

Abstract
Many exceptional advances have been made in mass spectrometry (MS)-based proteomics, with particular technical progress in liquid chromatography (LC) coupled to tandem mass spectrometry (LC-MS/MS) and isobaric labeling multiplexing capacity. Here, we introduce a deep-proteomics profiling protocol that combines 10-plex tandem mass tag (TMT) labeling with an extensive LC/LC-MS/MS platform, and post-MS computational interference correction to accurately quantitate whole proteomes. This protocol includes the following main steps: protein extraction and digestion, TMT labeling, 2-dimensional (2D) LC, high-resolution mass spectrometry, and computational data processing. Quality control steps are included for troubleshooting and evaluating experimental variation. More than 10,000 proteins in mammalian samples can be confidently quantitated with this protocol. This protocol can also be applied to the quantitation of post translational modifications with minor changes. This multiplexed, robust method provides a powerful tool for proteomic analysis in a variety of complex samples, including cell culture, animal tissues, and human clinical specimens.
Keywords: Biochemistry, Issue 129, bioinformatics, interference, isobaric labeling, LC-MS/MS, liquid chromatography, mass spectrometry, proteomics, TMT
Introduction
Advances in next-generation sequencing technology have led to a new landscape for studying biological systems and human disease. This has permitted a large number of measurements of the genome, transcriptome, proteome, metabolome, and other molecular systems to become tangible. Mass spectrometry (MS) is one of the most sensitive methods in analytical chemistry, and its application in proteomics has rapidly expanded after the sequencing of the human genome. In the proteomics field, the past few years have yielded major technical advances in MS-based quantitative analyses, including isobaric labeling and multiplexing capability combined with extensive liquid chromatography, in addition to instrumentation advances, allowing for faster, more accurate measurements with less sample material required. Quantitative proteomics have become a mainstream approach for profiling tens of thousands of proteins and posttranslational modifications in highly complex biological samples1,2,3,4,5,6.
Multiplexed isobaric labeling methods such as isobaric tag for relative and absolute quantitation (i.e., iTRAQ) and tandem mass tag (TMT) MS have greatly improved sample throughput and increased the number of samples that can be analyzed in a single experiment1,6,7,8. Along with other MS-based quantitation methods, such as label-free quantitation and stable isotope labeling with amino acids in cell culture (i.e., SILAC), the potential of these techniques in the proteomics field is considerable9,10,11. For example, the TMT method permits 10 protein samples to be analyzed together in 1 experiment by using 10-plex reagents. These structurally identical TMT tags have the same overall mass, but heavy isotopes are differentially distributed on carbon or nitrogen atoms, resulting in a unique reporter ion during MS/MS fragmentation of each tag, thereby enabling relative quantitation between the 10 samples. The TMT strategy is routinely applied to study biological pathways, disease progression, and cellular processes12,13,14.
Substantial technical improvements have enhanced liquid chromatography (LC) –MS/MS systems, both in terms of LC separations and MS parameters, to maximize protein identification without sacrificing quantitation accuracy. First-dimension separation of peptides by a separation technique with high orthogonality to the second dimension is critical in this type of shotgun proteomics method to achieve maximum results20. High-pH reversed-phase liquid chromatography (RPLC) provides better performance than does conventional strong cation-exchange chromatography20. When high-pH RPLC is combined with a second dimension of low-pH RPLC, both analytical dynamic range and protein coverage are improved, resulting in the ability to identify the bulk of expressed proteins when performing whole-proteome analyses15,16,17,18. Other technical advances include small C18 particles (1.9 µm) and extended long column (~1 m)19. Furthermore, other notable improvements include new versions of mass spectrometers with rapid scan rates, improved sensitivity and resolution20, and sophisticated bioinformatics pipelines for MS data mining21.
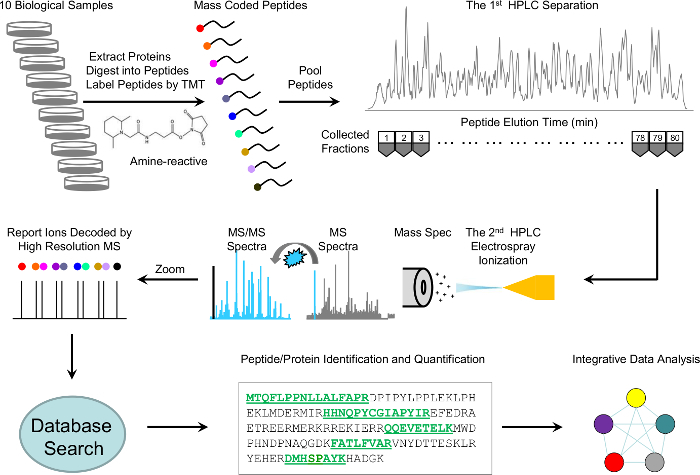
Here, we describe a detailed protocol that incorporates the most recent methodologies with modifications to improve both sensitivity and throughput, while focusing on quality control mechanisms throughout the experiment. The protocol includes protein extraction and digestion, TMT 10-plex labeling, basic pH and acid pH RPLC fractionation, high-resolution MS detection, and MS data processing (Figure 1). Moreover, we implement several quality control steps for troubleshooting and evaluating experimental variation. This detailed protocol is intended to help researchers new to the field routinely identify and accurately quantitate thousands of proteins from a lysate or tissue.
Protocol
CAUTION: Please consult all relevant safety data sheets (i.e., MSDS) before use. Please use all appropriate safety practices when performing this protocol.
NOTE: A TMT 10-plex isobaric label reagent set is used in this protocol for the proteome quantitation of 10 samples.
