

Abstract
In recent years, the zebrafish embryo has emerged as a popular model to study developmental biology due to traits such as ex utero embryo development and optical transparency. In particular, the zebrafish embryo has become an important organism to study vertebrate kidney organogenesis as well as multiciliated cell (MCC) development. To visualize MCCs in the embryonic zebrafish kidney, we have developed a combined protocol of whole-mount fluorescent in situ hybridization (FISH) and whole mount immunofluorescence (IF) that enables high resolution imaging. This manuscript describes our technique for co-localizing RNA transcripts and protein as a tool to better understand the regulation of developmental programs through the expression of various lineage factors.
Keywords: Developmental Biology, Issue 129, Cilia, zebrafish, multiciliated cell, fluorescent in situ hybridization, immunofluorescence, pronephros, kidney
Introduction
Over the past several decades, the zebrafish (Danio rerio) has emerged as a prime model organism to study developmental biology. The embryos develop outside of the mother and are optically transparent. Also, the formation of vital organs such as the eye, kidney, and forebrain occurs rapidly, with structures formed by just 24 h post fertilization (hpf). Importantly, the zebrafish genome is highly conserved with mammals1,2,3. Additionally, zebrafish and mammalian organs have similar anatomy and physiology. The zebrafish embryonic kidney, or pronephros, demonstrates the value of the model system for examining gene function during early nephrogenesis and fate determination of conserved epithelial cell populations of the vertebrate nephron4,5,6,7,8,9,10. Similarly, the zebrafish embryo has become increasingly important in examining the ontogeny of MCCs11,12,13,14,15,16,17.
As their name suggests, MCCs are epithelial cells characterized by a bundle of motile cilia located on the apical surface17. In the zebrafish, MCCs function in fluid flow and are dispersed in a "salt-and-pepper" like fashion throughout the middle of each nephron of the pronephros by 24 hpf11,12,13,14,15,16,17. Although they have only been noted in a handful of human kidney disease cases18,19,20,21, MCCs are prevalent in other mammalian tissues such as the brain and trachea22,23,24, which poses a host of challenges for experimental design. Elegant studies in various vertebrate models including zebrafish have demonstrated a conserved pathway of MCC fate, with the Notch signaling pathway as an inhibitor of MCC development12,17,25,26,27. Therefore, the zebrafish pronephros provides an easily accessible model to study the genetic mechanisms of MCC development in vivo11,12,13,14,15,16,17.
Both transparency and easy genetic manipulation of zebrafish embryos have proven to be invaluable traits when studying the genetic and molecular pathways that regulate cell fate, tissue growth, and development of the early embryo1,2,3. As such, traditional techniques to visualize protein and gene transcripts, such as in situ hybridization and whole mount IF, have been applied to and optimized for the zebrafish16,28,29,30,31,32,33,34,35,36,37,38. By combining popular protocols for FISH and IF, it is possible to label and analyze MCCs in vivo16,28,37,38.
Protocol
The following protocol uses zebrafish adults maintained and cared for by the Center for Zebrafish Research at the University of Notre Dame. All methods for working with zebrafish adults and embryos were approved by the Institutional Animal Care and Use Committee.
1. Embryo Fixation
Collect zebrafish embryos using previously described methods39,40. At 24 hpf, add 500 µL of non-specific protease mixture solution (50 mg/mL) to the E3 in an embryo dish Incubate at room temperature (RT).
Once embryos begin breaking out of the chorion, remove all liquid from the embryo dish.
Wash the embryos 2 - 3 times with fresh E3 by adding approximately 20 mL of E3, gently swirling the E3 in the dish, and then decanting the fluid into a liquid waste receptacle.
To euthanize the embryos before fixing, add 2 mL of 0.2% tricaine to the E3.
Transfer the embryos into a 5 mL glass vial using a plastic Pasteur pipette. Remove most of the tricaine/E3 solution using the pipette after the embryos have settled to the bottom of the vial.
Fix 24 hpf embryos with 5 mL of 4% paraformaldehyde (PFA) for 2 - 4 h at RT, without agitation. CAUTION: PFA is toxic and PFA solutions should be handled under a chemical hood while the researcher is wearing eye protection, gloves, and a lab coat. Preparation of the PFA fixative from the granulated PFA powder should be performed with exceptional care, as the granulated PFA tends to disperse through static charge. NOTE: Embryos can fix in 4% PFA overnight at 4 °C. Prepare 4% PFA by heating 1x phosphate buffered saline (PBS) to a boil while continually stirring the solution to completely dissolve the granulated PFA. Once the solution has cooled back down to RT, the 4% PFA is filter sterilized by passing it through a 0.45 µm disposable system for vacuum filtration, then and aliquoted into 15 mL or 50 mL portions for storage at -20 °C.
