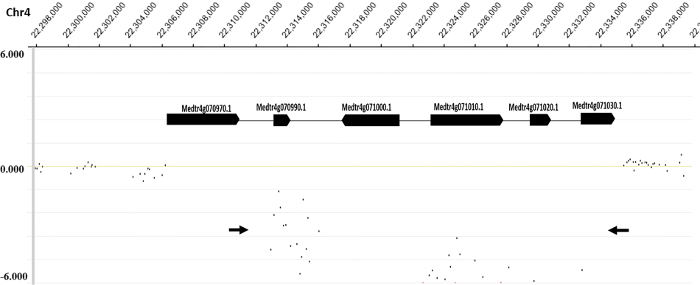
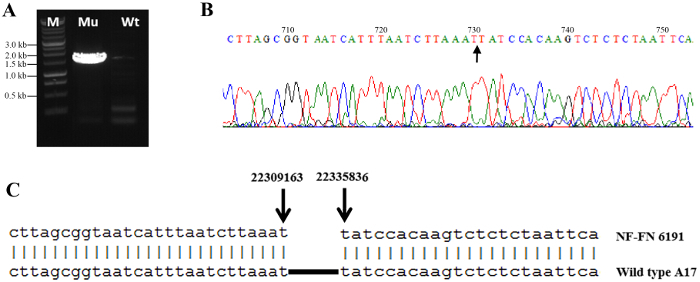
Figure 1: Workflow of array comparative genomic hybridization (aCGH) analysis of M. truncatula mutant. (A) Preparation of plant materials: M. truncatula wild type (Jemalong A17) and FN6191 mutant were grown for three to four weeks in greenhouse. (B) Isolation of high quality genomic DNA: One gram of young leaves from single plants was used to isolate genomic DNA. DNA quality was determined using a spectrophotometer and gel electrophoresis. (C) DNA labeling and purification. Genomic DNAs of reference and experimental samples were labeled with Cyanine 3 (Cy3) and Cy5 using a DNA labeling kit and purified using a purification column. (D) Hybridization, washing, and scanning of 1 x 1 M M. truncatula CGH array. (E) CGH analysis: Log2 ratios of experimental/reference signals that are less than or equal to -2.5 or greater than or equal to 2.5 are considered as putative deletions and duplications, respectively. Please click here to view a larger version of this figure.
Figure 2: Array based comparative genomic hybridization analysis of copy number variations in M. truncatula hyper nodulation mutant FN6191. Shown are log2 mutant/wild type ratios of probes on all eight chromosomes. A consecutive 73-probe region on chromosome 4 was identified as the deleted region in the mutant (arrow). Please click here to view a larger version of this figure.
Figure 3. Identification of the deleted region in M. truncatula FN6191 mutant. A close-up view of the region on chromosome 4, in which 73 microarray probes exhibited significantly reduced log2 mutant/wild type ratios, and six mapped genes including SUNN (Medtr4g070970). Arrows indicate the location and direction of primers for PCR amplification of deletion borders. Please click here to view a larger version of this figure.
Figure 4: Confirmation of deletion borders in M. truncatula FN6191 mutant. (A) PCR amplification of deletion borders: A 1.5 kb product was amplified from the FN6191 mutant using primers flanking the deletion borders (lane 1; Mu). The same set of primers did not amplify any products from WT (M. truncatula Jemalong A17) due to a size limitation of PCR (lane 2; Wt). Lane M, 1 kb ladder. (B) Sequences of the deletion junction in the FN6191 mutant. An arrow denotes the deletion junction. (C) Sequencing results showed that the deletion borders (arrows) are located at the coordinates 22309163 and 22335836 on chromosome 4. Please click here to view a larger version of this figure.
| Labeling mix 1 | Per reaction x sample # (μL) | Labeling mix 2 | Per reaction x reference # (μL) |
| ddH2O | 6.0 x | ddH2O | 6.0 x |
| 5x Buffer | 10.0 x | 5x Buffer | 10.0 x |
| 10x dNTPs | 5.0 x | 10x dNTPs | 5.0 x |
| Cyanine 5 | 3.0 x | Cyanine 3 | 3.0 x |
| Exo-Klenow | 1.0 x | Exo-Klenow | 1.0 x |
| Total | 25 x | Total | 25 x |
Table 1: Labeling Mixes.