1. Preparation of Cells/Tissues
NOTE: It is critical to collect samples in minimal time at low temperature to keep proteins in their original biological state.
Wash adherent cells (e.g., HEK 293 cells) on a 10 cm plate twice with 10 mL of ice cold phosphate-buffered saline (PBS). Scrape and collect cells (~2 x 106 cells) in 1 mL of PBS. Transfer to 1.5 mL tubes and remove PBS after centrifugation at 600 x g for 5 min at 4 °C. Store cell pellets at -80 °C until lysis. NOTE: A pilot experiment is often performed to examine protein yield. Approximately 1 mg of proteins can be extracted from 10 x 106 mammalian cells or from a 15 cm plate with 80% confluence.
Alternatively, lyse adherent cells on their plate by adding lysis buffer directly to the plate after washing. Collect lysates into 1.5 mL tubes and use the protocol description in step 2.5.
Collect suspension cells in 15 mL tubes, wash twice with 10 mL of ice cold PBS, transfer into 1.5 mL tubes, and centrifuge at 600 x g to remove PBS. Remove PBS from the wash steps completely to maintain the desired concentration of lysis buffer ingredients. Store cell pellets at -80 °C until lysis.
Collect and weigh tissues quickly. Keep tissues in liquid nitrogen immediately after dissection and store at -80 °C. NOTE: The protein yield of tissues is approximately 5% to 10 % of the tissue weight
Use homogenous tissue sizes and anatomic regions for the 10 samples to reduce sample heterogeneity. Remove blood contamination by rinsing samples twice with 1 mL of ice cold PBS to avoid highly abundant blood proteins affecting protein quantitation and downstream data analysis.
2. Protein Extraction, Quality Control Western Blotting, In-solution Digestion, and Peptide Desalting
NOTE: Handling each of the 10 samples the same way during any step before the pooling of TMT-labeled samples is essential to reduce variation.
Make lysis buffer (50 mM HEPES [pH 8.5], 8 M urea, and 0.5% sodium deoxycholate) the day of the experiment. NOTE: Partial protein denaturation during sample lysis affects protein digestion efficiency.
Add lysis buffer so that the ratio of buffer to sample volume is 10:1 and ~20% of the final volume of glass beads (0.5 mm diameter) to cell pellets or tissues (e.g., 10: 1 volume ratio between buffer and samples) to achieve a final protein concentration of 5 to 10 mg/mL. Keep all 10 samples the same concentration by considering the number of cells used or tissue weight of each sample.
Maintain urea concentration at 8 M by adding solid urea to the sample. Remember to consider the volume of the cell pellet or tissue when calculating the amount of solid urea to add to the sample to achieve the 8 M concentration. NOTE: Un-fresh urea buffer can introduce artificial chemical modification (i.e., protein carbamylation) and negatively affect the number of peptides identified. Urea occupies a considerable volume (100 mg of urea occupies ~73 µL in solution).
Keep the insoluble debris in the lysate to allow digestion of the insoluble proteins23.
Lyse the samples in a blender at 4 °C at speed 8 for 30 s, rest for 5 s, and repeat 5 times or until the samples are homogenized. The samples can also be lysed by vortexing for 30 s at room temperature (RT) with a 30 s cooling period on ice for ~10 cycles. Adjust lysis conditions as necessary, depending on the sample type.
Mix the lysate well without centrifugation and make 2 small aliquots (~15 µL each). Use 1 aliquot to measure protein concentration and 1 aliquot to perform positive control validation by performing western blot analysis. Keep the remaining large aliquot for the proteomics analysis.
Measure protein concentration by a standard protein quantitation assay or by a Coomassie-stained short sodium dodecyl sulfate polyacrylamide gel (10%)22 with bovine serum albumin (BSA) as a standard. Use a BSA standard with a high degree of known concentration accuracy. To achieve the highest level of accuracy, perform amino acid analysis of the BSA standard. NOTE: Other accurate standards may be available. The protein concentrations can also be estimated by cell numbers or tissue weights. Although 1 mg of protein (100 µg/sample) is recommended, the analysis may be performed with as little as 100 µg of protein (10 µg/sample).
Add 100% acetonitrile (ACN) to achieve a final concentration of 10% ACN and LysC protease at an enzyme-to-substrate ratio of 1:100 (w/w) to digest proteins under highly denaturing conditions at RT for 2 h1. Add dithiothreitol (DTT) to a final concentration of 1 mM to reduce disulfide bonds in proteins and incubate for 1 h. Perform LysC digestion at a final concentration of 7.2 M urea.
Dilute samples to 2 M urea with 50 mM HEPES (pH 8.5) and further digest samples with trypsin at RT for 3 h or overnight at a trypsin to protein ratio of 1:50 (w/w). Use approximately 2 µg of trypsin for 100 µg of protein and a trypsin concentration of approximately 10 ng/µL. NOTE: Factors that can lead to inefficient trypsin digestion are pH issues, inactive trypsin, or the use of an inappropriate ratio of enzyme to substrate.
Add DTT to yield 1 mM and further reduce peptides for 2 h at RT.
Add iodoacetamide (IAA) to achieve 10 mM. Incubate at RT for 30 min in the dark to alkylate Cys-containing peptides.
Quench unreacted IAA by adding DTT to achieve 30 mM and incubate for 30 min at RT.
Check the efficiency of trypsin digestion by examining a small aliquot of each sample. Desalt by using a 10 µL pipette tip with chromatography media embedded in the dead space, according to the manufacturer's protocol. Analyze each sample by LC-MS/MS. LC-MS/MS parameters are the same as in step 5, with the exception that the gradient is 10 min23.
