

Summary
The system L neutral amino acid transporter (LAT; LAT1, LAT2, LAT3, or LAT4) has multiple functions in human biology, including the cellular import of S-nitrosothiols (SNOs), biologically active derivatives of nitric oxide (NO). SNO formation by hemoglobin within red blood cells (RBC) has been studied, but the conduit whereby a SNO leaves the RBC remains unidentified. Here we hypothesized that SNO export by RBCs may also depend on LAT activity, and investigated the role of RBC LAT in modulating SNO-sensitive RBC-endothelial cell (EC) adhesion. We used multiple pharmacologic inhibitors of LAT in vitro and in vivo to test the role of LAT in SNO export from RBCs and in thereby modulating RBC-EC adhesion. Inhibition of human RBC LAT by type-1-specific or nonspecific LAT antagonists increased RBC-endothelial adhesivity in vitro, and LAT inhibitors tended to increase post-transfusion RBC sequestration in the lung and decreased oxygenation in vivo. A LAT1-specific inhibitor attenuated SNO export from RBCs, and we demonstrated LAT1 in RBC membranes and LAT1 mRNA in reticulocytes. The proadhesive effects of inhibiting LAT1 could be overcome by supplemental L-CSNO (S-nitroso-L-cysteine), but not D-CSNO or L-Cys, and suggest a basal anti-adhesive role for stereospecific intercellular SNO transport. This study reveals for the first time a novel role of LAT1 in the export of SNOs from RBCs to prevent their adhesion to ECs. The findings have implications for the mechanisms of intercellular SNO signaling, and for thrombosis, sickle cell disease, and post-storage RBC transfusion, when RBC adhesivity is increased.
Keywords: transfusion, nitric oxide, S-nitrosothiol, erythrocyte, cysteine
Introduction
Mechanisms of red blood cell (RBC) adhesion to the endothelium have been investigated in sickle cell disease, retinal vein thrombosis, diabetes mellitus, malarial infection, and conventional RBC storage, among other settings. A variety of RBC and endothelial cell (EC) adhesion receptors, including ICAM-4 (intracellular adhesion molecule 4, also known as the Landsteiner-Weiner (LW) adhesion molecule), phosphatidyl serine, laminin, and integrins have been causally implicated. Nitric oxide (NO) and its derivatives can influence RBC-EC adhesion, reminiscent of the ability of NO to prevent or reverse leukocyte adhesion to endothelial cells (1). Specifically, inhibition of NOS production promoted endothelial adhesion of RBCs, and NO donors attenuated the excess adhesivity of RBCs in diabetes, malarial infection, sickle cell disease, and blood storage (2-6). The mechanistic basis of the antiadhesive effect of NO or its derivatives is uncertain, but could reflect inhibitory protein S-nitrosylation or downregulation (6) of either an adhesion receptor or a downstream element of signal transduction. Indeed, the depressed levels of NO derivatives, including S-nitrosothiols (SNOs), in banked RBCs might contribute to the excess adhesivity of banked RBCs. But how SNOs are exported from the RBC remains undetermined. The deoxygenation-linked interaction between RBC SNO-hemoglobin (SNO-Hb) and anion exchanger 1 (AE1) localizes a SNO group to the inner RBC membrane (7), but the conduit by which SNOs are subsequently exported from the RBC membrane is still unknown.
To test whether and how normal human RBCs can export anti-adhesive SNO, we studied the system L neutral amino acid transporter, type 1 (LAT1) which in several cell types imports SNO across the cell membrane (8, 9). (LAT1 and LAT2, belonging to the LAT family, are heterodimeric proteins, each comprising a constant heavy chain 4F2hc (SLC3A2) and a variable light chain. LAT1 is a heterodimer comprised of the heavy chain and a light chain encoded by SLC7A5 (together also known as the glycoprotein CD98). LAT1 is known to transport large (hence “system L”), neutral amino acids, including branched-chain and aromatic amino acids, i.e., leucine, isoleucine, valine, phenylalanine, tyrosine, and tryptophan (10). LATs can also transport L-DOPA, the thyroid hormones T3 and T4, and the system-L-specific inhibitor 2-aminobicyclo-(2,2,1)-heptane-2-carboxylic acid (BCH) (11) in a gradient-dependent manner (12). Finally, LATs have been shown to import the small SNO, S-nitroso-L-cysteine (CSNO), in endothelial cells, lung epithelial cells, vascular smooth muscle cells, RBCs, and other cell types (8, 9, 13, 14). LAT1 operates in a sodium-independent manner as an obligatory 1-to-1 exchanger (i.e., as an antiporter), with the affinity for influx about 100-fold greater than for efflux (11). Although the role of LAT in the import of SNO has been studied in various cells and tissues, the role of SNO export by LAT has not been tested in any cell type to our knowledge. We therefore tested the role of SNO export and LAT1 in preventing the basal adhesion of RBCs to endothelial cells. We propose a novel function of LAT1, namely a role in the cellular export of CSNO and modulation of RBC-endothelial adhesion in both in vitro and in vivo settings.
Methods
Chemicals and reagents
Chemicals and reagents were purchased from Sigma except where otherwise noted.
Red blood cell (RBC) preparation and labeling
Using an IRB-approved protocol, fresh whole blood was obtained aseptically from healthy adult human donors via Transfusion Services of Duke University Medical Center. The RBCs were separated by centrifugation and washed three times with isotonic PBS, pH 7.4, containing Ca++ and Mg++ [0.01 % (w/v) each, required for adhesion studies], labeled with fluorescent dye PKH26, and allowed to incubate for 3 minutes. Isotonic PBS containing 1% (w/v) BSA was added, and after one minute, the labeled cells were washed three times in PBS while minimizing exposure to light. Finally 5–10 µL of labeled RBCs were suspended in 3 mL of PBS for adhesion assays.
HUVEC culture
Human umbilical vein endothelial cells (HUVECs, Lonza Clonetics Endothelial Cell Systems) were expanded to passage four or five. The cells were then plated on glass slides precoated with a 2% gelatin solution and grown to confluence in Lonza Clonetics EBM-2 media with EGM-2 supplements and fungizone. The cells were incubated at 37°C at 5% CO2.
RBC-EC adhesion assays
The HUVECs, grown to confluence on glass slides, were placed in a graduated-height flow chamber. The height was measured at 7 different points along the chamber. The RBC sample was introduced to the chamber at a rate of 1.5 mL min−1. The RBCs were then allowed to dwell for 5–10 minutes, and the number of cells at each location (height) was recorded. After the static phase, 5–10 minutes of fluidic flow was conducted with PBS at flow rates calculated to produce the desired shear stress range of ~1–10 dynes/cm2. Following exposure to flow, the number of adhered cells at each location was counted. Shear stress and percent adhesion were calculated at each height. The method has been described previously (15). No effort was made either to superoxygenate or deoxygenate (expose to hypoxic gas) the perfusion medium or flow chamber.
