

Abstract
Local microirradiation with lasers represents a useful tool for studies of DNA-repair-related processes in live cells. Here, we describe a methodological approach to analyzing protein kinetics at DNA lesions over time or protein-protein interactions on locally microirradiated chromatin. We also show how to recognize individual phases of the cell cycle using the Fucci cellular system to study cell-cycle-dependent protein kinetics at DNA lesions. A methodological description of the use of two UV lasers (355 nm and 405 nm) to induce different types of DNA damage is also presented. Only the cells microirradiated by the 405-nm diode laser proceeded through mitosis normally and were devoid of cyclobutane pyrimidine dimers (CPDs). We also show how microirradiated cells can be fixed at a given time point to perform immunodetection of the endogenous proteins of interest. For the DNA repair studies, we additionally describe the use of biophysical methods including FRAP (Fluorescence Recovery After Photobleaching) and FLIM (Fluorescence Lifetime Imaging Microscopy) in cells with spontaneously occurring DNA damage foci. We also show an application of FLIM-FRET (Fluorescence Resonance Energy Transfer) in experimental studies of protein-protein interactions.
Keywords: Cellular Biology, Issue 129, DNA damage, confocal microscopy, FLIM-FRET, PCNA, 53BP1, p53
Introduction
DNA damage leads to the appearance of DNA lesions consisting of cyclobutane pyrimidine dimers (CPDs), 8-oxo-7,8-dihydro-2'-deoxyguanosine, and single-strand or double-strand breaks1,2. γ-rays are the form of ionizing radiation with the highest energy and high penetrance, thus this source of radiation is widely used in radiotherapy3. On the other hand, experimentally induced DNA damage caused by UV lasers mimics natural exposure to UV light. UVA microirradiation, as a microscopic method, represents an experimental tool for studying DNA damage in individual living cells. Microirradiation was used for the first time 40 years ago in order to reveal the organization of chromosome regions4,5. This technique is highly dependent on either the functional properties of confocal microscopes or the technical limits of modern nanoscopy. To induce DNA lesions, cells can be presensitized by 5' bromodeoxyuridine (BrdU) or Hoechst 33342 prior to UV irradiation. Bártová et al.6 previously described the presensitization step, and recently we optimized this microirradiation technique in order to avoid cell death, or apoptosis. For example, the use of a 405-nm UV laser (without Hoechst 33342 presensitization) leads to the induction of 53BP1-positive double-strand breaks (DSBs) at the expense of cyclobutane pyrimidine dimers (CPDs). On the other hand, presensitization steps combined with UV microirradiation induce very high levels of CPDs and DSBs simultaneously7,8. This methodology is difficult to apply to the study of a single DNA repair pathway.
With microirradiation, it is possible to analyze protein recruitment, kinetics, and interaction at DNA lesions in living cells. An example of this method was published by Luijsterburg et al.9 for heterochromatin protein 1β, and we recently showed for the first time that the pluripotency factor Oct4 and a protein associated with Cajal bodies, coilin, are recruited to UV-induced DNA lesions6,10. Protein kinetics at these DNA lesions can also be studied using the FRAP (Fluorescence Recovery After Photobleaching)11,12,13 or FRET (Fluorescence Resonance Energy Transfer) techniques14,15. These methods have the potential to reveal simple diffusion of proteins at DNA lesions or protein-protein interactions. A useful tool for additional characterization of proteins is FLIM (Fluorescence Lifetime Imaging Microscopy) or its combination with FRET technology (FRET-FLIM)16. These methods enable the study of processes in living cells that are stably or transiently expressing the protein of interest tagged by a fluorescent molecule17. Here, an example of exponential decay time (τ) for GFP-tagged p53 protein and its interaction partner, mCherry-tagged 53BP1, playing an important role in DNA damage response18,19 is shown. The parameter τ, the lifetime of the fluorochrome provided by FLIM calculations, is specific for a given fluorescence dye, its binding abilities, and its cellular environment. Therefore, this method can show us distinctions between protein subpopulations, their binding abilities, and their functional properties after, for example, DNA damage.
Here, an outline of the methodological approaches of the advanced microscopy techniques that is used in our laboratory to study time-specific protein recruitment, kinetics, diffusion, and protein-protein interactions at the site of microirradiated chromatin is presented. The step-by-step methodology for the induction of local DNA lesions in living cells, and a description of methodologies useful for studies of DNA-damage-related events at locally induced DNA lesions caused by UV lasers are provided.
Protocol
1. Cultivation of Cell Lines
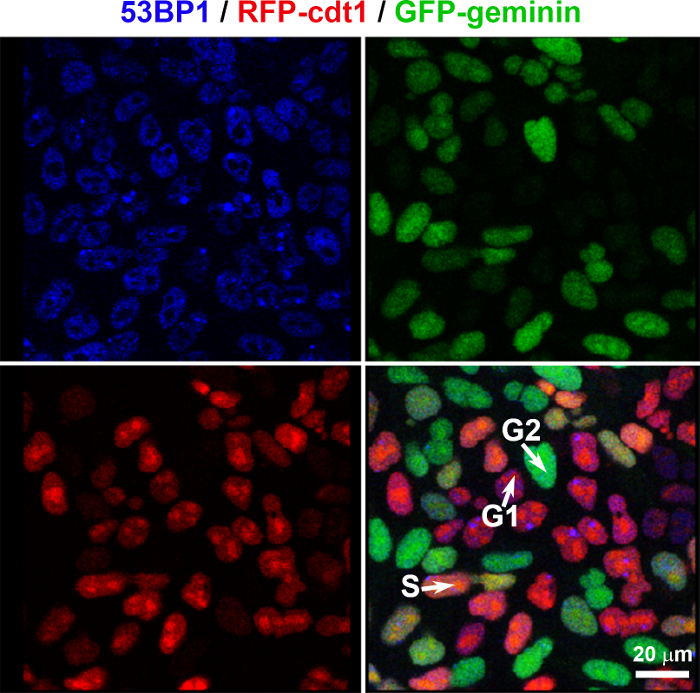
- HeLa-derived cell lines NOTE: Use HeLa cervical carcinoma cells: either HeLa cells stably expressing histone H2B tagged with GFP or HeLa-Fucci cells expressing RFP-Cdt1 in the G1 and early S phases and GFP-geminin in the S/G2-M phases (Figure 1).