Perform a database search for MS raw data (see more detail in step 6) by using 4 missed cleavages as a search parameter. NOTE: If the number of missed cleavages is >10%, it may indicate incomplete trypsin digestion. This will negatively affect downstream results. This is a critical quality control (QC) step.
Digest samples again if the missed cleavage is >10% by adding an additional 10 µL of trypsin to the samples.
Acidify the samples by adding trifluoroacetic acid (TFA) to achieve 1% concentration. Measure the pH by using a pH strip. Verify that the pH is between 2 and 3. Add more TFA drop wise (1 µL) as needed until the correct pH is achieved.
Centrifuge at 20,000 x g at RT for 10 min and collect the supernatant.
Wash 0.5 to 60 µg capacity spin columns containing C18 reverse-phase resin with 0.25 mL of methanol if using 100 µg samples. Wash 0.03 to 30 µg capacity spin columns with 0.25 mL of 60% ACN/0.1% TFA if using 10 µg samples. Centrifuge spin columns at 500 x g for 30 s.
Equilibrate columns with 0.5 mL of 0.1% TFA and remove by centrifuging at 500 x g for 30 s.
Load samples on columns and bind samples to columns by centrifugation at 100 x g for 3 min or until all the sample has passed through the column.
Add 0.5 mL of 0.1% TFA to columns and centrifuge at 500 x g for 30 s.
Add 125 µL of 60% ACN/0.1% TFA to each column and elute by centrifuging at 100 x g for 3 min. Ensure all the solution has passed through the column.
Dry the eluents in a vacuum concentrator. Ensure the samples are completely dry and no liquid remains in the tubes. Store at -80 °C until further analysis.
3. TMT Labeling of Peptides
NOTE: It is critical to ensure that all samples are fully labeled by TMT reagents. Several factors (e.g., amount of TMT reagents used, pH value, and accuracy of protein quantitation) can affect TMT labeling efficiency, which will negatively alter all downstream results.
Reconstitute each desalted peptide sample in 50 µL of 50 mM HEPES buffer (pH 8.5). Check the pH and ensure that it is between 7 and 8. NOTE: Sample may be acidic if not dried completely after the desalting step, which will affect the labeling efficiency.
Keep ~1 µg of each unlabeled sample for a subsequent TMT labeling efficiency test, as described in step 3.3.
Dissolve TMT reagents in anhydrous ACN and label peptide samples by following the manufacturer's instructions. Incubate reagents at RT for 1 h.
Perform a QC TMT labeling efficiency test by desalting ~1 µg of each TMT-labeled and -unlabeled sample by using 10 µL pipette tips with embedded chromatography media according to the manufacturer's protocol. Analyze TMT-labeled and unlabeled samples by LC-MS/MS. NOTE: LC-MS/MS parameters are the same as in section 5, with the exception that the gradient is 10 min.
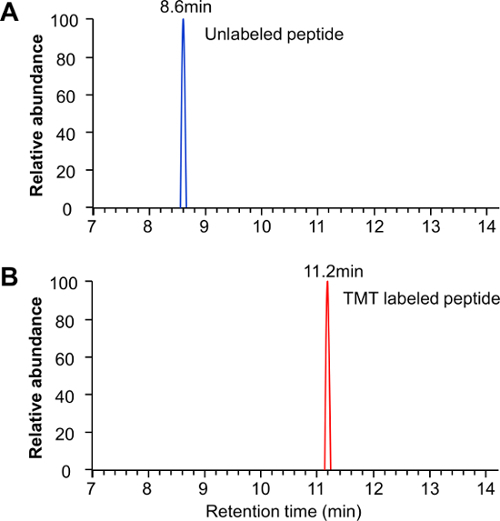
Examine the raw data to ensure that unlabeled peptides are not detected in the labeled samples, ensuring complete labelling efficiency (Figure 2). Do this for 6 to 10 separate peptides to verify the labeling efficiency.
Quench the reaction by adding 4 µL of 5% hydroxylamine and incubate for 15 min.
Mix a small and equal volume (2 µL) aliquot of each TMT-labeled sample. Desalt by using 10 µL pipette tips with embedded chromatography media. Analyze the samples by LC-MS/MS, as described in step 3.4. Use the JUMP software program (an open-source database search algorithm that converts tandem MS raw files to a list of peptides and proteins) to determine the relative concentration ratio of all 10 samples by the TMT tag intensities of all identified peptides. NOTE: This QC premix ratio test is helpful to determine the correct ratio for equal mixing before the final pooling, as pipetting errors can occur that will change concentrations and the protein quantitation step may not be always accurate. It is also the best way to ensure that the amount of each of the 10 samples used for the final mix are all at an equal ratio, as described in step 3.5.
Repeat as many times as necessary to achieve a 1:1 ratio across all 10 samples.
Equally mix the 10 samples according to the results of the premix ratio test. Use the sample with the lowest concentration as the baseline to adjust the volumes of the other 9 samples.
Remove the byproducts of the quenching reaction of the pooled TMT-labeled sample by desalting.
Wash 1 mL solid-phase extraction cartridges containing 50 mg sorbent per column with 1 mL/0.25 mL of methanol if using 100 µg samples. Wash 0.5- to 60-µg capacity C18 spin columns with 1 mL/0.25 mL of 60% ACN/0.1% TFA if using 10 µg samples. Centrifuge columns at 500 x g for 30 s.
Equilibrate columns with 0.5 mL of 0.1% TFA and remove by centrifuging at 500 x g for 30 s.
Load samples on columns and bind by centrifugation at 100 x g for 3 min or until all of the sample has passed through the column.