Remove the 4% PFA and wash embryos twice with 1X phosphate buffered saline with 0.1% Tween-20 (PBST) at RT. Wash the embryos once with 5 mL of 100% methanol (MeOH) at RT. Add 5 mL of fresh 100% MeOH to the embryos and incubate at -20 °C for at least 20 min. NOTE: Embryos can be stored in 100% MeOH at -20°C until ready for embryo preparation.
2. Embryo preparation and hybridization
Rehydrate the embryos by washing once in 5 mL of 50% MeOH in 1x PBST for 5 min at RT. Continue rehydration by washing the embryos in 5 mL of 30% MeOH in 1x PBST for 5 min at RT. Wash the embryos in 5 mL of 1x PBST twice for 5 min each at RT.
Permeabilize the embryos by incubating them in 5 mL of a solution of 1:1,000 proteinase K (pK) (10 mg/mL) in 1x PBST for 2 min at RT. NOTE: Older embryos can be incubated longer than 2 min in the pK concentration stated above, however a concentration of 5 mg/mL and incubation time less than 2 min is suggested for embryos younger than 24 hpf.
Remove pK solution and wash immediately in 5 mL of 1x PBST twice at RT. Fix the embryos in 5 mL of 4% PFA at RT for at least 20 min. NOTE: Embryos can fix in 4% PFA overnight at 4 °C.
Remove PFA and wash twice in 5 mL of 1x PBST at RT.
Transfer embryos in 5 mL of 1x PBST into flat bottom microcentrifuge tubes placed upright into a microcentrifuge rack at RT to facilitate interactions between each embryo and the subsequent hybridization solution.
Wash the embryos twice in 1.5 mL of hybridization solution (Hyb+) at RT. Add 1.5 mL of fresh Hyb+ and incubate embryos at 70 °C in a hybridization oven for 4-6 h.
Replace the 1.5 mL of Hyb+ with a volume of probe solution (10 µL RNA probe/500 µL Hyb+) sufficient to fully cover the embryos. NOTE: Synthesize antisense RNA probe using previously described methods39.
Hybridize the probe overnight in the hybridization oven at 70 °C with the individual tubes positioned upright in the microcentrifuge rack, without shaking.
3. Hot Washes and Blocking
NOTE: Hot washes are performed by putting the appropriate solution on the embryos and then incubating in the hybridization oven at 70 °C. To keep the washing solutions at 70 °C, place 50 mL tubes of each solution in the hybridization oven when the probe/Hyb+ mixture is added.
Remove the probe. Wash the embryos twice at 70 °C in 1.5 mL of 50% formamide/2x saline-sodium citrate (SSC) buffer for 20-30 min each. NOTE: The probe can be stored in Hyb+ at -20 °C and re-used at a later date.
Wash the embryos once at 70 °C in 1.5 mL of 2x SSC for 15 min. Wash the embryos twice at 70 °C in 1.5 mL of 0.2X SSC for 20-30 min each. Replace 0.2x SSC with 1.5 mL of blocking reagent and incubate overnight at 4 °C. NOTE: It is possible to incubate blocking reagent at RT for 4 h instead of overnight.
4. Antibody Incubation and Maleic Acid Buffer Washes
NOTE: Keep embryos protected from ambient light.
Replace the blocking reagent with 0.2 mL of anti-DIG-POD in blocking reagent at a ratio of 1:1,000 and incubate for 3 h under foil or another light-blocking cover at RT. NOTE: Alternatively, the antibody can incubate overnight at 4 °C.
After 3 h, wash the embryos in 1.5 mL of maleic acid buffer 2-4 times for 10-15 min each at RT. Incubate the embryos overnight at 4 °C in 1.5 mL of fresh maleic acid buffer. NOTE: It is not necessary to incubate in maleic acid buffer overnight, but doing so decreases probe background.
5. Probe Detection and Removal of Peroxidase
NOTE: Keep embryos protected from ambient light.