- For cultivation of all HeLa-derived cell lines, use Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum and appropriate antibiotics at 37 °C in a humidified atmosphere containing 5% CO2. Replace the medium 2 - 3 times per week.
- Remove culture medium, and rinse the cells using 1x PBS to eliminate all traces of serum that contains trypsin inhibitor. Perform this step in a biohazard hood at room temperature.
- Add 1 mL of prewarmed Trypsin-EDTA solution to cover the cell layer, and keep cells at 37 °C. Trypsinize cells until cell layer is dispersed (usually 3 - 5 min).
- Add 3 mL of prewarmed complete growth medium to inactivate Trypsin-EDTA, and disperse the medium by gently pipetting several times. NOTE: This procedure must be performed in a biohazard hood in order to protect the operator from contaminants and to maintain optimal cell cultivation conditions.
- Count cells using an automatic cell counter or Bürker chamber. Dilute cell suspension to 1 × 105 cells/mL and pipet 5 mL into a new dish. Incubate cells at 37 °C and 5% CO2.
- Cultivation of mouse embryonic stem cells (mESCs), D3 cell line
- Culture mESCs on cell culture dishes coated with 0.2% gelatin and mESC maintenance medium. Incubate cells at 37 °C and 5% CO2. NOTE: Preparing the mESCs cultivation/maintenance medium in 50 mL aliquots and storing it at 2 - 8 °C for up to 2 weeks is recommended. The mESC medium contains 75 mL ES Cell FBS, 5 mL Penicillin G and Streptomycin, 5 mL Non-Essential Amino Acids (10 mM) (final concentration 0.1 mM), 50 µL mouse Leukemia Inhibitor Factor (mLIF; 100 µg/mL) (final concentration 10 ng/mL), 430 µL MTG (1-Thioglycerol; 1:100 dilution in DMEM) (final concentration 100 µM) and high glucose DMEM medium up to 500 mL.
- Aspirate the mESC cultivation medium from the culture dish and rinse the cells once with 1x PBS.
- Add 0.5 mL Trypsin-EDTA and incubate at 37 °C just until the cells begin to detach from the dish. Then, inactivate the Trypsin by washing the cells with 2 mL of mESC cultivation medium supplemented with 15% fetal bovine serum.
- Use a pipette to dislodge the remaining cells from the culture dish. NOTE: It is important to obtain single cells, because cell clumps promote differentiation of the cells.
- Split cells 1:10 onto gelatinized dishes with mESC maintenance medium. NOTE: If the density of mESC cells is too low, they do not grow well; if the density is too high, differentiation is promoted.
- Ensure that mESCs are maintained in an undifferentiated state by performing mESC passage every second day by splitting cells from one Petri dish into four new dishes. Inspect the mESC colonies with an inverted microscope under 100X or 200X magnification, and, if there is evidence of spontaneous mES cell differentiation, perform Western blots to assess Oct4 protein depletion. NOTE: With optimal cell passaging, spontaneous differentiation of mESCs in vitro can be reduced.
2. Cell Transfection
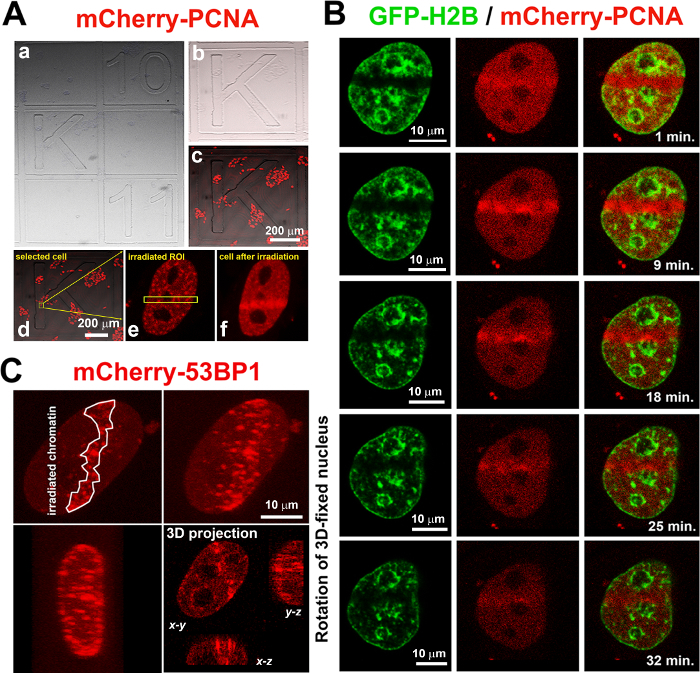
- Seed cells 48 h before experiments on gridded microscopy dishes (diameter 35 mm) in order to find microirradiated cells according to their coordinates on the Petri dish. NOTE: This is an example in which microirradiation is the first experimental step and immunostaining is the second (Figure 2A).
- For seeding, use a concentration of 5 × 104 cells/mL for HeLa-derived cell lines (or 1 × 104 cells/mL for D3 mES cells). Use up to 2 mL complete medium per dish. During cultivation and microirradiation, maintain cells in a thermostat or cultivation chamber at 37 °C and 5% CO2.
- Count cells using an automatic cell counter. NOTE: Do not use a higher concentration of cells. Especially in the case of densely packed colonies of D3 mESCs, high cell density prevents an identification of locally microirradiated cells after immunostaining.
Prepare plasmid DNA and transfection reagent of choice as follows. Prepare Solution A using 2 - 3 µg of selected plasmid DNA (see the Table of Materials for details) and 150 µL 1x PBS. Prepare Solution B using 3.5 µL transfection reagent in 150 µL 1 × PBS. Ensure that each solution is well mixed by careful pipetting. NOTE: The optimal ratio of DNA and transfection reagent must be empirically determined for each cell line and individual plasmid.
At 24 h before experimental observation of transiently transfected living cells, combine solution A and solution B without vortexing. Incubate the combined solutions at room temperature for 15 - 20 min. In a dropwise manner, homogeneously disperse this complete transfection mixture (300 µL, as described in point 2.2) on the cell layer growing in the microscopy dish.
Incubate cells at 37 °C and 5% CO2 for 4 - 6 h, then replace the transfection mixture with fresh medium. Cultivate cells under standard conditions. NOTE: When using a 405-nm laser, do not presensitize the cells with any reagent, but when using a 355-nm laser, perform cell presensitization with 10 µM 5-bromo-2'-deoxy-uridine (BrdU) 7,8. Presensitization time depends on the cell type and the length of the cell cycle. For HeLa cells, add BrdU 16 to 20 h before local microirradiation. In the case of mESCs, add BrdU 6 to 8 h before microirradiation.