Add 0.5 mL of 0.1% TFA to columns and centrifuge at 500 x g for 30 s.
Add 125 µL of 60% ACN/0.1% TFA to each column and elute by centrifuging at 100 x g for 3 min. Ensure all of the solution has passed through the column.
Dry the eluents in a vacuum concentrator. Ensure the samples are completely dry and no liquid remains in the tubes. Store at -80 °C until further analysis.
4. Extensive High-resolution, Basic pH LC Prefractionation
Set up 2 connected C18 columns containing bridged ethylene hybrid particles (4.6 mm x 25 cm, totaling 50 cm; 3.5 µm particle size) and use a microliter flow high-performance LC pump for fractionation. NOTE: The use of 2 columns increases peptide load and does not necessarily improve chromatography. For samples containing ≤200 µg of peptides, 1 column is sufficient.
Wash the 100-µL loop with subsequent additions of 300 µL each of methanol, water, and buffer A (10 mM ammonium formate, pH 8.0).
Wash the columns with 100 µL of equally mixed isopropanol, methanol, ACN, and water and equilibrate the column in 95% buffer A for 2 h.
Solubilize the desalted pooled TMT-labeled peptide sample in 65 µL of buffer A. Verify that the sample pH is ~8.0. If still acidic, use ammonium hydroxide to adjust pH to 8.0.
Fractionate sample by using the following gradient with buffer B (buffer A plus 90% ACN): 15% to 20% for 15 min, 20% to 35% for 100 min, and 35% to 50% for 30 min. Set the fraction collector to collect fractions every 2 min including loading time. Set the flow rate to 0.4 mL/min. See reference29 and Figure 1 for a representative chromatogram.
Collect a total of 80 fractions and dry 40 fractions (every other fraction) with a vacuum concentrator to complete dryness.
Use the 40 dried samples for LC-MS/MS analysis.
Analyze all 80 fractions by LC-MS/MS to achieve ultra-deep proteome coverage.
5. LC-MS/MS Preparation and Parameters
NOTE: In TMT-based quantitation, peptide ions are isobaric and appear as 1 mass in an MS1 scan. However, they are quantitated according to the intensity of reporter ions (10 unique reporter ions) in the MS/MS scan after the peptide ion has been fragmented with higher energy collision dissociation (HCD). The TMT reporter ion ratios may be suppressed from co-elution of TMT-labeled ions24. Narrowing the ion isolation window25, gas-phase purification26, the MultiNotch MS3 method27, or extensive fractionation with multidimensional LC and long gradients (4-8 h)28 are alternative approaches.
Pack 75 µm inner diameter (ID) empty columns with 1.9 µm of C18 resin to 30 to 40 cm length (~1.3 µL bed volume).
Heat the columns at 65 °C with a butterfly portfolio heater to reduce backpressure and prevent over pressuring of the ultra-high-pressure liquid chromatography (UPLC) system. Coil the column 2 to 3 times within the butterfly heater to ensure that the whole column bed length is heated. Tape the column to the inside of the heater being careful not to tape over the temperature sensor inside the heater. NOTE: The butterfly portfolio heater is mounted and taped onto a custom-made piece of plexiglass attached to the XYZ stage of the instrument.
Prepare buffer A (3% dimethyl sulfoxide and 0.2% formic acid) and buffer B (buffer A plus 67% ACN) and wash the columns thoroughly with 95% buffer B. Then, fully equilibrate columns in 95% buffer A (at least 3 column volumes). Use a flow rate of ~0.25 µL/min and backpressure of 280 to 320 bar.
Examine the quality of the LC-MS/MS system by running 100 ng of rat brain peptides (or standard of our choice) twice before analyzing the TMT-labeled samples. Check the following parameters frequently to ensure high-quality data collection: survey MS signal intensity (between e8 and e9 for base peak intensity), quality of MS/MS spectra (a good representation of y and b ions), peak width (12-22 s), overall retention time, LC system pressure (pressure rise >50 bars indicates a dirty column or clogged emitter tip), and run-to-run variability (identical peaks should be <30 s between runs). NOTE: To resolve the near-isobaric reporter ions of the 10-plex TMT reagents (6.32 mDa difference), the MS/MS resolution must be set to at least 30,000 at 400 m/z. HCD is typically used for TMT10-plex reporter ion fragmentation, as it is not subject to the one-third rule that applies to collision induced dissociation (CID) in ion trap instruments.
Clean and calibrate the MS instrument regularly and use fresh LC buffers to improve the system performance.
Reconstitute the dried peptides from the basic pH separation in 5% TFA.
Load ~0.2 µg on the column while flowing 5% buffer A.
Elute with 15% to 45% of buffer B in 150 min. As a starting point, use the following gradient: time 0, buffer B 5%; time 2, buffer B 15%; time 135, buffer B 45%, time 145, buffer B 70%; and time 150, buffer B 95%. Adjust the gradient slightly for early and late basic pH RPLC fractions after reviewing base peak chromatograms.
Operate the mass spectrometer in data-dependent mode with a survey scan in the ion trap mass analyzer (400-1600 m/z; 60,000 resolution; 1 x 106 automatic gain control (AGC) target; 50 ms maximum ion time; and centroid mode). Perform 20 MS/MS high-resolution scans (60,000 resolution, 1 x 105 AGC target, ~100 ms maximum ion time, 35 HCD normalized collision energy, 0.4 m/z isolation window, and 20 s dynamic exclusion). NOTE: Electron transfer dissociation (ETD) is not recommended for TMT10-plex reagents because ETD cleavage sites are different from HCD, resulting in reporter ion overlap. In addition, ETD is not effective as is HCD because tryptic peptides typically generate +2 charged states, and ETD is more efficient at higher charge states.