Remove maleic acid buffer and replace with 1.5 mL of 1x PBS. Wash embryos twice in 1.5 mL of 1x PBS for 5 min each at RT. Incubate embryos in 0.2 mL of Cy3 fluorescent staining solution for 60 min at RT. NOTE: Incubation time may vary for different probes.
Wash the embryos once with 1.5 mL of the following percent MeOH in 1X PBS for 10 min at RT: 30% MeOH, 50% MeOH, 75% MeOH and 100% MeOH.
Incubate the embryos with 1.5 mL of 1% H2O2 in MeOH for 30 min at RT. Wash the embryos once with 1.5 mL of the following MeOH solutions in 1x PBS for 10 min at RT: 75% MeOH, 50% MeOH, and 30% MeOH. Wash the embryos twice for 5 min each at RT in 1.5 mL of 1x PBS. NOTE: Embryos can be stored in PBS overnight at 4°C.
6. Immunofluorescence
NOTE: Keep embryos protected from ambient light.
Wash the embryos in 1.5 mL of ddH2O for 5 min at RT. Wash the embryos in 1.5 mL of pre-chilled acetone (kept at -20 °C) for 7 min at RT. Wash the embryos once in 1.5 mL of ddH2O for 5 min at RT. Wash the embryos once in 1.5 mL of 1x PBST with 1% DMSO (PBDT) for 5 min at RT.
Incubate the embryos in 1.5 mL of PBDT + 10% fetal bovine serum (FBS) on a rocker at RT for 2 h.
Remove PBDT + FBS and replace with 1.5 mL of primary antibodies, anti-acetylated tubulin (produced from mouse) and anti-γ-tubulin (produced from rabbit), diluted together at 1:400 in PBDT + 1% FBS. Incubate the embryos overnight at 4 °C. NOTE: Incubation times may vary depending on the primary antibody. Testing of different time intervals may be necessary to optimize labeling.
7. Secondary Antibody
NOTE: Keep embryos protected from ambient light.
To remove excess unbound antibody from the embryos, wash in 1.5 mL of PBDT + 1% FBS + 0.1 M NaCl for 1 min at RT on a rocker.
Wash the embryos 5 times in 1.5 mL of PBDT + 1% FBS + 0.1 M NaCl for 30 min each on a rocker at RT. Wash the embryos in 1.5 mL of PBDT + 1% FBS for 30 min at RT on a rocker.
Incubate the embryos overnight at 4 °C in 200 µL of the secondary antibodies, Alexa 488 goat anti-mouse IgG and Alexa 647 goat anti-rabbit IgG, diluted together in PBDT at 1:500.
8. DAPI Staining
NOTE: Keep embryos protected from ambient light.
Wash the embryos quickly at RT in 1.5 mL of PBDT two times. Replace PBDT with 1.5 mL of DAPI, diluted 1:15000 in 1X PBST, and incubate 15 min at RT on a rocker.
Wash the embryos in 1.5 mL of PDBT + 1% FBS + 0.1 M NaCl three times at RT for 15-20 min each. Store embryos in 1.5 mL of PBDT at 4 °C until ready to mount and image.
9. Mounting and Imaging for Zebrafish Pronephros
Lay embryo lateral on glass slide in a small volume, approximately 10 µL, of PBDT.
Squeeze the embryo behind the eyes with a pair of fine forceps to remove the head and some of the yolk ball.
Use the fine forceps to gently scrape away the remaining yolk ball from the embryo body. Remove the dissociated yolk and extra liquid from the slide with a thin tissue.
Add a drop of mounting media to the remaining tail of the embryo and position so the tail is laid laterally. Place a coverslip on top of the tail to flatten the sample, then place in a covered slide box.
Image kidney cilia at 60X magnification on a confocal microscope using the following channels: DAPI in blue (laser set at 408.0 nm), FITC in green (laser set at 488.0 nm), Cy3 dye-labeled in red (laser set at 561.0 nm), and Alexa 680 in white (laser set at 637.0 nm).