3. Induction of Local DNA Lesions and Confocal Microscopy
Place the dish on the microscope stage and record the position of the selected cells on the dish; the cell coordinates will be needed for further analysis (Figure 2Aa-c).
Find transfected cell(s) in a square labeled with a number or letter (Figure 2Ad-f, arrows in Ad show the location of the cell magnified in panels Ae and Af ). Obtain an image of the cell(s) using bright field microscopy and acquire a fluorescence image in order to identify the position of both the cell(s) and the labeled square (Figure 2A).
For image acquisition before irradiation, use a white-light laser (WLL) (470-670 nm in 1-nm increments) connected to the confocal microscope (or another available laser connected to the confocal microscope).
Use the following settings: 512 × 512-pixel resolution, pixel size 64 nm, 400 Hz, bidirectional mode (scanning in two directions), line average set to 8, zoom 8x to 12x. NOTE: Line average represents a given number of scans; the same line is scanned multiple times before continuing to the next line. Repeated scans of all lines produce the final frame of the image.
For image observation and acquisition, use a 63× oil objective with a numerical aperture of 1.4 and sequentially acquire fluorescence images. Eliminate cross-talk between the two fluorochromes by using the sequential scanning mode of the appropriate software. NOTE: All microscope software enables the setting of a so-called sequential scanning mode. The operator can select whether to acquire the fluorochromes' information simultaneously (e.g., in red-green-blue fluorescence) or, as mentioned here, individually (sequentially), fluorochrome by fluorochrome. Take into account the general information that the maximum excitation/emission for GFP is 395/509 nm and the maximum excitation/emission for mCherry is 587/610 nm (see fluorochromes used here in Figure 2B). Based on this information, select suitable excitation and emission filters for the desired fluorochromes. Filter selection and scanning procedures must be optimized for each confocal microscope.
For induction of DNA lesions, use 64 line scanning mode, switch off the WLL (in 'acquisition' mode), switch on the external UV source (on "UV laser" button), set the region of interest (ROI) (in 'acquisition' mode), and set the 355-nm laser to 100% power or, alternatively, use a 405-nm laser source. NOTE: Generally, laser power is adjusted by the appropriate laser adjustment button in image acquisition mode.
For local microirradiation, use lasers that emit in the near UVA spectrum. For microirradiation of ROI in the nucleus and for capture image, set the background to zero in image acquisition mode to regulate laser intensity outside the ROI; if the cell is irradiated properly, the signal of GFP-histone H2B will disappear, and, for example, mCherry-tagged PCNA protein will be recruited to DNA lesions immediately after irradiation (Figure 2B and Video 1). NOTE: A DNA injury induced in the ROI of one confocal section also appears in three-dimensional (3D) projection (see 3D rotation and the x-y, x-z, y-z projections in Figure 2C and Video 2). For descriptions of the complete technical parameters for 355-nm and 405-nm lasers, see Stixová et al.7
After the ROI has been irradiated, switch off the external UV source, switch off the ROI, switch on the WLL, change line scanning to 8, and find another cell for irradiation. For ROI selection, use the applicable button in the software's image acquisition mode. NOTE: mCherry-PCNA has a maximum accumulation peak at DNA lesions between 2 and 20 min (Figure 2B). To verify the result on exogenous protein levels, proceed with immunofluorescence staining immediately after local microirradiation. Select the irradiation time interval according to interest.
- Verify that an accumulation of exogenous proteins can be detected in the majority of microirradiated cells. For this verification, use the criteria in the following steps.
- Check that there is no overexpression of exogenous protein in transiently transfected cells. As a possible solution, choose cells with only a weak expression of fluorescent protein (e.g., mCherry-PCNA or mCherry-53BP1).
- Assess the phototoxicity of the WLL exposure used for image acquisition. NOTE: For phototoxicity testing, expose transfected cells to prolonged laser microirradiation and verify that cells proceed to mitosis as shown in Figure 3A-B. As a possible solution, reduce scanning time and number of confocal slices per cell, optimize 3D scanning by selection of the optimal axial step (0.3 µm is recommended) and set the laser's power at its minimum. Alternatively, phototoxicity can be eliminated using Trolox (6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid, a water-soluble analog of vitamin E) dissolved in the cultivation medium. For DNA-damage-related experiments, it is not recommended to use of Trolox due to its protective effect against UV radiation.
- In the case of irradiation with a 355-nm laser, verify sufficient incorporation of BrdU using an appropriate BrdU detection antibody from the BrdU incorporation kit (see Table of Materials). Since BrdU is incorporated into DNA during the S phase of the cell cycle, the majority of cells must proceed through the S-phase during presensitization. As a possible solution, consider the length of the cell cycle, which is cell-type specific.
- Analyze whether the DNA repair proteins of interest have an ability to recognize DNA lesions in specific cell cycle phase(s). A possible solution: To verify that recruitment of selected proteins to DNA lesions is restricted to specific cell cycle phases (G1/S/G2), use HeLa-Fucci cells. These cells express RFP-Cdt1 in the G1 and early S phases and GFP-tagged geminin in the S/G2-M cell cycle phases 22 (Figure 1) .
- To avoid apoptosis, cell death, select an appropriate laser source and laser power setting23. Keep in mind that irradiated cells should proceed to mitosis (Figure 3A-B, Video 3). NOTE: Unwanted apoptosis can be detected by Annexin V positivity, caspase 3, or lamin B cleavage. On the morphological level, cell detachment and formation of apoptotic bodies will be visible.

4. Immunofluorescence Staining
Rinse the cell layer (growing in microscope dishes) briefly with 1 × PBS and after rinsing, fix cells using 4% formaldehyde for 10 min (for HeLa-derived cell lines) or 20 min (for D3 ES cells) at room temperature. Rinse the dish with 1 × PBS. Caution: Formaldehyde causes serious eye damage, may cause an allergic skin reaction, and is also a potential carcinogen; thus, all experimental steps with formaldehyde should be performed in an extractor hood. NOTE: To avoid unnecessary bleaching of fluorescence signals, store the dish in a dark chamber during the incubation time necessary for immunofluorescence staining.