6. MS Data Analysis
NOTE: We describe data analysis with the JUMP software program. However, data analysis can be performed with other commercially available or free programs.
Process raw files from the mass spectrometer with the tag-based hybrid search engine JUMP21, which combines a pattern-based database search and a tag-based de novo sequencing to improve sensitivity and specificity.
Convert raw data to mzXML format and search MS2 spectra against the target-decoy UniProt human database (or other appropriate species-specific database) to calculate the false discovery rate (FDR).
Perform searches by using a 10 ppm mass tolerance for both precursor and fragment ions. Set other search parameters to fully tryptic restriction with 2 maximum missed cleavages, 3 maximum modification sites per peptide, and the assignment of a, b, and y ions to score the peptides. Set static modifications to TMT tags on Lys residues and N termini (+229.162 Da) and carbamidomethylation of Cys residues (+57.021 Da). Set dynamic modifications to include Met oxidation (+15.994 Da), which is a common peptide artifact resulting from sample handling.
Filter data by the following criteria: 7 amino acid minimum peptide length, m/z accuracy (e.g., 5 ppm), and matching scores (J score and deltaCn). Group peptides according to peptide length, trypticity, and charge state.
Use an additional level of filtering by matching scores to reduce protein FDR to below 1%. Proteins identified by a single spectral count require a higher matching score.
For peptides shared by multiple members of a protein family, cluster the matched protein members into 1 group. NOTE: According to the parsimony principle, the group is represented by the protein with the highest number of assigned peptides and other proteins matched by unique peptides1.
Quantitate proteins by summing reporter ion counts across all matched peptide spectrum matches (PSMs) by using a program built into the JUMP software suite.
For each accepted PSM, extract and correct the TMT reporter ion intensities according to the isotopic distribution of the labeling reagents and the loading bias, which is calculated from the global PSM quantitation data. Filter PSMs with low intensity and high noise level with user-defined thresholds during the quantitation.
Calculate a relative signal between each reporter ion and the average of all 10 reporter ions. Sum the relative signals of the PSMs for the identified proteins. Convert these relative signals to absolute signals by multiplying the averaged reporter ion intensity of the top 3 PSMs in corresponding proteins.
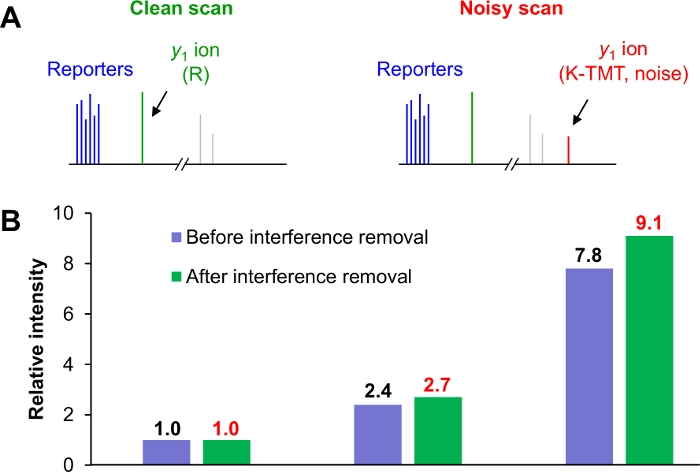
Use a post-MS computational approach to correct interference. Assuming that the y1 ion intensity is proportional to the reporter ion intensity, calculate the linear relationship from clean scans and derive the interference level from the contaminated y1ion intensity in noisy scans23. NOTE: For TMT-labeled tryptic peptides, K-TMT and R residues are 2 types of y1 ions (376.27574 Da and 175.11895 Da, respectively) in MS2 scans. If only 1 y1 ion is detected and is consistent with the identified peptide, the MS2 is deemed clean scan (Figure 3A). If both y1ions are detected, the MS2 is deemed a noisy scan.
Export the protein quantitation values to a spreadsheet software program for further analysis. Use pairwise comparisons or an analysis of variance to identify proteins that are differentially expressed.
7. MS Data Validation
NOTE: To evaluate the quality of MS data, at least 1 method of validation should be performed before proceeding with time-consuming biological experiments.
Manually examine the MS/MS spectra of proteins of interest to validate the peptide sequence and TMT reporter ion quantitation.
Use known protein quantitation data to confirm the MS results.
Verify protein changes with antibody-based approaches (e.g., western blot and immunohistochemistry analysis).
Chemically synthesize the peptides of interest and use them as internal standards to confirm the identification of native peptides. NOTE: Their MS/MS patterns and retention time during LC-MS/MS should be identical.
Verify protein changes with a targeted MS approach.
Representative Results
We used a previously described cross-species peptide mix to systematically analyze the effect of ratio compression in 3 major protocol steps, including pre-MS fractionation, MS settings, and post-MS correction23. The pre-MS fractionation was evaluated and optimized by using a combination of basic pH RPLC and acidic pH RPLC. For post-MS analysis, only species-specific peptides were considered. We used this interference model to examine a number of parameters in LC/LC-MS/MS, including the MS2 isolation window, online LC resolution, loading amount of the online acidic pH RPLC, and resolution of the offline high-pH RPLC (see Figure 3 in reference29).
To alter the resolution during the offline LC, we separated the isobaric labeled peptides into 320 fractions, and then combined selected fractions together to adjust the separation power. Offline LC resolution was tested by using a number of collected fraction subsets (1, 5, 10, 20, 40, 80, and 320), while monitoring the interference levels. We found that the interference levels decreased gradually from 16.4% to 2.8%, indicating that extensive fractionation during offline LC separation somewhat alleviated the problem of co-eluting peptides but was not capable of completely removing the interference.