Supplemental Table 1: Probe sequences deleted in M. truncatula FN6191 mutant. Please click here to download this file.
Discussion
We have developed an array-based CGH platform for the detection and characterization of fast neutron bombardment (FNB)-induced mutants in M. truncatula cv. Jemalong A17. To demonstrate the use of the array CGH method in detecting gene mutations, we performed aCGH analysis of the mutant FN6191, which exhibited a hyper-nodulation phenotype in contrast to wild type plants, when inoculated with S. meliloti Sm1021. For segmentation analysis, a segment was deemed significant if the log2 ratio mean of the probes within the segment was above the upper threshold or below the lower threshold for that given array comparison. The upper threshold for each comparison was determined to be the log2 ratio value of the 95th percentile of all data points. The lower threshold for each comparison was determined to be the log2 ratio value of the 5th percentile of all data points24.
Our analysis showed that significant copy number changes can be determined by retrieving segments with an average normalized log2 ratio greater than the average by 2.5 SD (duplication) or less than the average by 2.5 SD (deletion). Based on CGH analysis, we detected a deletion region with an estimated size of 22 kb on chromosome 4, encompassing the SUNN gene33. Sequencing results confirmed that the deletion size was 26.67 kb based on M. truncatula genome release v3.5. It has been shown that SUNN encodes a CLV1-like leucine-rich repeat receptor kinase that controls the nodule number in M. truncatula33. Our CGH results suggest that FN6191 is a new SUNN allele. Based on these results, we reasoned that the CGH method coupled with phenotypic and genetic analysis can be successfully used to rapidly identify candidate genes in FNB deletion mutants. Over the past several years, we have used this platform in identifying and characterizing numerous FNB mutants for gene functional studies in M. truncatula34,35. We have generated a database of copy number variations (CNVs) associated with FNB mutant lines36. In this database, deleted sequences from CGH analyses of over 100 confirmed symbiotic nodulation mutants have been mapped to the M. truncatula genome. A blast server has been set up to facilitate the search for deleted sequences in the mutant collection. The development and further expansion of the deletion database would be highly valuable for functional genomics research using M. truncatula FNB mutant resources.
The aCGH protocol has five major steps. Although all steps are important and should be carried out carefully as described earlier, the followings are especially critical: (1) DNA samples should not be degraded and should not be contaminated by RNA during the preparation step; (2) Test and reference DNA samples should be equally well prepared so that the labeled samples are of the same high quality; (3) Labeled DNA samples should not be exposed to a high intensity of light and a high level of ozone; and (4) it is important to keep Washing Buffer II at 37 °C during the washing step.
The coverage of the current M. truncatula genome array is primarily on the exonic regions and less on the intronic regions, and not at all in intergenic regions. The coverage in the latter regions can be improved if lesions in these regions are important for mutant phenotypes. On the other hand, the current probe design has less power in identifying small mutations and single nucleotide polymorphisms (SNPs). This limitation can be overcome by using CGH+SNP microarray design37. Overall, the genome array-based comparative genomic hybridization (aCGH) platform is a powerful tool for the analysis of copy number and structural variations in M. truncatula FNB mutants.
Disclosures
The authors declare no competing financial interests.
Acknowledgments
Funding of this work is provided in part by a grant from NSF Plant Genome Research (IOS-1127155).