Pharmacological LAT1 inhibition
We studied the effects of multiple, mechanistically distinct inhibitors of the transport function of LAT. Leu is a substrate for LAT and, at high concentration, competitively blocks the transport of other substrates. BCH, a synthetic amino acid analog, also acts competitively. Where indicated, fresh RBCs were incubated for 45 mins at 25 degrees C (25°C) with the non-subtype-specific LAT inhibitors BCH (10 mM) or L-Leucine (10 mM) (Figs. 1 and 6). In RBC-EC adhesion experiments using BCH and Leu, the competitive inhibitor was also included in the perfusion medium because we reasoned that when diluted the agent may be ineffective. Alternatively, the RBCs were incubated at 37°C with the LAT1-specific, noncompetitive inhibitor JPH-203 (5–50 nM), provided by J-Pharma and Drs. Wempe and Endou (16) in PBS/0.05% DMSO (Figs. 2, 4 and 5). Paired aliquots of RBCs from a given human blood donor were used for control and inhibitor-exposed preparations. Following RBC exposure to any of the LAT(1) inhibitors, cells were washed twice with 25°C PBS. In Figs. 1 and 2, the respective inhibitor was also included in the perfusion medium. In another series of experiments, the adhesion to HUVECs of RBCs treated first (or not) with JPH-203 (Figure 5) was assessed in the subsequent presence or absence of the small SNO, S-nitroso-L-cysteine (L-CSNO) at two different concentrations, 10 or 240 µM, in the perfusion medium. Alternatively, either unmodified L-Cys or the D isomer of CSNO, D-CSNO synthesized using D-Cys, was used (10 µM). In another series of experiments, fresh human RBCs exposed either to JPH-203 (1 uM) or its vehicle (0.1% DMSO) were suspended in PBS (with 0.1 mM DTPA) in order to measure hypoxia-induced SNO export (Fig. 4). The RBC suspension (20% hematocrit) was placed in a rotating glass tonometer and exposed to a hypoxic gas mixture (2% O2/5% CO2/balance N2) at 37 deg. C. After 25 mins, the suspension was sampled, gently centrifuged, and the supernatant (extracellular space) was assayed for NO adducts using mercury-coupled photolysis-chemiluminescence (MPC) as described (17). Briefly, supernatant exposed to inorganic mercury (HgCl2) or not was injected into a photolysis device. NO bound to Cys thiols (SNO), heme, or nitrosamine is detected downstream by ozone-based chemiluminescence of the evolved NO. SNO content, which is mercury labile, is the difference in NO content in the presence and absence of mercury. Nitrite is inefficiently detected by this technique. Supernatants were aliquoted and assayed with or without an additional filtration (10 kDa) step to discriminate low-mass (i.e., non-protein) from other SNOs.
Figure 1. Influence of treatment of RBCs with the LAT inhibitors BCH and L-leucine (L-Leu) on RBC-EC adhesion as a function of shear stress.
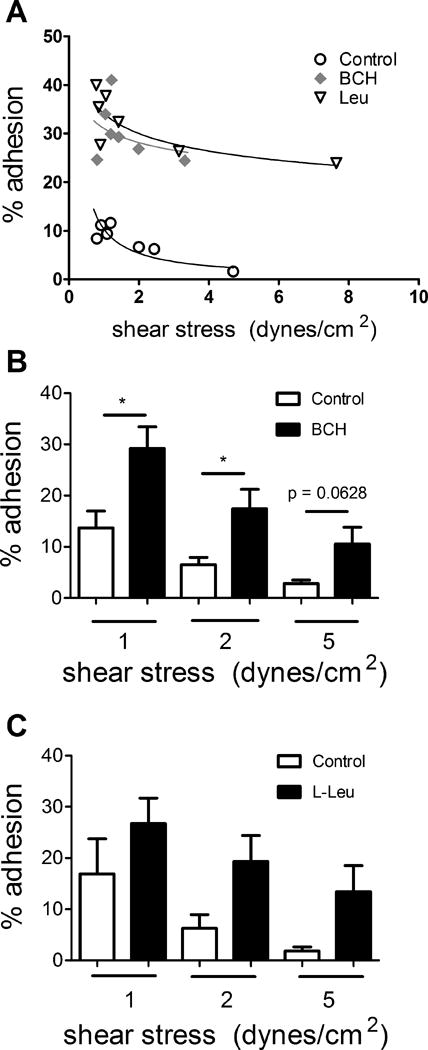
The adhesion of fresh human RBCs to HUVECs exposed or not to BCH or L-Leu was quantified in customized flow chambers at varying shear stress. One set of individual adhesion curves is shown in (A) and the mean results from n=4–9 experiments at 1, 2, or 5 dynes/cm2 (B, BCH) and (C, L-Leu). *, p<0.05 vs. Con (vehicle control) by paired t-test. #, p=0.0002 for comparison of full Con vs. BCH curves; ##, p = 0.0021 for Con vs. Leu curves (two-way ANOVA).
Figure 6. Pulmonary responses in nude mice to transfusion of fresh human RBCs treated or not with the LAT inhibitors L-Leu or BCH.
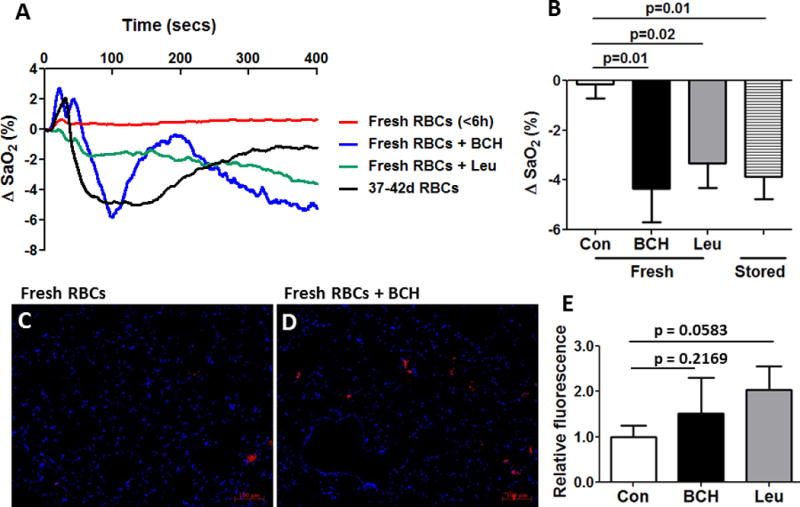
(A) Individual blood oxygenation tracings and (B) mean (± SEM; n=4) data are shown. The indicated p-values are for paired t-tests. Responses to RBCs stored 37–42 days are also shown. RBC transfusate exposure to either LAT inhibitor tended to promote transfused-RBC sequestration within the lungs, as seen in fluorescence photomicrographs (C,D); mean data for fluorescence intensity are summarized in (E). p=0.2169 for Con vs. BCH, and p=0.0583 for Con vs. Leu by paired t-test. n=4 pairs in lung images and n=6 pairs for blood oxygenation means.
Figure 2. Influence of the LAT1-specific inhibitor JPH-203 on RBC-EC adhesion as a function of shear stress.