Representative Results
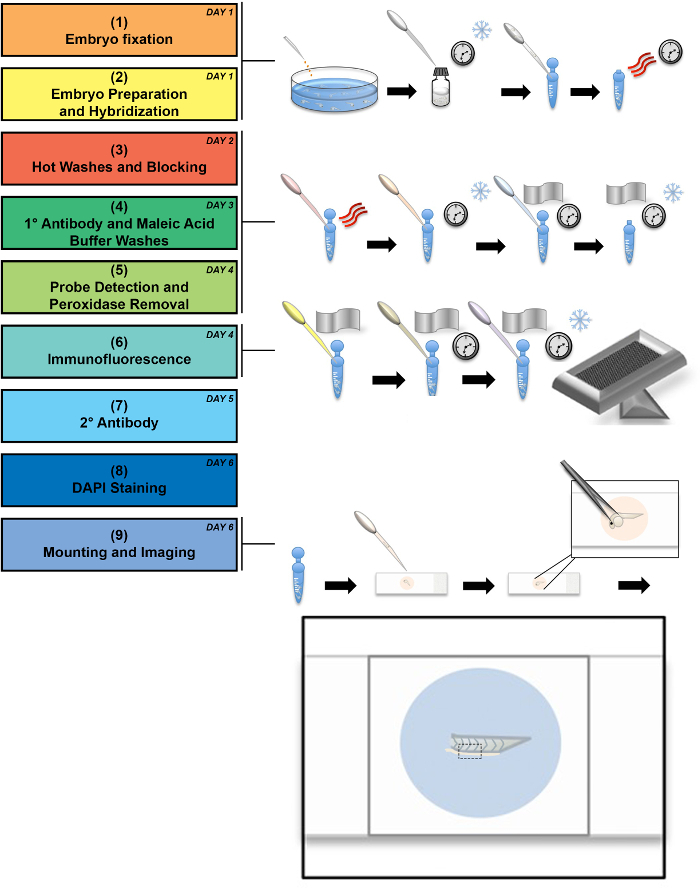
Wild-type zebrafish embryos were fixed at 24 hpf and immediately prepared as described above. Figure 1 depicts an experimental workflow along with selected illustrated stages. The workflow outlined in Steps 1-8 encompasses the processes of sample procurement, fixation, and manipulation of the fixed tissue to label endogenous transcripts with antisense riboprobes followed by immunofluorescence to label protein(s) of interest. Step 9 in the workflow refers to the mounting and imaging techniques specifically designed to optimize visualization of the zebrafish embryonic trunk. In the drawings that accompany Step 9, we illustrate the method of manipulation for positioning the tissue on a glass slide. In this step, the embryo head and yolk ball are removed, leaving the tail to be laterally positioned between a glass slide and coverslip. Removal of the yolk ball and head is suggested for the best imaging of the resident MCC population in the pronephros, as the yolk ball auto-fluoresces and obstructs the mounting process.
Representative images obtained using a confocal microscope are provided in Figure 2, which shows the same wild-type embryo at 60X magnification and a digital zoom at the same magnification. The top panels provide images of each individual channel, while the bottom two panels are the composite overlay of these imaging data. The white box (in the left column image) outlines the area that is the focus of the digital zoom (the right column image). Highlighted in the final panel of the digital zoom, nuclei were labeled in DAPI, and MCCs were detected based on an antisense riboprobe designed to recognize odf3b, where antibodies to γ-tubulin denote the basal bodies and α-tubulin denotes the cilia. In the digital zoom of the composite overlay, the yellow dashed circle indicates the perimeter of an MCC, which was identified due to the possession of odf3b transcripts, multiple basal bodies, and cilia. The adjacent mono-ciliated cell, labeled with a yellow dotted circle, was identified based on the phenotype of possessing a single basal body and a single cilium.