References
- Tang H, et al. An improved genome release (version Mt4.0) for the model legume Medicago truncatula. BMC Genomics. 2014;15:312. doi: 10.1186/1471-2164-15-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young ND, et al. The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature. 2011;480(7378):520–524. doi: 10.1038/nature10625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Li G, Chen R. Fast neutron bombardment (FNB) induced deletion mutagenesis for forward and reverse genetic studies in plants. In: da Silva JT, editor. Floriculture, Ornamental and Plant Biotechnology: Advances and Topical Issues. 1st ed. Isleworth, UK: Global Science Books; 2006. pp. 629–639. [Google Scholar]
- Rogers C, Wen J, Chen R, Oldroyd G. Deletion-based reverse genetics in Medicago truncatula. Plant Physiol. 2009;151(3):1077–1086. doi: 10.1104/pp.109.142919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso JM, et al. Five components of the ethylene-response pathway identified in a screen for weak ethylene-insensitive mutants in Arabidopsis. Proc Natl Acad Sci U S A. 2003;100(5):2992–2997. doi: 10.1073/pnas.0438070100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverstone AL, Ciampaglio CN, Sun T. The Arabidopsis RGA gene encodes a transcriptional regulator repressing the gibberellin signal transduction pathway. Plant Cell. 1998;10(2):155–169. doi: 10.1105/tpc.10.2.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Lassner M, Zhang Y. Deleteagene: a fast neutron deletion mutagenesis-based gene knockout system for plants. Comp Funct Genomics. 2002;3(2):158–160. doi: 10.1002/cfg.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolon YT, et al. Phenotypic and genomic analyses of a fast neutron mutant population resource in soybean. Plant Physiol. 2011;156(1):240–253. doi: 10.1104/pp.110.170811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Men AE, et al. Fast Neutron Mutagenesis of Soybean (Glycine soja L.) Produces a Supernodulating Mutant Containing a Large Deletion in Linkage Group H. Genome Letters. 2002;1(3):147–155. [Google Scholar]
- Hoffmann D, Jiang Q, Men A, Kinkema M, Gresshoff PM. Nodulation deficiency caused by fast neutron mutagenesis of the model legume Lotus japonicus. J Plant Physiol. 2007;164(4):460–469. doi: 10.1016/j.jplph.2006.12.005. [DOI] [PubMed] [Google Scholar]
- Li X, et al. A fast neutron deletion mutagenesis-based reverse genetics system for plants. Plant J. 2001;27(3):235–242. doi: 10.1046/j.1365-313x.2001.01084.x. [DOI] [PubMed] [Google Scholar]
- Bourcy M, et al. Medicago truncatula DNF2 is a PI-PLC-XD-containing protein required for bacteroid persistence and prevention of nodule early senescence and defense-like reactions. New phytol. 2013;197(4):1250–1261. doi: 10.1111/nph.12091. [DOI] [PubMed] [Google Scholar]
- Chen J, et al. Control of dissected leaf morphology by a Cys(2)His(2) zinc finger transcription factor in the model legume Medicago truncatula. Proc Natl Acad Sci U S A. 2010;107(23):10754–10759. doi: 10.1073/pnas.1003954107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge L, et al. Increasing seed size and quality by manipulating BIG SEEDS1 in legume species. Proc Natl Acad Sci U S A. 2016;113(44):12414–12419. doi: 10.1073/pnas.1611763113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalo P, et al. Nodulation signaling in legumes requires NSP2, a member of the GRAS family of transcriptional regulators. Science. 2005;308(5729):1786–1789. doi: 10.1126/science.1110951. [DOI] [PubMed] [Google Scholar]
- Oldroyd GE, Long SR. Identification and characterization of nodulation-signaling pathway 2, a gene of Medicago truncatula involved in Nod actor signaling. Plant Physiol. 2003;131(3):1027–1032. doi: 10.1104/pp.102.010710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J, et al. Regulation of compound leaf development in Medicago truncatula by fused compound leaf1, a class M KNOX gene. Plant Cell. 2011;23(11):3929–3943. doi: 10.1105/tpc.111.089128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujimoto Y, et al. Arabidopsis TOBAMOVIRUS MULTIPLICATION (TOM) 2 locus encodes a transmembrane protein that interacts with TOM1. EMBO J. 2003;22(2):335–343. doi: 10.1093/emboj/cdg034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, et al. A nodule-specific protein secretory pathway required for nitrogen-fixing symbiosis. Science. 2010;327(5969):1126–1129. doi: 10.1126/science.1184096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejjani BA, Shaffer LG. Application of array-based comparative genomic hybridization to clinical diagnostics. J Mol Diagn. 2006;8(5):528–533. doi: 10.2353/jmoldx.2006.060029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerson JJ, Cardoso-Moreira M, Borevitz JO, Long M. Natural selection shapes genome-wide patterns of copy-number polymorphism in Drosophila melanogaster. Science. 2008;320(5883):1629–1631. doi: 10.1126/science.1158078. [DOI] [PubMed] [Google Scholar]