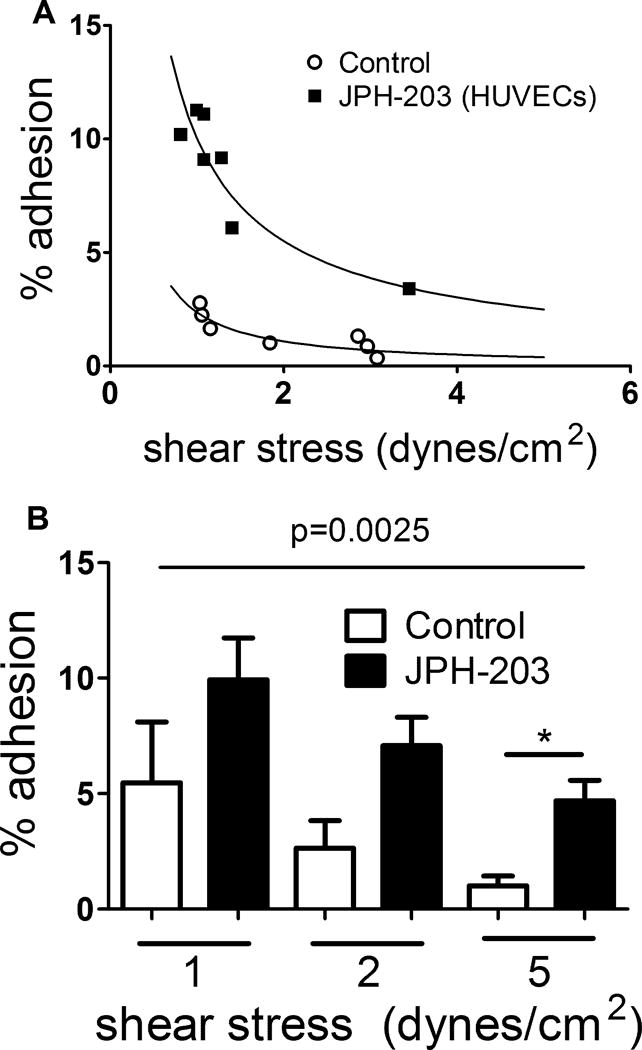
The adhesion to HUVECs of fresh human RBCs exposed or not to JPH-203 (50 nM) was quantified in customized flow chambers at varying shear stress. JPH-203 (50 nM) was also included in the perfusion medium. One set of individual adhesion curves is shown in (A) and the mean results from n=5 experiments each at 1, 2, or 5 dynes/cm2 in (B). *, p<0.05 vs. Con (vehicle control) by paired t-test; comparison of Con vs. JPH-203 curves is by two-way ANOVA.
Figure 4.
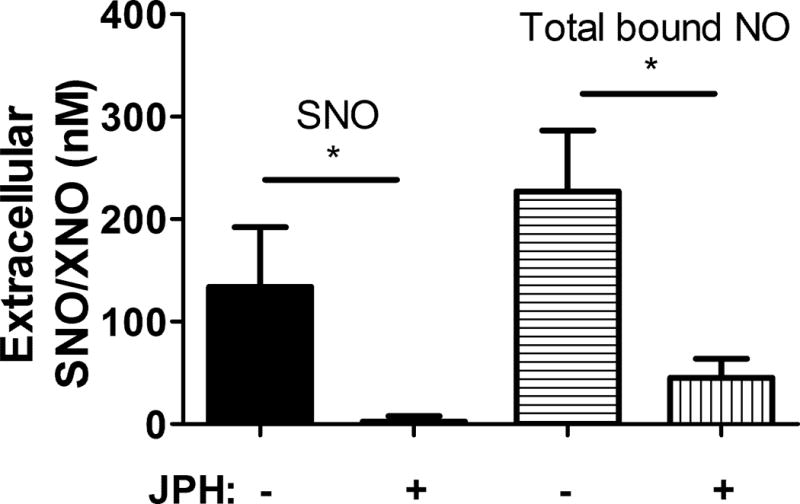
RBC treatment with the LAT1 inhibitor JPH-203 blocked SNO export in hypoxia, and significantly attenuated the export of all NO adducts. n=4. *, p<0.01 by paired T-test.
Figure 5. Influence on RBC-EC adhesion of treatment of RBCs alone with the LAT1-specific inhibitor JPH-203 (50 nM) with or without the addition of L-CSNO, D-CSNO, or L-Cys.
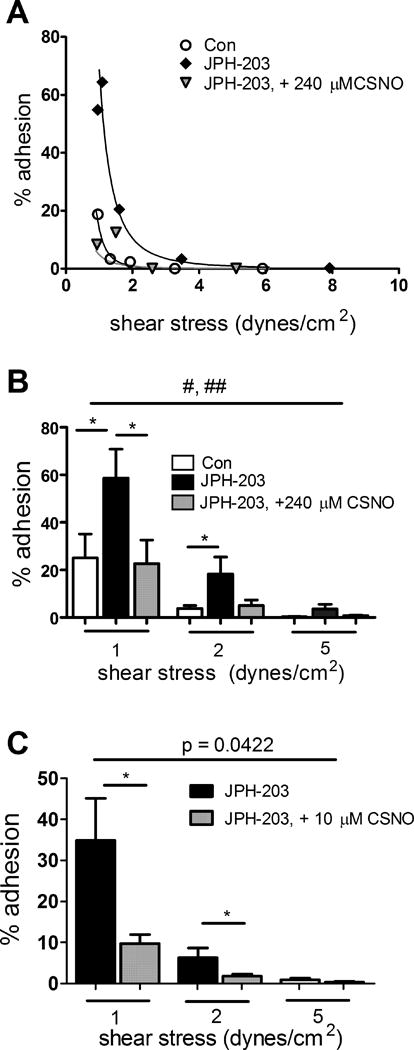
One set of individual adhesion curves is shown in (A) and the mean results from n=9 experiments using 240 µM L-CSNO at 1, 2, or 5 dynes/cm2 in (B). *, p<0.05 for the pair-wise comparison at the indicated shear stress (paired t-test). #, p=0.0004 for comparison of full Con vs. JPH curves (by two-way ANOVA); ##, p=0.0024 for JPH vs JPH + L-CSNO curves. (C-E), RBCs were treated with JPH-203, 50 nM, with or without the subsequent addition of (C), L-CSNO (n=6 pairs); (D), D-CSNO (n=6 pairs); or (E), L-Cys (n=6 pairs), 10 µM final in each case. With L-CSNO only, the differences at 1 and 2 dynes/cm2 were significant (*, p<0.05, paired t-test), and the curves differed significantly as indicated by two-way ANOVA.
RT-PCR for LAT1 mRNA
Total RNA was isolated using TRIZOL (Invitrogen). Reverse transcription was performed using iScript cDNA synthesis kit (Bio-Rad) according to the manufacturer’s instructions with forward primer 5′-atcgggaagggtgatgtgt-3′ and reverse primer 5′-ggacagggtggtgaagtagg-3′. Conditions for amplification were: 94°C for 3 min, 60°C for 1 min and 72°C for 2 min; then 36 cycles of 94°C for 1 min, 60°C for 1 min and 72°C for 2 min; and a 10-min final extension at 72°C. The product was separated on a 2% agarose gel and stained with GelRed Nucleic Acid ethidium bromide (Phenix).
LAT1 Western blotting
RBCs membranes were prepared in RIPA lysis buffer. 50 µg was electrophoresed on 10% SDS-PAGE gels, and the proteins were electrophoretically transferred to PVDF membrane, followed by blocking in 5% nonfat milk in TBS-T for 1 hour at room temperature. The primary antibody, anti-LAT1 mouse monoclonal (Santa Cruz Biotechnology, #sc-374232) was applied at 1:200 dilution overnight at 4°C. The blot was washed with TBS-T and incubated with the secondary antibody, goat anti-mouse IgG horseradish peroxidase (Santa Cruz, #sc-2005) at 1:5000 dilution. Enhanced chemiluminescence (ECL, Amersham) was used according to the manufacturer’s instructions.