Next, we evaluated the effect of the MS2 isolation window on interference. We determined experimentally that the interference was approximately proportional to the size of the isolation window, in agreement with previous studies29,30. For example, a 4-fold difference of window size (1.6 to 0.4 Da) resulted in a ~4-fold difference of inference level (14.4% to 3.7%). We also determined the optimal loading amount on the column for MS analysis and used this information to further negate interference. We reduced the interference from 9.4% to 3.3% by loading the proper amount of sample. Loading too much sample may lead to peak broadening31 and therefore raise the interference level, resulting in reduced protein quantitation accuracy. Lastly, we optimized the online RPLC resolution by changing the gradient lengths (1, 2, and 4 h) and using a long column (~45 cm). We found that a 4-h gradient nearly eliminated the interference (down to 0.4%) but also added instrument time. The optimized parameters revealed a narrow isolation window (0.4 Da), ~100 ng of sample loaded on column, and midlevel fractionation (~40 x 2 h, 3.3 days). We also showed that by using a computer-based post-MS correction strategy we improved quantitative precision when determining protein ratios (Figure 3B).
Our results indicate that the use of optimized LC-MS parameters and post-MS correction can provide a thorough proteomic profile and virtually eliminate the interference that is often produced by isobaric labeling techniques for protein quantitation.
Figure 1: Scheme of whole-proteome profiling analysis by TMT-LC/LC-MS/MS. Ten biological samples were lysed, digested, and labeled with 10 different TMT tags, pooled equally, and fractionated into 80 fractions by offline basic pH reverse-phase liquid chromatography (LC). Every other fraction was further separated by acidic RPLC and analyzed online with a high-resolution mass spectrometer. The MS/MS raw files were processed and searched against a Uniprot database for peptide and protein identification. Proteins were quantitated according to the relative intensities of the TMT tags. Finally, the identified and quantitated proteins were submitted for integrative data analysis. Please click here to view a larger version of this figure.
Figure 2: TMT labeling efficiency examination. Both TMT-labeled and their corresponding unlabeled samples were analyzed by LC-MS/MS separately to examine TMT labeling efficiency. For full TMT labeling, (A) the unlabeled peptide peak was completely absent in the labeled sample, (B) whereas the TMT-labeled peptide was present only in labeled sample. Please click here to view a larger version of this figure.
Figure 3: Computational approach for interference removal after MS data collection. (A) All MS2 scans were divided into clean (left panel) and noisy scans (right panel). Noisy scans exhibit both y1 ions of K and R (1 from a target peptide and the others from contaminating peptides). (B) Escherichia coli peptides were individually labeled with 3 different TMT reagents and pooled at 1:3:10 ratios. Approximately 20-fold more rat peptides were added as background. The summed relative intensities of the E. coli peptides in the 3 groups revealed that y1 ion-based correction is more accurate for TMT-based quantitation. Please click here to view a larger version of this figure.
Discussion
We describe a high-throughput protocol for the quantitation of proteins with a 10-plex isobaric labeling strategy, which has been implemented successfully in several publications12,13,14,32. In this protocol, we can analyze up to 10 different biological protein samples in 1 experiment. We can routinely identify and quantitate well over 10,000 proteins with high confidence. Although isobaric labeling is an effective technology to quantitate proteins, it is limited by ratio compression that can lead to quantitative inaccuracy. Our protocol using various strategies, such as the optimization of LC/LC and MS/MS settings in combination with y1 ion-based post-MS correction, effectively removes most interference effects and therefore considerably enhances the precision of protein quantitation.
For protein quantitation, the JUMP suite of programs functions in a number of ways. We measured the isotopic impurity for every batch of purchased reagents, which differs slightly from the reported values in vendor-provided certificates. The measured isotopic impurity is used for correcting the intensities of reporter ions in the JUMP software. A protein is typically quantitated by many PSMs with various absolute intensities that are influenced by multiple factors, such as ionization efficiency. Because the influence of the factors is unknown, mass spectra reveal only the relative abundances of reporter ions in a TMT assay. The relative abundances of reporter ions are calculated by dividing the intensity of each reporter ion by the average intensity of all 10 reporters. To provide the best representative "absolute signal" of the protein, we calculated an average of the relative intensities of 3 PSMs with the highest absolute intensities from the protein and the average of the strongest absolute intensity to estimate the absolute signal of the protein. We defined these steps as rescaling. The y1 correction is universally applicable to any TMT assays; however, in some cases we could not correct for peptides with MS/MS spectra containing the y1 ion. However, we found that ~90% of spectra have the y1 ions from both lysine and arginine. To compensate for the interference caused by co-eluting peptides that have the same C-terminal residues as the identified peptide, the estimated interference level was essentially doubled and used for correction. This assumption was based on empirical evidence that we have collected over numerous TMT experiments, in which we observed the same intensity level for y1 in K- and R-containing peptides.
Interference can be reduced by many methods other than what we have described in this protocol, such as the MS3 multinotch approach. All of these methods are valid and have their individual merits. Ultimately, the methodology used depends on a number of factors, including but not limited to sample amount, instrument availability (i.e., instrument type and time required for analyzing samples), desired results (i.e., number of proteins identified), and quantitation accuracy needed.