- Gong JM, et al. Microarray-based rapid cloning of an ion accumulation deletion mutant in Arabidopsis thaliana. Proc Natl Acad Sci U S A. 2004;101(43):15404–15409. doi: 10.1073/pnas.0404780101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guryev V, et al. Distribution and functional impact of DNA copy number variation in the rat. Nat Genet. 2008;40(5):538–545. doi: 10.1038/ng.141. [DOI] [PubMed] [Google Scholar]
- Haun WJ, et al. The composition and origins of genomic variation among individuals of the soybean reference cultivar Williams 82. Plant Physiol. 2011;155(2):645–655. doi: 10.1104/pp.110.166736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Infante JJ, Dombek KM, Rebordinos L, Cantoral JM, Young ET. Genome-wide amplifications caused by chromosomal rearrangements play a major role in the adaptive evolution of natural yeast. Genetics. 2003;165(4):1745–1759. doi: 10.1093/genetics/165.4.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MR, Maydan JS, Flibotte S, Moerman DG, Baillie DL. Oligonucleotide Array Comparative Genomic Hybridization (oaCGH) based characterization of genetic deficiencies as an aid to gene mapping in Caenorhabditis elegans. BMC Genomics. 2007;8:402. doi: 10.1186/1471-2164-8-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakshmi B, et al. Mouse genomic representational oligonucleotide microarray analysis: detection of copy number variations in normal and tumor specimens. Proc Natl Acad Sci U S A. 2006;103(30):11234–11239. doi: 10.1073/pnas.0602984103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra RM, et al. A Ca2+/calmodulin-dependent protein kinase required for symbiotic nodule development: Gene identification by transcript-based cloning. Proc Natl Acad Sci U S A. 2004;101(13):4701–4705. doi: 10.1073/pnas.0400595101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios G, et al. Characterization of hemizygous deletions in citrus using array-comparative genomic hybridization and microsynteny comparisons with the poplar genome. BMC Genomics. 2008;9:381. doi: 10.1186/1471-2164-9-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skvortsov D, Abdueva D, Stitzer ME, Finkel SE, Tavare S. Using expression arrays for copy number detection: an example from E. coli. BMC Bioinformatics. 2007;8:203. doi: 10.1186/1471-2105-8-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner JD, et al. Quantitative trait locus mapping and DNA array hybridization identify an FLM deletion as a cause for natural flowering-time variation. Proc Natl Acad Sci U S A. 2005;102(7):2460–2465. doi: 10.1073/pnas.0409474102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi J, Chen Y, Nakashima J, Wang SM, Chen R. Medicago truncatula esn1 defines a genetic locus involved in nodule senescence and symbiotic nitrogen fixation. Mol Plant Microbe Interact. 2013;26(8):893–902. doi: 10.1094/MPMI-02-13-0043-R. [DOI] [PubMed] [Google Scholar]
- Schnabel E, Journet EP, de Carvalho-Niebel F, Duc G, Frugoli J. The Medicago truncatula SUNN gene encodes a CLV1-like leucine-rich repeat receptor kinase that regulates nodule number and root length. Plant Mol Biol. 2005;58(6):809–822. doi: 10.1007/s11103-005-8102-y. [DOI] [PubMed] [Google Scholar]
- Horvath B, et al. Loss of the nodule-specific cysteine rich peptide, NCR169, abolishes symbiotic nitrogen fixation in the Medicago truncatula dnf7 mutant. Proc Natl Acad Sci U S A. 2015;112(49):15232–15237. doi: 10.1073/pnas.1500777112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, et al. An antimicrobial peptide essential for bacterial survival in the nitrogen-fixing symbiosis. Proc Natl Acad Sci U S A. 2015;112(49):15238–15243. doi: 10.1073/pnas.1500123112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble Research Institute. Medicago truncatula Mutant Database. 2017. Available from: https://medicago-mutant.noble.org/mutant/FNB.php.
- Burton R. SNP genotyping with the next generation of CGH microarray. MLO Med Lab Obs. 2013;45(7) [PubMed] [Google Scholar]