Murine model of RBC transfusion
All experiments in mice were approved in advance by our IACUC. Nude mice were anesthetized with intraperitoneal sodium pentobarbital (100 mg/kg). Tracheostomy was performed, and the mice were mechanically ventilated (7 mcL/g body weight, 150 breaths/min) as we have described (18). After equilibration, mice were transfused over ~15 seconds via a tail-vein catheter with either fresh, untreated human RBCs (<6 hours old) (control group) or with fresh RBCs, treated with a LAT inhibitor (BCH or Leucine). Paired aliquots of RBCs from a given human blood donor were used for control and inhibitor-exposed RBC preparations. The transfusate was a 35%-hematocrit RBC suspension in PBS, and 10 µL/g body weight was administered. The transfusate RBCs in each case were pre-labeled with the fluorescent cell membrane dye PKH-26 using the method described previously (18). Percent arterial Hb oxygen saturation (SaO2) was measured by mouse-specific pulse oximetry (MouseOx, Oakmont, PA) before and at multiple time points after transfusion. The mice were sacrificed and the lungs were perfused with PBS and harvested. Frozen sections of the lungs were obtained. Red fluorescence signal, corresponding to the PKH-26 dye applied to the RBC transfusates, was measured in sections of harvested lung, reflecting the sequestration of RBCs.
Statistical analyses
Paired t-tests or analysis of variance were used, as indicated. A P<0.05 was considered statistically significant. Comparison of the curves of percent adhesion vs. shear stress was performed using the Extra sum-of-squares F test after curve fitting to a power model. Numbers of experiments given are the number of unique biological samples studied.
Results
RBCs treated with the LAT inhibitors BCH or L-Leu adhere excessively to HUVECs
The adhesion to HUVECs of RBCs incubated with the LAT inhibitors L-leucine (L-Leu) or BCH, and suspended in perfusion medium containing Leu or BCH (to minimize the egress from treated RBCs of these competitive inhibitors), respectively, was consistently higher compared to that of RBCs incubated only with vehicle. One set of individual curves, illustrating the relationship between shear stress and the % adhesion to HUVECs of fresh RBCs exposed or unexposed to the LAT antagonists, is shown in Fig. 1A. A power regression fit was used to model the adhesion to HUVECs of fresh RBCs treated or not with various inhibitors of LAT1. The overall shear stress-vs.-percent adhesion curves differed significantly between Con and BCH conditions (p<0.0005). The effects of BCH were also statistically significant (p < 0.05) at shear stresses of 1 and 2 dynes/cm2 but did not reach significance at 5 dynes/cm2 (Fig. 1B; p= 0.0628; n=9). The adhesion to HUVECs of RBCs exposed to L-Leu was typically greater than of vehicle-treated RBCs, but the results were not statistically significant (n=4; Fig. 1C). The overall shear stress-vs.-adhesion curves differed significantly (p=0.0021) between Con and Leu conditions.
RBC LAT1 inhibition using JPH-203 promotes RBC-EC adhesion
LAT1-specific inhibition has been demonstrated previously in other cell types using a compound, JPH-203 (19). We therefore studied the adhesion of fresh vehicle- or JPH-203-treated RBCs to HUVECs. RBC-EC adhesion was increased by pretreatment of RBCs with JPH-203 (and its inclusion in the perfusion medium) relative to untreated RBCs, illustrated by the individual adhesion curves in Fig 2A. The overall shear stress-vs.-adhesion curves for JPH-203-exposed RBCs differed significantly from that for control RBCs (p=0.0243). The result was also statistically significant at a shear stress of 5 dynes/cm2, with trends toward significantly increased adhesion at lower shear stresses as shown in Fig 2B. We measured supernatant free Hb as an index of hemolysis of exposed RBCs, and found that neither JPH-203 (0.104 ±0.0141% (mean ± SEM) hemolysis) nor L-Leu (0.101 ± 0.028%) induced significantly greater hemolysis than did the vehicle (0.087 ± 0.005%).
LAT1 is present in human RBCs and LAT1 mRNA in human reticulocytes
We performed Western blotting of RBC membrane preparations and stained with an anti-LAT1 antibody sc-374232. As illustrated in Fig. 3, LAT1 is present in RBCs and its abundance was unaltered by conventional RBC storage. To determine whether mRNA for LAT1 is present in RBC precursors, we isolated reticulocytes from the blood of sickle cell disease patients using anti-CD71 antibodies and magnetic beads, and performed RT-PCR on lysates. The results demonstrate the presence of LAT1 message (expected size 249; Fig. 3B).
Figure 3. LAT1 is present in fresh and stored human RBCs.
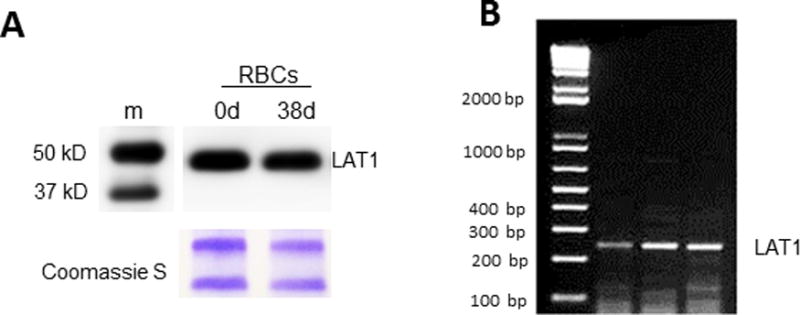
(A), Immunoblots detecting LAT1 (Santa Cruz Ab sc-374232) in human RBC membranes stored either 0 days or 38 days. Lower panel shows Coomassie protein staining after gel was stripped (loading control). Results are typical of n=4 experiments. (B), mRNA in reticulocytes. Magnetic beads and CD71 (transferrin) antibodies were used to isolate reticulocytes and RT-PCR was performed for LAT1 message. LAT1 mRNA is detected in reticulocytes. Left lane, PCR ladder; other lanes are sample replicates.
The LAT1 inhibitor JPH-203 inhibits SNO export by RBCs
In order to test whether SNO export from RBCs can be inhibited by a LAT inhibitor, we measured SNO in the supernatant of RBCs during hypoxia. SNO export was consistently detected from control (vehicle-treated) RBCs, but was minimal from JPH-203-exposed RBCs (Fig. 4). Quantitatively similar results were seen regardless of whether or not (data not shown) the supernatant was first filtered (10 kDa cutoff) to remove the protein fraction, suggesting that the exported species is a low-mass SNO. The total bound NO (Including SNO, heme-NO, nitrosamine) appearing in the supernatant also fell significantly and to a comparable extent when RBCs were pre-exposed to the LAT1 inhibitor (Fig. 4).