Permeabilize cells with 0.1% Triton X-100 dissolved in H2O for 8 min, and then incubate the cells for 12 min with 1× PBS containing 0.1% saponin and 0.1% Triton X-100. After incubation, wash the cells in 1× PBS two times for 15 min.
Incubate cells with 1% BSA in 1× PBS for 60 min to block unspecific binding of selected antibodies. Wash cells in 1× PBS for 15 min after incubation.
Dilute primary antibody at a ratio of 1:100 (or 1:200) (this step must be optimized for individual antibodies) in 1% BSA in 1 × PBS, using 25 µL of this solution per dish, and cover the cells with a coverslip. Incubate the cells with the solution containing the primary antibody in a humidified chamber overnight at 4 °C.
On the following day, wash cells in 1 × PBS two times for 5 min.
Dilute the secondary antibody at a ratio of 1:100 (or alternatively 1:200) in 1% BSA in 1 × PBS, using 100 µL of this solution per dish, and cover the cells with a coverslip. Incubate the cells with the solution containing the secondary antibody for 60 min. After incubation, wash the cells in 1 × PBS three times for 5 min.
Mount the coverslip with a drop of mounting medium, and seal the coverslip with nail polish or glue to prevent drying and movement during microscopic observation. Store the dish in the dark at 4 °C.
5. Fluorescence Lifetime Image (FLIM) Microscopy
Start the FLIM software. Open the "Application Suite" of the software and switch it to the "FLIM" mode. Make sure that the WLL is operating in pulsed mode. NOTE: The operator must switch a hardware button to reach pulse mode.
Place the dish containing the stained cells (stained in section 4) on the microscope stage in the same orientation as during local microirradiation, and find the irradiated cells according to the grids on the Petri dish. Perform image acquisition by confocal microscope. NOTE: Use the following settings: 1024 × 1024 pixels, 400 Hz, bidirectional mode, 16 lines, zoom 8x to 12x.
- Before beginning FLIM measurement, perform a preliminary test to show a real-time record of exponential decay, by clicking on "Setup FLIM" and "Acquisition" and pressing "Run FLIM test". Optimize the FLIM parameters for the sample by assessing the two main parameters as follows.
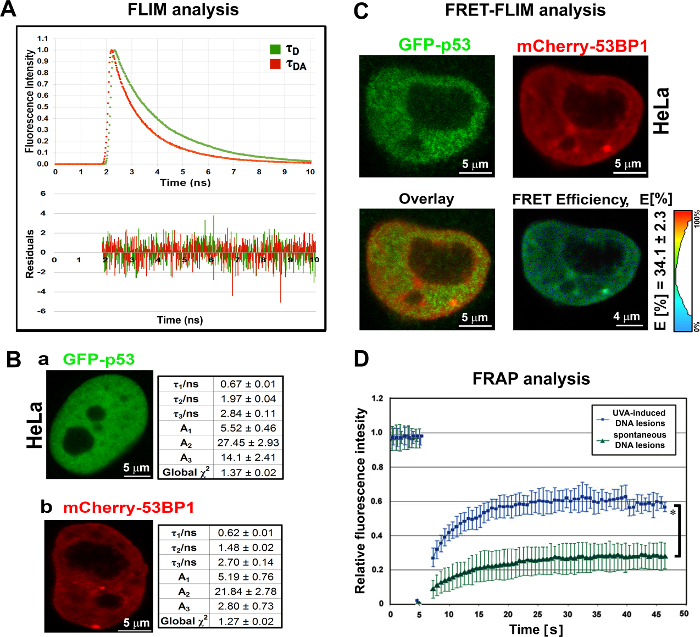
- Optimize the repetition rate of the WLL with the excitation wavelength tuned to the lifetime of the sample (see Note below). In the confocal microscope software, adjust the laser intensity with the appropriate button, which is generally called "laser adjustment setting" in the 'image acquisition' menu. Then, calculate a decay curve (or histogram showing all photon counts) using the FLIM software (see Figure 4A-B). NOTE: Use the WLL in pulse mode for excitation. The software must be equipped with a pulse picker for WLL, which allows selection of the repetition rate (80, 40, 20, or 10 MHz). The excitation wavelength in the case of the fluorescence donor, GFP, is 488 nm. The decay curve is recorded using the FLIM software workspace. The parameter (τD, τDA) shows exponential decay times (e.g., fluorochrome lifetime); parameter (A) represents the amplitude of fluorochrome decay (see examples of FLIM data for GFP-tagged p53 and mCherry-tagged 53BP1 in Figure 4A-B).
- Check the count repetition rate using the software acquisition tool (select repetition rate mode). Select the brightest pixels in the sample; the curve for the brightest pixel must be below 10% of the overall fluorescence intensity.
Click on "Measurement" and press "Run FLIM".
6. Donor Lifetime Using a FLIM Script and Calculation of FRET Efficiency
Start the FRET-FLIM software and open the 'Donor' workspace.
Select the "Analysis"/"Imaging" tab in the menu and start the FLIM script by clicking "Start". For optimal FRET-FLIM settings, use well-known interacting protein partners. NOTE: The FRET-FLIM efficiency for well-known interacting partners GFP-p53 and mCherry-53BP1 was (34.1 ± 2.3)% (Figure 4C).
Use only the Donor emission channel (channel 1 or 2) and press "Calculate Fast FLIM". NOTE: The image and the graph will both be updated.
In the FRET-FLIM software, set the intensity scale (0 - 4000 counts) and the lifetime scale (0 - 5 ns) manually in the software. With this approach, visualize the cell of interest and set the FRET-FLIM measurement. NOTE: This setting eliminates differences in fluorescence intensity among individual cells in the cell population.
In the decay form, choose the fitting model. NOTE: We recommend n-exponential reconvolution and selection of model parameter "n". An optimal fit has the following criteria: the fitted curve overlays the decay curve well and the χ2- value is equal to 1 (Figure 4B).
Select the "Threshold"/"Use Threshold" and set the threshold to 75. Start "Initial Fit". NOTE: The SPT64 software calculates the average donor lifetime in the absence of FRET ("Amplitude weighted average lifetime", τAv.Amp)
7. FRET Efficiency Using the FLIM-FRET Script
Select the Donor/Acceptor workspace and then open "Analysis"| "Imaging"| "Lifetime FRET Image".