To ensure the most accurate results, it is important to heed the quality control steps throughout the protocol. These include using accurate standards for sample quantitation (e.g., BSA that has undergone amino acid analysis), testing the labelling efficiency of the TMT reagent, determining the efficiency of trypsin digestion, performing a premix ratio test to ensure an equal mixing of all 10 samples, and using a software to correct any loading bias. In cases in which a known protein or multiple proteins are expected to exhibit expression changes between individual samples, it is important to perform western blot analysis after the initial cell lysis to confirm that these changes are present and can be detected. This ensures that the samples represent the biology to be tested and saves time, money, and effort before carrying out the entire TMT protocol. If a particular sample is expected to not express certain protein(s), it is also important to use western blot analysis to confirm their absence after lysis before continuing with the protocol. The premix ratio test is used when most proteins are expected to not change across the 10 biological samples. We also removed known contaminating proteins, such as keratins, so they would not negatively affect our correction. Our quantitation method corrected for any errors that may have occurred from pipetting errors, etc. When possible, we specifically used pipette volumes greater than 5 µL to reduce errors. We performed multiple rounds of this premix test to ensure that we obtained an accurate 1:1 mix. It is important to note that we did not perform this type of premix ratio test when using immunoprecipitation samples for the protocol, as we expected a large percentage of the proteins to change. In such cases, the premix test would skew the results. This is also true of any experiment in which at least 1 of the 10 samples is expected to vary greatly in protein expression (empty vector, proteasome inhibition, etc.) In such cases, it is highly recommended to use replicates to facilitate quantitation statistics. We typically recommend performing 3 replicates for these types of samples. Ultimately, the number of replicates is based on the expected variability of each sample.
With some fine-tuning, this robust protocol can be used as a general proteomic pipeline to investigate proteins, protein pathways, disease progression, and other biological functions. The protocol presented here provides a powerful technique to obtain deep protein coverage and alleviate ratio suppression during quantification. With minor modifications, this protocol can be readily adapted to quantitate protein posttranslational modifications, such as phosphorylation, ubiquitination, and acetylation33,34. These types of comprehensive proteomics projects can be integrated with genomics, transcriptomics, and possibly metabolomics for a systems biology approach to understand biological systems and facilitate novel discoveries of molecular mechanisms, biomarkers, and therapeutic targets in disease13,28,35,36,37,38.
We have demonstrated a method for accurately quantitating over 10,000 proteins from whole-cell or tissue lysates. We expect this method to be widely applicable to many biological systems.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The authors thank all other lab and facility members for helpful discussion. This work was partially supported by NI H grants R01GM114260, R01AG047928, R01AG053987, and ALSAC. The MS analysis was performed in the St. Jude Children's Research Hospital Proteomics Facility, partially supported by NIH Cancer Center Support grant P30CA021765. The authors thank Nisha Badders for help with editing the manuscript.
References
- Pagala VR, et al. Quantitative protein analysis by mass spectrometry. Methods Mol Biol. 2015;1278:281–305. doi: 10.1007/978-1-4939-2425-7_17. [DOI] [PubMed] [Google Scholar]
- Altelaar AF, Munoz J, Heck AJ. Next-generation proteomics: towards an integrative view of proteome dynamics. Nat Rev Genet. 2013;14(1):35–48. doi: 10.1038/nrg3356. [DOI] [PubMed] [Google Scholar]
- Rauniyar N, Yates JR., 3rd Isobaric labeling-based relative quantification in shotgun proteomics. J Proteome Res. 2014;13(12):5293–5309. doi: 10.1021/pr500880b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross PL, et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics. 2004;3(12):1154–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- Aebersold R, Mann M. Mass-spectrometric exploration of proteome structure and function. Nature. 2016;537(7620):347–355. doi: 10.1038/nature19949. [DOI] [PubMed] [Google Scholar]
- Bai B, et al. Deep Profiling of Proteome and Phosphoproteome by Isobaric Labeling. Extensive Liquid Chromatography, and Mass Spectrometry. Methods Enzymol. 2017;585:377–395. doi: 10.1016/bs.mie.2016.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlister GC, et al. Increasing the multiplexing capacity of TMTs using reporter ion isotopologues with isobaric masses. Anal Chem. 2012;84(17):7469–7478. doi: 10.1021/ac301572t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everley RA, Kunz RC, McAllister FE, Gygi SP. Increasing throughput in targeted proteomics assays: 54-plex quantitation in a single mass spectrometry run. Anal Chem. 2013;85(11):5340–5346. doi: 10.1021/ac400845e. [DOI] [PubMed] [Google Scholar]
- Bai B, et al. Integrated approaches for analyzing U1-70K cleavage in Alzheimer's disease. J Proteome Res. 2014;13(11):4526–4534. doi: 10.1021/pr5003593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai B, et al. U1 small nuclear ribonucleoprotein complex and RNA splicing alterations in Alzheimer's disease. Proc Natl Acad Sci U S A. 2013;110(41):16562–16567. doi: 10.1073/pnas.1310249110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thongboonkerd V, LaBaer J, Domont GB. Recent advances of proteomics applied to human diseases. J Proteome Res. 2014;13(11):4493–4496. doi: 10.1021/pr501038g. [DOI] [PubMed] [Google Scholar]