L-CSNO, but not L-Cys or D-CSNO, rescues the proadhesive effect of JPH-203
When RBCs alone (and not the HUVECs) were exposed to JPH-203, increases in RBC-EC adhesion were again seen across a range of shear stresses. Figure 5A shows results from one set of individual of experiments, and Figure 5B illustrates the mean data at shear stresses of 1, 2, or 5 dynes/cm2. The proadhesive effect of JPH-203 reached statistical significance at shear stresses of 1 and 2 dynes/cm2 but not at 5 dynes/cm2. The overall shear stress-vs.-adhesion curves for JPH-203-exposed RBCs again differed significantly from that for Con RBCs (p<0.0024 for comparison of fitted curves). Additional experiments were designed to confirm the mechanistic basis of the effects of LAT1 inhibition. We reasoned that if the proadhesive effect of exposing RBCs to LAT inhibitors does in fact involve the inhibition of SNO export by the RBC, then applying L-CSNO (S-nitroso-L-cysteine, a small SNO) to the fluid used in the flow chamber and perfusing the endothelial cells should overcome the increased adhesivity associated with LAT inhibition. The proadhesive effect of JPH-203 was in fact significantly attenuated by CSNO at 1 dyne/cm2 but not at 2 or 5 dynes/cm2 (Fig. 5B). This effect was also observed at a much lower concentration, 10µM CSNO, with a significant attenuation of RBC adhesion to endothelium at 1 and 2 dynes/cm2 (Fig. 5C).
In order to test whether the ability of L-CSNO to surmount the proadhesive effect of JPH-203 was stereospecific (and therefore consistent with movement through an amino acid transporter), we also studied the ability of the corresponding D isomer (D-CSNO synthesized from D-Cys) to attenuate increases in RBC-EC adhesion after RBC exposure to JPH-203. The results indicate that D-CSNO did not significantly attenuate the adhesion of JPH-treated RBCs (Fig. 5D). Additionally, we tested whether the ability of L-CSNO to reverse the increased adhesion of RBCs treated with JPH-203 might be attributable to an effect of L-Cys (liberated by in vitro decomposition of L-CSNO), by measuring the ability of L-Cys to overcome the increased adhesivity of JPH-203-treated fresh human RBCs. L-Cys, in contrast to L-CSNO, did not significantly attenuate the adhesion of JPH-treated RBCs to HUVECs (Fig. 5E). Taken together, these findings indicate that the ability of L-CSNO to reverse adhesion of JPH-203-exposed RBCs is stereospecific, consistent with involvement of an amino acid transporter, and is not the result of the decomposition of L-CSNO to parent L-Cys.
In experiments examining the stability of CSNO in the presence of varying concentrations of JPH-203, CSNO decay over time was not accelerated (but rather was slowed) in the presence of JPH-203 (Fig. S1), suggesting that the CSNO effect we observed in adhesion assays was not the result of a degradative interaction between CSNO and JPH-203. Taken together, these results suggest that the mechanism by which these LAT(1) inhibitors promote RBC adhesion to ECs is at least in part through inhibiting the export by RBCs of a small-molecule SNO (such as CSNO).
Leu or JPH-203 does not alter the deformability of RBCs
In order to determine whether the effects of the LAT(1) inhibitors might be mediated in part through rheological changes, we assayed, using an ektacytometer, the deformability of RBCs treated with Leu, JPH-203, or dilute DMSO (0.1%), the vehicle for JPH-203. Deformability did not differ significantly (Fig. S2), and was similar to that of healthy human RBCs in reports we and others have published previously (18, 20).
Transfusion of RBCs treated with a LAT inhibitor promotes lung morbidity
In order to determine whether SNO transport by RBC LAT may prevent the endothelial adhesion of transfused RBCs, we transfused human RBCs stored <6 hours and exposed to vehicle, L-Leu (10 mM), or BCH (10 mM) while measuring systemic blood oxygenation, reflecting gas exchange by the lung. Alternatively, we studied the adhesion of human RBCs stored for 35–42-days after transfusion into nude mice. All RBCs were first exposed to a cell-surface fluorescent red label. Transfusion of RBCs stored for 35–42 days, but not those stored <6 hours, elicited decreases in blood oxygenation (Fig 6A and 6B) and increased red fluorescence within the lung (Fig 6C, 6D, 6E), consistent with endothelial adhesion or “sequestration” of RBCs (18). Similarly, pre-transfusion incubation with LAT inhibitors depressed SaO2 relative to untreated fresh-RBC controls (Fig 6A and 6B), with RBC sequestration tending to increase (particularly with Leu exposure) within the lungs (Fig. 6C–E). The trend approached but did not reach statistical significance (p=0.2169 for BCH, p=0.0583 for Leu).
Discussion
Our results show that inhibition of the amino acid transporter LAT promoted increased RBC-EC adhesion in vitro, tended to do so within the lung microvasculature in vivo, and depressed blood oxygenation in vivo. RBC exposure to either subtype-1-specific or nonspecific LAT inhibitors, in concentrations known to inhibit canonical LAT activity (21, 22), promoted RBC-EC adhesion. The LAT1-specific, noncompetitive inhibitor JPH-203 also inhibited SNO export from RBCs. Although the extent of baseline and post-treatment RBC-EC adhesion and the effect sizes of the inhibitors varied considerably among experimental series, the proadhesive effect of inhibiting LAT was consistent. Taken together, the results are consistent with LAT-mediated SNO export from the RBC acting to limit the adhesion of RBCs basally. These findings are the first to identify cellular SNO export as a function of LAT1, and suggest for the first time that the conduit for RBC SNO export is LAT1. Because excessive and injurious RBC adhesivity to vascular endothelial cells may contribute morbidity and mortality in settings including transfusion, atherosclerosis, thrombosis or sickle cell disease, this study may have important clinical implications (18, 23-26).
We used a well-established, non-static in vitro adhesion model in which RBCs were allowed to incubate on the surface of endothelial cells (ECs), then exposed RBC-EC preparations incrementally to a physiologically relevant range of shear stresses of fluidic flow, simulating in vivo conditions in order to produce physiologically relevant intercellular adhesion events. Strictly speaking, our in vitro experiments examine the ability of RBCs to maintain adhesion to HUVECs established under static conditions, as fluid flow is initiated and raised incrementally so as to increase the shear stress. We selected the range of shear stresses based on those encountered in the human microcirculation in the lung and elsewhere (27, 28). Postcapillary venules, where shear stress can be very low, are particularly susceptible to the adverse sequelae of RBC or leukocyte adhesion. Inasmuch as flow itself may influence the behavior of adhesion receptors, this nonstatic model is likely to be pertinent to clinical consequences of RBC adhesion. Increases in RBC adhesivity to endothelium during blood banking are well documented (18, 24, 29), but the direct role of post-transfusion RBC-EC adhesion in the morbidity or limited benefit of RBC transfusion is uncertain. Both endothelial cells (9) and RBCs are now known to contain LAT1. Our focus was on LAT1 activity in the RBC. Pharmacological LAT(1) inhibition promoted the adhesion of RBCs, and this increased adhesivity could be overcome with added L-CSNO, but not with D-CSNO or L-Cys. The stereospecificity of the (L)-CSNO rescue effect is consistent with transporter-dependent import by endothelial cells of antiadhesive L-CSNO. We did not study the effects of very low CSNO concentrations (low micromolar or submicromolar) in these experiments, and because physiological levels of CSNO are lower (30), our findings do not bear directly on signaling by CSNO. Whether close apposition of RBCs to ECs in vivo may enable high local fluxes of CSNO is unknown. The LAT inhibitors Leu and JPH-203 did not alter RBC deformability, meaning that it is unlikely that impaired flexibility could have contributed to the proadhesive effect we demonstrated in vitro and the sequestration of transfused RBCs in vivo. Together, these data suggest that EC import of SNO derived from the RBC acts to prevent RBC-EC adhesion.