Select only the Donor as the active channel and adjust Pixel Binning depending on the photon number/pixel. Then, run the software mode called "Calculate FastFLIM". If the photon number/pixel in the image is low, increase the Pixel Binning to 4 points.
Set the intensity to 0 - 200 counts and the lifetime to 0 - 5 ns.
Activate "Use Threshold" and enter a threshold of 75.
Set the Fitting model to "Multi-Exp. Donor", enter the model parameter "n", enter τAv.Amp (step 6.6) as a τD, and activate "Initial Fit".
Set the parameters BkgrDec, ShiftIRF, and BkgrIRF to constant valueS by removing the mark. NOTE: Set the majority of parameters to remain constant as much as possible. This approach reduces statistical fluctuations. In this case, do not press "Initial fit" again. Descriptions of the mentioned parameters follow: IRF = Instrument Response Function, BkgrDec = Background from the exponential fit, ShiftIRF = IRF shift from exponential reconvolution fit, BkgrIRF = IRF background from exponential reconvolution fit. All three parameters are calculated and can be fixed using the software for further analysis.
Press "Calculate FRET"; a FRET image (plotted in the FLIM image area) and FRET efficiency histogram are calculated, and a FRET distance histogram is plotted (Figure 4C). When the image analysis is finished, press "Save Result". NOTE: During the FRET experiments, it is necessary to consider that fluorescent proteins exhibit reversible transitions between fluorescent (bright) and non-fluorescent (dark) states. This characteristic is called "blinking", and FRET efficiency is affected by this phenomenon (an explanation of this effect was provided by Vogerl et al.24).
8. FRAP Analysis
- Place the dish on the microscope stage, find the transfected cells expressing the fluorescently tagged protein of interest, and perform image acquisition using bright field microscopy with the standard microscope lamp (see Figure 2Aa-b) and fluorescence microscopy modes.
- For image acquisition, use the white-light laser (WLL) connected to the confocal microscope (or a laser of choice, according to the selected fluorochrome). Use the following settings: 512 × 512 pixels, 1000 Hz, bidirectional mode, line average 1, zoom 8x to 12x.
- Acquire at least 15 prebleaching images (at ~10% laser power) and define the region of interest (ROI) in the image acquisition menu.
For photobleaching experiments, use an argon laser (488 nm). Apply the following settings: frame resolution 512 × 512 pixels, 1000 Hz, bidirectional mode, zoom 8x to 10x. NOTE: Fluorochromes including GFP, mCherry, and RFP can be excited by an argon laser working at 488 nm or 514 nm.
- Use the FRAP software mode of the microscope of choice. During the photobleaching, set the argon laser's power to maximum using the laser adjustment button. Perform image acquisition at 0.256 s intervals, at 10% laser power. NOTE: For postbleaching steps, use the standard image acquisition mode of the confocal microscope of choice.
- Monitor fluorescence recovery time after photobleaching (up to 45-50 s after the bleaching procedure). NOTE: As shown in Figure 4D, the recovery time after photobleaching for mCherry-tagged 53BP1 is distinct at spontaneous DNA lesions and UVA-irradiation-induced foci. mCherry-53BP1 at UVA lesions recovered rapidly in comparison to mCherry-53BP1 accumulations at spontaneously occurring DNA repair foci; see Figure 4D and Foltankova et al.25
Export the data from the microscope software (or software of choice) to a spreadsheet and perform statistical analysis.
Representative Results
Using advanced confocal microscopy, we observed an accumulation of mCherry-tagged 53BP1 and mCherry-PCNA proteins at DNA lesions. Analyses were performed by local microirradiation of living cells. To recognize the nuclear distribution patterns of DNA-repair-related proteins in individual cell cycle phases, we used the Fucci cellular system, by which it is possible to determine the G1, early S, and G2 phases of the cell cycle (Figure 1). The biological application of the Fucci cellular model that we published in Suchankova et al.26 shows that 53BP1 was recruited to locally induced DNA lesions in the G1, S, and G2 phases of the cell cycle, which was associated with the function of this protein during non-homologous end joining (NHEJ), one of the main mechanisms leading to double-strand break repair. On the other hand, this experimental system showed us the individual cellular level that the PCNA protein, which is linked to the homologous recombination repair pathway (HRR), recognizes DSBs in the late S and G2 phases of the cell cycle 27. For studies on DNA repair machinery, we also optimized irradiation conditions in order to ensure that cell cultures continued to undergo mitosis after exposure to UV lasers (Figure 3). Microirradiated cells undergoing mitosis prove that, in these experimental conditions, DNA repair proceeds in a relatively physiological manner and cells have not been injured by the lasers, which at higher intensities induce apoptosis. Here, we also show how to apply FRET-FLIM analysis to the characterization of DNA repair proteins. This advanced technology enables us to study local protein-protein interactions in the cell nucleus or, alternatively, protein interactions in nucleolar regions such as the nucleolus, nuclear lamina, or clusters of heterochromatin. For beginners in this technology, we would like to recommend optimizing this FRET-FLIM technique by using well-known interacting protein partners such as p53 and 53BP1 (Figure 4A-C). In general, a knowledge of protein-protein interaction leads to understanding how protein complexes regulate processes like replication, gene activation, silencing, or DNA repair. In addition, a very useful tool for studies of protein kinetics is the FRAP method, which shows the characteristics of local protein diffusion and mobility (Figure 4D). Taken together, here we provide methodological instructions for applying advanced confocal microscopy techniques in DNA repair studies.
Figure 1: Formation of DNA repair foci can be studied in HeLa-Fucci cells expressing RFP-cdt1 (red) in the G1/early S phases and GFP-geminin (green) in the S/G2 phases of the cell cycle. Spontaneously occurring DNA lesions positive for 53BP1 protein (blue; Alexa 405 staining), were studied in the G1 (red), early S (orange; expression of both RFP-cdt1 and GFP-geminin), and G2 (green) phases of the cell cycle. Please click here to view a larger version of this figure.