- Churchman ML, et al. Efficacy of Retinoids in IKZF1-Mutated BCR-ABL1 Acute Lymphoblastic Leukemia. Cancer Cell. 2015;28(3):343–356. doi: 10.1016/j.ccell.2015.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, et al. Joint mouse-human phenome-wide association to test gene function and disease risk. Nat Commun. 2016;7:10464. doi: 10.1038/ncomms10464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertz JL, et al. Sequential elution interactome analysis of the Mind bomb 1 ubiquitin ligase reveals a novel role in dendritic spine outgrowth. Mol Cell Proteomics. 2015;14(7):1898–1910. doi: 10.1074/mcp.M114.045898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Shen Y, Camp DG, 2nd, Smith RD. High-pH reversed-phase chromatography with fraction concatenation for 2D proteomic analysis. Expert Rev Proteomics. 2012;9(2):129–134. doi: 10.1586/epr.12.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, et al. An off-line high pH reversed-phase fractionation and nano-liquid chromatography-mass spectrometry method for global proteomic profiling of cell lines. J Chromatogr B Analyt Technol Biomed Life Sci. 2015;974:90–95. doi: 10.1016/j.jchromb.2014.10.031. [DOI] [PubMed] [Google Scholar]
- Batth TS, Francavilla C, Olsen JV. Off-line high-pH reversed-phase fractionation for in-depth phosphoproteomics. J Proteome Res. 2014;13(12):6176–6186. doi: 10.1021/pr500893m. [DOI] [PubMed] [Google Scholar]
- Song C, et al. Reversed-phase-reversed-phase liquid chromatography approach with high orthogonality for multidimensional separation of phosphopeptides. Anal Chem. 2010;82(1):53–56. doi: 10.1021/ac9023044. [DOI] [PubMed] [Google Scholar]
- Wang H, et al. Systematic optimization of long gradient chromatography mass spectrometry for deep analysis of brain proteome. J Proteome Res. 2015;14(2):829–838. doi: 10.1021/pr500882h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert AS, et al. The one hour yeast proteome. Mol Cell Proteomics. 2014;13(1):339–347. doi: 10.1074/mcp.M113.034769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, et al. JUMP: a tag-based database search tool for peptide identification with high sensitivity and accuracy. Mol Cell Proteomics. 2014;13(12):3663–3673. doi: 10.1074/mcp.O114.039586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Duong DM, Peng J. Systematical optimization of reverse-phase chromatography for shotgun proteomics. J Proteome Res. 2009;8(8):3944–3950. doi: 10.1021/pr900251d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu M, et al. Extensive Peptide Fractionation and y1 Ion-Based Interference Detection Method for Enabling Accurate Quantification by Isobaric Labeling and Mass Spectrometry. Anal Chem. 2017;89(5):2956–2963. doi: 10.1021/acs.analchem.6b04415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting L, Rad R, Gygi SP, Haas W. MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat Methods. 2011;8(11):937–940. doi: 10.1038/nmeth.1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitski MM, et al. Delayed fragmentation and optimized isolation width settings for improvement of protein identification and accuracy of isobaric mass tag quantification on Orbitrap-type mass spectrometers. Anal Chem. 2011;83(23):8959–8967. doi: 10.1021/ac201760x. [DOI] [PubMed] [Google Scholar]
- Wenger CD, et al. Gas-phase purification enables accurate, multiplexed proteome quantification with isobaric tagging. Nat Methods. 2011;8(11):933–935. doi: 10.1038/nmeth.1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlister GC, et al. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal Chem. 2014;86(14):7150–7158. doi: 10.1021/ac502040v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou F, et al. Genome-scale proteome quantification by DEEP SEQ mass spectrometry. Nat Commun. 2013;4:2171. doi: 10.1038/ncomms3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitski MM, et al. Delayed fragmentation and optimized isolation width settings for improvement of protein identification and accuracy of isobaric mass tag quantification on Orbitrap-type mass spectrometers. Anal. Chem. 2011;83(23):8959–8967. doi: 10.1021/ac201760x. [DOI] [PubMed] [Google Scholar]
- Ahrne E, et al. Evaluation and Improvement of Quantification Accuracy in Isobaric Mass Tag-Based Protein Quantification Experiments. J Proteome Res. 2016;15(8):2537–2547. doi: 10.1021/acs.jproteome.6b00066. [DOI] [PubMed] [Google Scholar]
- Xu P, Duong DM, Peng JM. Systematical Optimization of Reverse-Phase Chromatography for Shotgun Proteomics. J Proteome Res. 2009;8(8):3944–3950. doi: 10.1021/pr900251d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan H, et al. Integrative Proteomics and Phosphoproteomics Profiling Reveals Dynamic Signaling Networks and Bioenergetics Pathways Underlying T Cell Activation. Immunity. 2017;46(3):488–503. doi: 10.1016/j.immuni.2017.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Na CH, Tan H, Peng J. Global ubiquitination analysis by SILAC in mammalian cells. Methods Mol Biol. 2014;1188:149–160. doi: 10.1007/978-1-4939-1142-4_11. [DOI] [PubMed] [Google Scholar]
- Tan H, et al. Refined phosphopeptide enrichment by phosphate additive and the analysis of human brain phosphoproteome. Proteomics. 2015;15(2-3):500–507. doi: 10.1002/pmic.201400171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, et al. JUMPg: an Integrative Proteogenomics Pipeline Identifying Unannotated Proteins in Human Brain and Cancer Cells. J Proteome Res. 2016;17(7):2309–2320. doi: 10.1021/acs.jproteome.6b00344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, et al. Assessing the clinical utility of cancer genomic and proteomic data across tumor types. Nat Biotechnol. 2014;32(7):644–652. doi: 10.1038/nbt.2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesvizhskii AI. Proteogenomics: concepts, applications and computational strategies. Nat Methods. 2014;11(11):1114–1125. doi: 10.1038/nmeth.3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagerberg L, et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteomics. 2014;13(2):397–406. doi: 10.1074/mcp.M113.035600. [DOI] [PMC free article] [PubMed] [Google Scholar]