Our approach to LAT(1) inhibition was multifaceted. The in vitro study used 3 chemically and mechanistically distinct known LAT inhibitors. BCH and L-Leu are competitive LAT inhibitors, and JPH-203 has been shown to inhibit LAT1 specifically, with little inhibition of LAT2 at concentrations similar to ours (16). We selected concentrations previously demonstrated to inhibit LAT activity, typically measured by the cellular uptake of 14C- or 3H-L-Leu (8, 16, 21, 31). Endothelial cell uptake of CSNO has been shown to depend on LAT activity (9). Given the need to include competitive inhibitors in the perfusion medium (to prevent their loss from RBCs), we cannot exclude that the proadhesive effect of BCH and Leu in adhesion assays was due to an effect on EC LAT. However, given the multiple inhibitors used and the general agreement in the direction of findings (albeit with unexplained variation of the statistical significance of the results) with all inhibitors, it is unlikely that the increased adhesion seen was due to an effect other than RBC LAT1 inhibition. Further supporting the assertion that the effect of increased adhesion after LAT(1) inhibition is RBC-dependent is the observation that loading the RBCs with JPH-203, a non-competitive, LAT1-specific inhibitor, potently (at 50 nM) increased adhesivity even when the inhibitor was absent from the solution bathing the ECs. We showed that LAT1 is present in healthy human RBCs and that LAT1 mRNA is present in human reticulocytes. In vivo, we found a trend toward increased RBC sequestration and significantly decreased oxygenation in the lung after transfusion of RBCs treated with LAT inhibitors, consistent with findings in mice transfused with RBCs deficient in SNO after storage (18, 20). These respiratory changes could be expected to contribute to post-transfusion lung injury and dysfunction, which are seen excessively more frequently in patients transfused with more or older RBCs in randomized clinical trials (32-34). Our findings are consistent with impaired gas exchanged due to LAT1- and SNO-sensitive RBC adhesion to ECs, but other possible mechanisms could contribute, such as disrupted vasoregulation by the treated RBCs. The alternative mechanism of impaired RBC deformability was excluded.
One limitation of our study is that we did not determine definitively the specific mechanism whereby cellular functions involving LAT1 and SNO influence adhesion. Possible mechanisms include general amino acid transport as is necessary for cellular protein synthesis, specific cell uptake of L-arginine (the substrate for NO synthesis), and LAT-mediated SNO import or export. We reason that, given the lack of new protein synthesis in the mature RBC, it is unlikely that depressed new protein synthesis via compromised amino acid uptake secondary to LAT inhibition was responsible. LAT (particularly LAT2) is capable of transporting L-arginine (35), and inhibition of its uptake could therefore in theory limit NO production by NO synthase (NOS) in the RBC or EC. However, intracellular RBC SNO content was unaltered by LAT inhibitors (data not shown). Finally, LAT1 has been demonstrated to be competent for transcellular transport of the NO derivative S-nitroso-L-cysteine (L-CSNO, also termed SNO-Cys) (8, 9). NO and its derivatives such as S-nitrosothiols (SNOs) can act as anti-adhesive molecules (1, 2, 36). We found that the proadhesive effect of the LAT1 inhibitor JPH-203 on RBCs was reversed when authentic L-CSNO (10 or 240 µM) was included in the perfusion medium. This finding strengthens the link between LAT1 and RBC SNO export and argues against a nonspecific or SNO-independent effect of the LAT antagonists. Specifically, the LAT system may play a critical role in cellular adhesion in vivo as a transporter of NO derivatives. We therefore suggest that the proadhesive effect of LAT1 inhibition arises because of suppressed transport of L-CSNO from the RBC to the endothelial cell. We did not definitively identify L-CSNO as the exported SNO, but LAT1 is known to import L-CSNO, functions as an antiporter, and as an amino acid transporter is unable to transport a larger molecule such as the tripeptide glutathione or S-nitrosoglutathione (8). Furthermore, the ability of L-CSNO to reverse the proadhesive effect of JPH-203 applied to RBCs was not replicated by equimolar D-CSNO or L-Cys. Our data provides support for the assertion that the SNO export function of the LAT1 transporter may be important in its own right, as opposed to simply passively balancing the import function. Future work is needed to examine the mechanistic basis of the antiadhesive effect of LAT1 activity and SNO transport, including whether relevant adhesion receptors are S-nitrosylated.
We show for the first time that LAT1 activity appears to be necessary for the export of endogenous SNO from human RBCs. The LAT1-specific inhibitor JPH-203 blocked the hypoxia-induced export of low-mass SNOs. Prior work has demonstrated the export of GSNO from SNO-loaded RBCs, and the export of S-nitrosylated protein disulfide isomerase (SNO-PDI) from the surface of nitrite-exposed RBCs (37, 38). However, our findings are the first to show the export of native SNO in unmodified RBCs, and its dependence on LAT1 function.
LAT1 has been shown to play a role in atherogenesis and inflammatory diseases (39), vasculoproliferative disorders (40), neoplastic disorders (40-42), and recently was shown to play a role in integrin-mediated cell adhesion (43). The LAT1-dependent modulation of RBC adhesivity that we show underscores a potential role for disturbed SNO signaling or altered LAT1 function in the morbidity and mortality associated with red blood cell (RBC) transfusion that is seen even after RBC units are stored for durations as short as a few days (1). Randomized clinical trials demonstrating no benefit, or even harm, from more liberal (vs. restrictive) RBC transfusion strategies in a variety of pediatric and adult anemic populations suggest that the therapeutic margin could be improved (20, 44). RBC storage lesions that may contribute to pathophysiology and thus disappointing clinical outcomes include excess erythrocyte adhesion (18, 24, 29), depressed erythrocytic vasoactivity, decreased deformability, increased osmotic fragility, hemolysis, microparticle accumulation, and spheroechinocyte formation (20, 45, 46). Prior work has shown that the ability of RBCs to export vasoregulatory mediators, including SNOs and ATP, declines with the duration of RBC storage (18, 20, 47). Intracellular red blood cell (RBC) S-nitrosothiols (SNOs) decline early during storage (20) and these changes could contribute to disappointing outcomes with transfusion for moderate anemia (34, 44) because RBC SNOs are required for RBC-induced vasodilation (46, 48). Although RBC SNO can be restored ex vivo after blood banking (5, 46), little is known of the effect of storage on LAT1 activity. Our findings implicating LAT1 as a conduit for the export of SNO from RBCs raise the question of whether post-storage restoration of RBC SNOs, which can improve RBC deformability and reduce adhesivity (5), might be further enhanced by measures that optimize LAT1 stability and activity. The relative efficacy of RBC SNO restoration, as opposed to or in combination with other antiadhesive approaches, remains to be determined.
What is known on this topic
Increased adhesivity of red blood cells (RBCs) to endothelium characterizes blood storage, sickle cell disease, and thrombosis. These states can also be characterized by deficiency of S-nitrosothiols (SNOs), which are nitric oxide (NO) derivatives.
Red blood cells export NO derivatives, but the SNO export conduit is unknown.
L-type amino acid transporters can import SNOs, but their role in SNO export has not been tested.
What this paper adds
A LAT1 antagonist inhibited SNO export from RBCs.
Inhibiting LAT(1) promoted RBC adhesivity to endothelial cells (ECs) in vitro, decreased blood oxygenation, and tended to increase RBC sequestration in lungs in vivo.