Figure 2: Recruitment of mCherry-tagged PCNA at DNA lesions over time. (A) Cells expressing mCherry-PCNA (red), were located on a gridded microscope dish in selected regions (gray). After fixation and immunostaining, irradiated cells (yellow arrows) were located according to registered coordinates (see the gray letter K as an example) (Aa-c). The yellow frame and arrows (Ad) show the cell that was microirradiated and magnified in panels Ae-f. (B) Accumulation of mCherry-PCNA (red) was studied in HeLa cells stably expressing GFP-tagged histone H2B (green). Cells were monitored immediately after local microirradiation for up to 35 min (see Video 1). (C) Microirradiated cells were analyzed by 3D confocal microscopy, and protein accumulation at DNA lesions (e.g., mCherry-53BP1) was found in all three dimensions (x-y, x-z, and y-z) although only the midsection of the cell nucleus was microirradiated (see 3D-cell rotation in Video 2 3D projections in Figure 2C). Please click here to view a larger version of this figure.
Figure 3: Cells microirradiated by a 405-nm laser diode proceed normally through the cell cycle.(A) After irradiation using a 405-nm diode laser, HeLa cells (stably expressing GFP-histone H2B; green) undergo mitosis (see also Video 2). Yellow arrows and frames indicate irradiated ROI in selected cells. White frames show mitosis.(B) Under the same experimental conditions, mitosis in irradiated cells was also observed by time lapse microscopy in HeLa cells stably expressing GFP-H2B (green) and transiently expressing mCherry-PCNA (red). Please click here to view a larger version of this figure.
Figure 4: FLIM and FRET-FLIM analysis of GFP-p53 and mCherry-53BP1. Fluorescence Resonance Energy Transfer (FRET) detected by FLIM and application of Fluorescence Recovery After Photobleaching(FRAP) at various DNA lesions.(A-C) Averaged (n=10) lifetimes of GFP-tagged p53 in the absence and presence of the acceptor (mCherry-53BP1), measured in whole nonreplicating HeLa cell nuclei. (A) Representative fluorescence decay curves and residuals studied in HeLa cells transiently expressing GFP-p53 (donor-only, τD) or GFP-p53 and mCherry-53BP1 (donor – acceptor, τDA). (B) Averaged lifetimes (τ1 -τ3) and amplitudes (A1 - A3) of (a) GFP-p53 and (b) mCherry-53BP1 measured in whole HeLa cell nuclei. χ2 values were also calculated and are shown. (C) Summary of FRET efficiency for the well-known interacting partners GFP-p53 and mCherry-53BP1. Scale bars = 4-5 µm. FRET-FLIM result showing ~35% FRET efficiency for well-known interacting partners GFP-tagged p53 and mCherry-tagged 53BP1. (D)Recovery kinetics of mCherry-53BP1 studied at spontaneously occurring DNA lesions (green curve) and UVA-induced DNA lesions (blue curve). mCherry at DNA lesions was bleached to the level of the background (dispersed form of protein of mCherry-53BP1 protein), and the background fluorescence was subtracted from each value. Data, shown as relative fluorescence intensity of mCherry-53BP1, are presented as the means ± standard error. Student's t-test revealed a statistically significant difference between two types of DNA lesions (* shows p ≤0.05). Please click here to view a larger version of this figure.
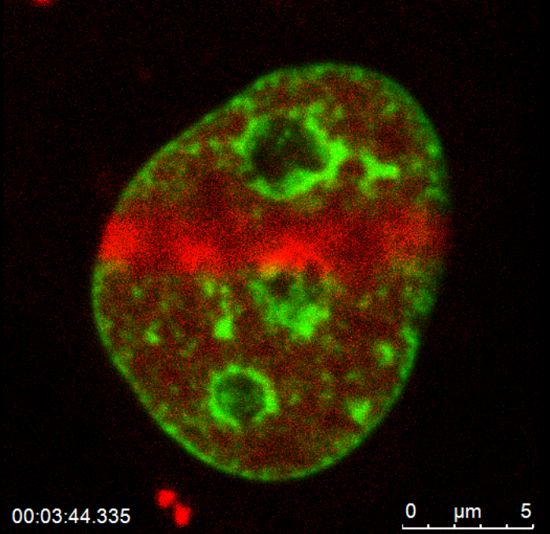 Video 1: Recruitment of mCherry-tagged PCNA protein (red fluorescence) to DNA lesions induced by local microirradiation in HeLa cells stably expressing GFP-tagged histone H2B (green fluorescence).
Please click here to download this video.
Video 1: Recruitment of mCherry-tagged PCNA protein (red fluorescence) to DNA lesions induced by local microirradiation in HeLa cells stably expressing GFP-tagged histone H2B (green fluorescence).
Please click here to download this video.
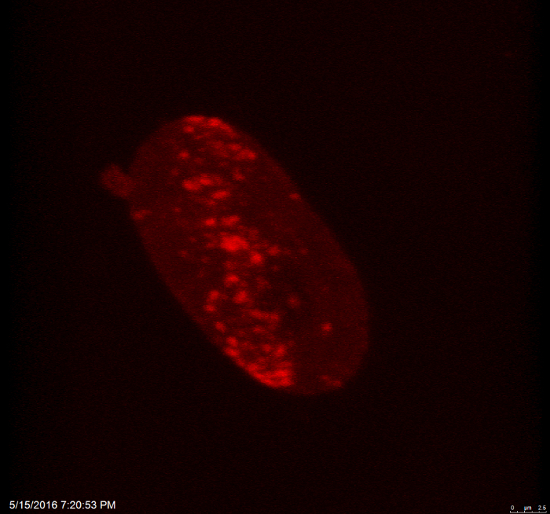 Video 2: Accumulation of mCherry-tagged 53BP1 protein (red fluorescence) at DNA lesions induced by local microirradiation in HeLa cells. The cell nucleus is shown in 3D space using a software mode that enables the visualization of cell rotation in space. Please click here to download this video.
Video 2: Accumulation of mCherry-tagged 53BP1 protein (red fluorescence) at DNA lesions induced by local microirradiation in HeLa cells. The cell nucleus is shown in 3D space using a software mode that enables the visualization of cell rotation in space. Please click here to download this video.
 Video 3: Microirradiated HeLa cells stably expressing GFP-tagged histone H2B (green fluorescence) proceed through mitosis. The irradiated region is characterized by the depletion of GFP-H2B (black regions are irradiated strips inside the cell nuclei). Please click here to download this video.
Video 3: Microirradiated HeLa cells stably expressing GFP-tagged histone H2B (green fluorescence) proceed through mitosis. The irradiated region is characterized by the depletion of GFP-H2B (black regions are irradiated strips inside the cell nuclei). Please click here to download this video.
Discussion
Microscopy techniques represent basic tools in research laboratories. Here, a brief description of the methods used for the study of protein recruitment and kinetics at DNA lesions is presented. We especially noted our experimental experience in the field of local microirradiation of living cells, and we discuss the study of protein kinetics by FRAP and protein-protein interaction at DNA lesions by acceptor-bleaching FRET28 and its advanced modification FRET-FLIM (Figure 4A-D). The methods shown here are essential tools for a true understanding of DNA repair processes, especially protein kinetics in living cellular systems, as inspected by advanced confocal microscopes6,7,8,28. These methods are of immense utility to future medicine in dealing with the effects of radiotherapy on tissues or characterizing tumor cell morphology. To optimize this method, we would like to recommend starting with well-known interacting protein partners, as shown in Figure 4C.
It is well known that protein recruitment to DNA lesions is highly dynamic and time-dependent; thus, the application of time-lapse confocal microscopy after cell irradiation is required not only in the field of basic science but also in clinics26. Locally induced DNA lesions due to laser microirradiation9,28,29 represent genomic regions where it is possible to study protein recruitment, protein-protein, or protein-DNA binding and interaction. The starting point of these methods is that the recruitment of exogenous proteins must be verified by appropriate antibodies on the endogenous level. Next, the critical step of such experiments is that over-transfected cells can provide false-positive results; thus, the best decision is to establish cell lines stably expressing the fluorescently tagged proteins of interest. Moreover, due to the phototoxic effects of the lasers used for image acquisition, either the conditions for scanning must be optimized or Trolox compound must be dissolved in the cell cultivation medium. Additionally elimination of the presensitization step before irradiation is recommended. After local microirradiation, DNA repair must proceed such that the cells undergo mitosis (Figure 3A-B and Video 3). Induction of apoptosis after laser irradiation is a less valuable process from the view of optimal DNA repair23. It is also evident that some DNA repair proteins recognize DNA damage only in specific cell cycle phases (e.g., Bártová et al.30). Thus, the use of the HeLa-Fucci cellular system, showing the cells in the individual phases of interphase, is a useful tool for studies of DNA damage response. In addition, cell cycle phases can be recognized according to the nuclear distribution pattern of the PCNA protein31.
It is well known that the FRAP and FRET methods represent very useful tools for investigating the characteristics of proteins that are recruited to DNA lesions7,8,32. Moreover, the FLIM technique32 can reveal information about the dynamic conformational changes of the proteins that accumulate at DNA lesions. The use of this method is important because FLIM measures dynamic cellular processes directly; thus, steady-state fluorescence intensity is measured over time. FLIM approaches increase image contrast by eliminating background fluorescence. This is an advantage of FRET-FLIM measurement in comparison with conventional acceptor-bleaching FRET. The fluorescence lifetimes measured by FLIM provide information about the fractions of fluorescently tagged proteins and show how heterogenic probes are due to environmental conditions. FLIM is also the best and most reliable biophysical approach to performing FRET for protein-protein interaction in living cells32,33.
Taken together, all of the described methods have the potential to reveal new mechanisms of the DNA damage response in human cells, which is important especially for radiotherapeutic approaches. For example, multiphoton FLIM has been applied to obtain high-resolution images in medicine, where it is called multiphoton tomography34. This highly sophisticated microscopic method can be applied in clinics for cancer diagnosis using histochemical samples and analyzed with high resolution. FLIM methods can additionally provide a precise analysis of tumor cell surfaces according to their autofluorescence. One disadvantage of the FLIM-FRET method is its highly sophisticated software background, which must be fully understood by the microscope operator. Further biological relevance of FLIM data, in general and in DNA repair processes, will certainly be revealed in the near future.
Disclosures
The authors declare that there are no conflicts of interest.
Acknowledgments
This work was supported by the Grant Agency of the Czech Republic, project P302-12-G157. Experiments were also supported by the Czech-Norwegian Research Programme CZ09, which is supervised by Norwegian funds, and by the Ministry of Education, Youth and Sport of the Czech Republic (grant number: 7F14369).
References
- Cadet J, Mouret S, Ravanat JL, Douki T. Photoinduced damage to cellular DNA: direct and photosensitized reactions. Photochem Photobiol. 2012;88(5):1048–1065. doi: 10.1111/j.1751-1097.2012.01200.x. [DOI] [PubMed] [Google Scholar]
- Cooke MS, et al. Immunochemical detection of UV-induced DNA damage and repair. J Immunol Methods. 2003;280(1-2):125–133. doi: 10.1016/s0022-1759(03)00269-2. [DOI] [PubMed] [Google Scholar]
- Lahtz C, et al. Gamma irradiation does not induce detectable changes in DNA methylation directly following exposure of human cells. PLoS One. 2012;7(9):e44858. doi: 10.1371/journal.pone.0044858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremer T, et al. Detection of chromosome aberrations in the human interphase nucleus by visualization of specific target DNAs with radioactive and non-radioactive in situ hybridization techniques: diagnosis of trisomy 18 with probe L1.84. Hum Genet. 1986;74(4):346–352. doi: 10.1007/BF00280484. [DOI] [PubMed] [Google Scholar]
- Zorn C, Cremer T, Cremer C, Zimmer J. Laser UV microirradiation of interphase nuclei and post-treatment with caffeine. A new approach to establish the arrangement of interphase chromosomes. Hum Genet. 1976;35(1):83–89. doi: 10.1007/BF00295622. [DOI] [PubMed] [Google Scholar]