The increased adhesivity in vitro could be overcome with extracellular SNO. The results suggest that SNO export from RBCs acts basally to prevent RBC-EC adhesion.
Acknowledgments
We thank Dr. Hitoshi Endou (J-Pharma Inc., Japan) for the kind gift of JPH-203, and Drs. Richard Auten, Mardee Delahunty, Milena Batchvarova, and Marilyn Telen for scientific advice. We thank Dr. Richard Whorton for scientific advice and an early critical reading of the manuscript.
Funding: NIH T32 HL-098099, R01s HL-107608 and GM-113838; and VA Merit BX-003478.
Footnotes
The authors declare that no conflict of interest exists.
References
- 1.Kubes P, Suzuki M, Granger DN. Nitric oxide: an endogenous modulator of leukocyte adhesion. Proc Natl Acad Sci U S A. 1991;88(11):4651–5. doi: 10.1073/pnas.88.11.4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Serirom S, Raharjo WH, Chotivanich K, et al. Anti-adhesive effect of nitric oxide on Plasmodium falciparum cytoadherence under flow. Am J Pathol. 2003;162(5):1651–60. doi: 10.1016/S0002-9440(10)64299-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grossin N, Wautier MP, Wautier JL. Red blood cell adhesion in diabetes mellitus is mediated by advanced glycation end product receptor and is modulated by nitric oxide. Biorheology. 2009;46(1):63–72. doi: 10.3233/BIR-2009-0519. [DOI] [PubMed] [Google Scholar]
- 4.Space SL, Lane PA, Pickett CK, et al. Nitric oxide attenuates normal and sickle red blood cell adherence to pulmonary endothelium. Am J Hematol. 2000;63(4):200–4. doi: 10.1002/(sici)1096-8652(200004)63:4<200::aid-ajh7>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 5.Riccio DA, Zhu H, Foster MW, et al. Renitrosylation of banked human red blood cells improves deformability and reduces adhesivity. Transfusion. 2015;55(10):2452–63. doi: 10.1111/trf.13189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gutsaeva DR, Montero-Huerta P, Parkerson JB, et al. Molecular mechanisms underlying synergistic adhesion of sickle red blood cells by hypoxia and low nitric oxide bioavailability. Blood. 2014;123(12):1917–26. doi: 10.1182/blood-2013-06-510180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pawloski JR, Hess DT, Stamler JS. Export by red blood cells of nitric oxide bioactivity. Nature. 2001;409(6820):622–6. doi: 10.1038/35054560. [DOI] [PubMed] [Google Scholar]
- 8.Li S, Whorton AR. Identification of stereoselective transporters for S-nitroso-L-cysteine: role of LAT1 and LAT2 in biological activity of S-nitrosothiols. J Biol Chem. 2005;280(20):20102–10. doi: 10.1074/jbc.M413164200. [DOI] [PubMed] [Google Scholar]
- 9.Li S, Whorton AR. Functional characterization of two S-nitroso-L-cysteine transporters, which mediate movement of NO equivalents into vascular cells. Am J Physiol Cell Physiol. 2007;292(4):C1263–71. doi: 10.1152/ajpcell.00382.2006. [DOI] [PubMed] [Google Scholar]
- 10.Morimoto E, Kanai Y, Kim DK, et al. Establishment and characterization of mammalian cell lines stably expressing human L-type amino acid transporters. J Pharmacol Sci. 2008;108(4):505–16. doi: 10.1254/jphs.08232fp. [DOI] [PubMed] [Google Scholar]
- 11.Verrey F, Closs EI, Wagner CA, et al. CATs and HATs: the SLC7 family of amino acid transporters. Pflugers Archiv-European Journal of Physiology. 2004;447(5):532–42. doi: 10.1007/s00424-003-1086-z. [DOI] [PubMed] [Google Scholar]
- 12.Christensen HN. Role of Amino Acid Transport and Countertransport in Nutrition and Metabolism. Physiological Reviews. 1990;70(1):43–77. doi: 10.1152/physrev.1990.70.1.43. [DOI] [PubMed] [Google Scholar]
- 13.Granillo OM, Brahmajothi MV, Li S, et al. Pulmonary alveolar epithelial uptake of S-nitrosothiols is regulated by L-type amino acid transporter. Am J Physiol Lung Cell Mol Physiol. 2008;295(1):L38–43. doi: 10.1152/ajplung.00280.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Broniowska KA, Zhang Y, Hogg N. Requirement of transmembrane transport for S-nitrosocysteine-dependent modification of intracellular thiols. J Biol Chem. 2006;281(45):33835–41. doi: 10.1074/jbc.M603248200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zennadi R, Hines PC, De Castro LM, et al. Epinephrine acts through erythroid signaling pathways to activate sickle cell adhesion to endothelium via LW-alphavbeta3 interactions. Blood. 2004;104(12):3774–81. doi: 10.1182/blood-2004-01-0042. [DOI] [PubMed] [Google Scholar]
- 16.Wempe MF, Rice PJ, Lightner JW, et al. Metabolism and pharmacokinetic studies of JPH203, an L-amino acid transporter 1 (LAT1) selective compound. Drug metabolism and pharmacokinetics. 2012;27(1):155–61. doi: 10.2133/dmpk.dmpk-11-rg-091. [DOI] [PubMed] [Google Scholar]
- 17.McMahon TJ, Stamler JS. Concerted nitric oxide/oxygen delivery by hemoglobin. Methods Enzymol. 1999;301:99–114. doi: 10.1016/s0076-6879(99)01073-3. [DOI] [PubMed] [Google Scholar]
- 18.Zhu H, Zennadi R, Xu BX, et al. Impaired adenosine-5′-triphosphate release from red blood cells promotes their adhesion to endothelial cells: a mechanism of hypoxemia after transfusion. Crit Care Med. 2011;39(11):2478–86. doi: 10.1097/CCM.0b013e318225754f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oda K, Hosoda N, Endo H, et al. L-type amino acid transporter 1 inhibitors inhibit tumor cell growth. Cancer science. 2010;101(1):173–9. doi: 10.1111/j.1349-7006.2009.01386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bennett-Guerrero E, Veldman TH, Doctor A, et al. Evolution of adverse changes in stored RBCs. Proc Natl Acad Sci U S A. 2007;104(43):17063–8. doi: 10.1073/pnas.0708160104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hayashi K, Jutabha P, Kamai T, et al. LAT1 is a central transporter of essential amino acids in human umbilical vein endothelial cells. J Pharmacol Sci. 2014;124(4):511–3. doi: 10.1254/jphs.13255sc. [DOI] [PubMed] [Google Scholar]
- 22.Ritchie JW, Taylor PM. Role of the System L permease LAT1 in amino acid and iodothyronine transport in placenta. Biochem J. 2001;356(Pt 3):719–25. doi: 10.1042/0264-6021:3560719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Malek AM, Alper SL, Izumo S. Hemodynamic shear stress and its role in atherosclerosis. JAMA. 1999;282(21):2035–42. doi: 10.1001/jama.282.21.2035. [DOI] [PubMed] [Google Scholar]
- 24.Anniss AM, Sparrow RL. Variable adhesion of different red blood cell products to activated vascular endothelium under flow conditions. Am J Hematol. 2007;82(6):439–45. doi: 10.1002/ajh.20837. [DOI] [PubMed] [Google Scholar]