- Bartova E, et al. Recruitment of Oct4 protein to UV-damaged chromatin in embryonic stem cells. PLoS One. 2011;6(12):e27281. doi: 10.1371/journal.pone.0027281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stixova L, et al. HP1beta-dependent recruitment of UBF1 to irradiated chromatin occurs simultaneously with CPDs. Epigenetics Chromatin. 2014;7(1):39. doi: 10.1186/1756-8935-7-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stixova L, et al. Advanced microscopy techniques used for comparison of UVA- and gamma-irradiation-induced DNA damage in the cell nucleus and nucleolus. Folia Biol (Praha) 2014;60(Suppl 1):76–84. [PubMed] [Google Scholar]
- Luijsterburg MS, et al. Heterochromatin protein 1 is recruited to various types of DNA damage. J Cell Biol. 2009;185(4):577–586. doi: 10.1083/jcb.200810035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartova E, et al. Coilin is rapidly recruited to UVA-induced DNA lesions and gamma-radiation affects localized movement of Cajal bodies. Nucleus. 2014;5(3):460–468. doi: 10.4161/nucl.29229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franek M, et al. Advanced Image Acquisition and Analytical Techniques for Studies of Living Cells and Tissue Sections. Microsc Microanal. 2016;22(2):326–341. doi: 10.1017/S1431927616000052. [DOI] [PubMed] [Google Scholar]
- Lemmer P, et al. Using conventional fluorescent markers for far-field fluorescence localization nanoscopy allows resolution in the 10-nm range. J Microsc. 2009;235(2):163–171. doi: 10.1111/j.1365-2818.2009.03196.x. [DOI] [PubMed] [Google Scholar]
- Reits EA, Neefjes JJ. From fixed to FRAP: measuring protein mobility and activity in living cells. Nat Cell Biol. 2001;3(6):E145–E147. doi: 10.1038/35078615. [DOI] [PubMed] [Google Scholar]
- Lakowitz JR. Principles of Fluorescence Spectroscopy. 3rd ed. New York, USA: Springers Science+Business Media, LLC; 2006. [Google Scholar]
- Piston DW, Kremers GJ. Fluorescent protein FRET: the good, the bad and the ugly. Trends Biochem Sci. 2007;32(9):407–414. doi: 10.1016/j.tibs.2007.08.003. [DOI] [PubMed] [Google Scholar]
- Levitt JA, Matthews DR, Ameer-Beg SM, Suhling K. Fluorescence lifetime and polarization-resolved imaging in cell biology. Curr Opin Biotechnol. 2009;20(1):28–36. doi: 10.1016/j.copbio.2009.01.004. [DOI] [PubMed] [Google Scholar]
- Shimomura O, Johnson FH, Saiga Y. Extraction, purification and properties of aequorin, a bioluminescent protein from the luminous hydromedusan, Aequorea. J Cell Comp Physiol. 1962;59:223–239. doi: 10.1002/jcp.1030590302. [DOI] [PubMed] [Google Scholar]
- Iwabuchi K, Bartel PL, Li B, Marraccino R, Fields S. Two cellular proteins that bind to wild-type but not mutant p53. Proc Natl Acad Sci U S A. 1994;91(13):6098–6102. doi: 10.1073/pnas.91.13.6098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward IM, Minn K, van Deursen J, Chen J. p53 Binding protein 53BP1 is required for DNA damage responses and tumor suppression in mice. Mol Cell Biol. 2003;23(7):2556–2563. doi: 10.1128/MCB.23.7.2556-2563.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda T, Sullivan KF, Wahl GM. Histone-GFP fusion protein enables sensitive analysis of chromosome dynamics in living mammalian cells. Curr Biol. 1998;8(7):377–385. doi: 10.1016/s0960-9822(98)70156-3. [DOI] [PubMed] [Google Scholar]
- Walter J, Schermelleh L, Cremer M, Tashiro S, Cremer T. Chromosome order in HeLa cells changes during mitosis and early G1, but is stably maintained during subsequent interphase stages. J Cell Biol. 2003;160(5):685–697. doi: 10.1083/jcb.200211103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaue-Sawano A, et al. Tracing the silhouette of individual cells in S/G2/M phases with fluorescence. Chem Biol. 2008;15(12):1243–1248. doi: 10.1016/j.chembiol.2008.10.015. [DOI] [PubMed] [Google Scholar]
- Sorokin DV, et al. Localized movement and morphology of UBF1-positive nucleolar regions are changed by gamma-irradiation in G2 phase of the cell cycle. Nucleus. 2015;6(4):301–313. doi: 10.1080/19491034.2015.1075111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel SS, Nguyen TA, van der Meer BW, Blank PS. The impact of heterogeneity and dark acceptor states on FRET: implications for using fluorescent protein donors and acceptors. PLoS One. 2012;7(11):e49593. doi: 10.1371/journal.pone.0049593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foltankova V, Matula P, Sorokin D, Kozubek S, Bartova E. Hybrid detectors improved time-lapse confocal microscopy of PML and 53BP1 nuclear body colocalization in DNA lesions. Microsc Microanal. 2013;19(2):360–369. doi: 10.1017/S1431927612014353. [DOI] [PubMed] [Google Scholar]
- Suchankova J, et al. Distinct kinetics of DNA repair protein accumulation at DNA lesions and cell cycle-dependent formation of gammaH2AX- and NBS1-positive repair foci. Biol Cell. 2015;107(12):440–454. doi: 10.1111/boc.201500050. [DOI] [PubMed] [Google Scholar]
- Bártová E, et al. PCNA is recruited to irradiated chromatin in late S phase and is most pronounced in G2 phase of the cell cycle. Protoplasma. 2017. [DOI] [PubMed]
- Krejci J, et al. Post-Translational Modifications of Histones in Human Sperm. J Cell Biochem. 2015;116(10):2195–2209. doi: 10.1002/jcb.25170. [DOI] [PubMed] [Google Scholar]
- Chou DM, et al. A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proc Natl Acad Sci U S A. 2010;107(43):18475–18480. doi: 10.1073/pnas.1012946107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sustackova G, et al. Acetylation-dependent nuclear arrangement and recruitment of BMI1 protein to UV-damaged chromatin. J Cell Physiol. 2012;227(5):1838–1850. doi: 10.1002/jcp.22912. [DOI] [PubMed] [Google Scholar]
- Smirnov E, et al. Separation of replication and transcription domains in nucleoli. J Struct Biol. 2014;188(3):259–266. doi: 10.1016/j.jsb.2014.10.001. [DOI] [PubMed] [Google Scholar]
- Bastiaens PI, Squire A. Fluorescence lifetime imaging microscopy: spatial resolution of biochemical processes in the cell. Trends Cell Biol. 1999;9(2):48–52. doi: 10.1016/s0962-8924(98)01410-x. [DOI] [PubMed] [Google Scholar]
- Day RN. Measuring protein interactions using Forster resonance energy transfer and fluorescence lifetime imaging microscopy. Methods. 2014;66(2):200–207. doi: 10.1016/j.ymeth.2013.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konig K, Uchugonova A, Breunig HG. High-resolution multiphoton cryomicroscopy. Methods. 2014;66(2):230–236. doi: 10.1016/j.ymeth.2013.07.006. [DOI] [PubMed] [Google Scholar]