- 25.Wautier MP, HÉRon E, Picot J, et al. Red blood cell phosphatidylserine exposure is responsible for increased erythrocyte adhesion to endothelium in central retinal vein occlusion. Journal of Thrombosis and Haemostasis. 2011;9(5):1049–55. doi: 10.1111/j.1538-7836.2011.04251.x. [DOI] [PubMed] [Google Scholar]
- 26.Wautier MP, El Nemer W, Gane P, et al. Increased adhesion to endothelial cells of erythrocytes from patients with polycythemia vera is mediated by laminin alpha5 chain and Lu/BCAM. Blood. 2007;110(3):894–901. doi: 10.1182/blood-2006-10-048298. [DOI] [PubMed] [Google Scholar]
- 27.van Bommel J, de Korte D, Lind A, et al. The effect of the transfusion of stored RBCs on intestinal microvascular oxygenation in the rat. Transfusion. 2001;41(12):1515–23. doi: 10.1046/j.1537-2995.2001.41121515.x. [DOI] [PubMed] [Google Scholar]
- 28.Lipowsky HH, Sheikh NU, Katz DM. Intravital microscopy of capillary hemodynamics in sickle cell disease. J Clin Invest. 1987;80(1):117–27. doi: 10.1172/JCI113036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luk CS, Gray-Statchuk LA, Cepinkas G, et al. WBC reduction reduces storage-associated RBC adhesion to human vascular endothelial cells under conditions of continuous flow in vitro. Transfusion. 2003;43(2):151–6. doi: 10.1046/j.1537-2995.2003.00310.x. [DOI] [PubMed] [Google Scholar]
- 30.Foster MW, McMahon TJ, Stamler JS. S-nitrosylation in health and disease. Trends Mol Med. 2003;9(4):160–8. doi: 10.1016/s1471-4914(03)00028-5. [DOI] [PubMed] [Google Scholar]
- 31.Brahmajothi MV, Tinch BT, Wempe MF, et al. Hyperoxia inhibits nitric oxide treatment effects in alveolar epithelial cells via effects on L-type amino acid transporter-1. Antioxid Redox Signal. 2014;21(13):1823–36. doi: 10.1089/ars.2013.5664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koch C, Li L, Figueroa P, et al. Transfusion and pulmonary morbidity after cardiac surgery. Ann Thorac Surg. 2009;88(5):1410–8. doi: 10.1016/j.athoracsur.2009.07.020. [DOI] [PubMed] [Google Scholar]
- 33.Koch CG, Li L, Sessler DI, et al. Duration of red-cell storage and complications after cardiac surgery. N Engl J Med. 2008;358(12):1229–39. doi: 10.1056/NEJMoa070403. [DOI] [PubMed] [Google Scholar]
- 34.Hebert PC, Wells G, Blajchman MA, et al. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. Transfusion Requirements in Critical Care Investigators, Canadian Critical Care Trials Group. N Engl J Med. 1999;340(6):409–17. doi: 10.1056/NEJM199902113400601. [DOI] [PubMed] [Google Scholar]
- 35.Rotoli BM, Closs EI, Barilli A, et al. Arginine transport in human erythroid cells: discrimination of CAT1 and 4F2hc/y+LAT2 roles. Pflugers Archiv: European journal of physiology. 2009;458(6):1163–73. doi: 10.1007/s00424-009-0692-9. [DOI] [PubMed] [Google Scholar]
- 36.Fox-Robichaud A, Payne D, Hasan SU, et al. Inhaled NO as a viable antiadhesive therapy for ischemia/reperfusion injury of distal microvascular beds. J Clin Invest. 1998;101(11):2497–505. doi: 10.1172/JCI2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jia L, Bonaventura C, Bonaventura J, et al. S-nitrosohaemoglobin: a dynamic activity of blood involved in vascular control. Nature. 1996;380(6571):221–6. doi: 10.1038/380221a0. [DOI] [PubMed] [Google Scholar]
- 38.Kallakunta VM, Slama-Schwok A, Mutus B. Protein disulfide isomerase may facilitate the efflux of nitrite derived S-nitrosothiols from red blood cells. Redox biology. 2013;1:373–80. doi: 10.1016/j.redox.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takabe W, Kanai Y, Chairoungdua A, et al. Lysophosphatidylcholine enhances cytokine production of endothelial cells via induction of L-type amino acid transporter 1 and cell surface antigen 4F2. Arteriosclerosis Thrombosis and Vascular Biology. 2004;24(9):1640–5. doi: 10.1161/01.ATV.0000134377.17680.26. [DOI] [PubMed] [Google Scholar]
- 40.Matsuyama R, Tomi M, Akanuma S, et al. Upregulation of L-Type Amino Acid Transporter 1 (LAT1) in Cultured Rat Retinal Capillary Endothelial Cells in Response to Glucose Deprivation. Drug metabolism and pharmacokinetics. 2012;27(3):317–24. doi: 10.2133/dmpk.dmpk-11-rg-122. [DOI] [PubMed] [Google Scholar]
- 41.Campbell WA, Thompson NL. Overexpression of LAT1/CD98 light chain is sufficient to increase system L-amino acid transport activity in mouse hepatocytes but not fibroblasts. Journal of Biological Chemistry. 2001;276(20):16877–84. doi: 10.1074/jbc.M008248200. [DOI] [PubMed] [Google Scholar]
- 42.Kanai Y, Segawa H, Miyamoto K, et al. Expression cloning and characterization of a transporter for large neutral amino acids activated by the heavy chain of 4F2 antigen (CD98) Journal of Biological Chemistry. 1998;273(37):23629–32. doi: 10.1074/jbc.273.37.23629. [DOI] [PubMed] [Google Scholar]
- 43.Rintoul RC, Buttery RC, Mackinnon AC, et al. Cross-linking CD98 promotes integrub-like signaling and anchorage-independent growth. Molecular Biology of the Cell. 2002;13(8):2841–52. doi: 10.1091/mbc.01-11-0530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lacroix J, Hebert PC, Hutchison JS, et al. Transfusion strategies for patients in pediatric intensive care units. N Engl J Med. 2007;356(16):1609–19. doi: 10.1056/NEJMoa066240. [DOI] [PubMed] [Google Scholar]
- 45.Donadee C, Raat NJ, Kanias T, et al. Nitric oxide scavenging by red blood cell microparticles and cell-free hemoglobin as a mechanism for the red cell storage lesion. Circulation. 2011;124(4):465–76. doi: 10.1161/CIRCULATIONAHA.110.008698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reynolds JD, Bennett KM, Cina AJ, et al. S-nitrosylation therapy to improve oxygen delivery of banked blood. Proc Natl Acad Sci U S A. 2013;110(28):11529–34. doi: 10.1073/pnas.1306489110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reynolds JD, Ahearn GS, Angelo M, et al. S-nitrosohemoglobin deficiency: a mechanism for loss of physiological activity in banked blood. Proc Natl Acad Sci U S A. 2007;104(43):17058–62. doi: 10.1073/pnas.0707958104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang R, Hess DT, Qian Z, et al. Hemoglobin betaCys93 is essential for cardiovascular function and integrated response to hypoxia. Proc Natl Acad Sci U S A. 2015;112(20):6425–30. doi: 10.1073/pnas.1502285112. [DOI] [PMC free article] [PubMed] [Google Scholar]