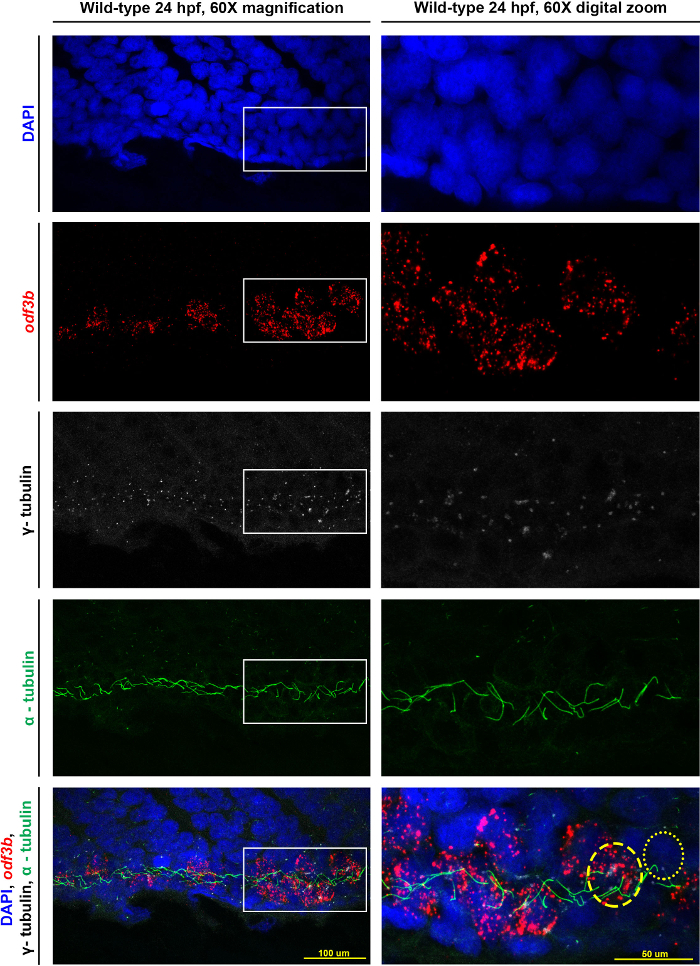
Figure 1: Schematic of experimental flowchart. This flowchart shows an experimental workflow, accompanied by illustrations of critical stages in which the snowflake indicates cold incubation, the three curved lines represent heat, and the clock represents long incubation times. The workflow can be performed in a minimum of 6 days (see day number in upper right corner of each stage in the process), though some steps can be performed over longer time periods, as noted in the protocol. A detailed depiction of the mounting process is drawn out, demonstrating the final mounted embryo tail between the glass slide and coverslip. A black dashed box outlines the area that is imaged at 60X magnification. Please click here to view a larger version of this figure.
Figure 2: Representative results for visualizing MCCs of the zebrafish pronephros. Maximum image projections of a 24 hpf wild-type zebrafish embryo at 60X magnification as well as a digital zoom on the confocal at 60X magnification of the same embryo. The white boxes indicate the area focused on for the zoom. Individual stains of DAPI (nuclei), odf3b (MCCs), γ-tubulin (basal bodies), and α-tubulin (cilia) are labeled and then merged together in the bottom two panels. In the digital zoom, we provide approximations of the cell locations as follows: an MCC is outlined by the dashed yellow circle, and the dotted yellow circle outlines a mono-ciliated cell. Please click here to view a larger version of this figure.
Discussion
The protocol detailed above is optimized for labeling MCCs in the pronephros with odf3b transcripts and α-tubulin in 24 hpf zebrafish embryos. For the best results, it is recommended to use freshly fixed and prepared embryos. Embryos that have been fixed and stored in either MeOH or Hyb+ at -20 °C for longer than one week can be used, but the probability of unwanted background staining increases with the time that the embryos are kept in storage.
Modifications and troubleshooting of this technique are necessary to tailor this method for other suites of particular markers, e.g. other antisense riboprobes and other antibodies. Many methods for adjusting the parameters of steps in the process of whole mount in situ hybridization have been previously documented by our group and others31,34,36,38,39. Likewise, each protein antibody will require some troubleshooting in terms of staining times, and approximate ranges of dilutions and incubation times for each primary antibody are best estimated by evaluation of documented results28,35. For embryos younger than 24 hpf, pK treatment should be shorter than 2 min, where embryos older than 24 hpf should be treated with pK for longer than 2 min. Also, embryos older than 24 hpf should be bleached of pigment before pK treatment.
After staining with the fluorescent staining solution, it is critical to remove any remaining stain, antibody, and peroxidases by performing the series of methanol and hydrogen peroxide washes. The PBS washes immediately following are vital to remove excess methanol from the embryos. Importantly, before proceeding with the IF antibodies, we have found that the acetone and deionized water washes make the embryos less likely to adhere to one another and/or the centrifuge tubes. In our experience, antibodies for specific transcription factors and other genes, though they may work efficiently in Western blots, do not work well for IF in the zebrafish. However, highly conserved, and abundant proteins, such as α-tubulin and β-catenin, do work well in the zebrafish for IF16,37.
With FISH, it is possible to visualize RNA transcripts of genes that do not yet have specific antibodies in the zebrafish. By combining FISH with IF, as demonstrated by this protocol, co-localization of RNA transcripts and protein can be seen in vivo (Figure 2). The flexible nature of the protocol enables quick troubleshooting for the visualization of various RNA transcripts and proteins in numerous tissues and time points. When combined with the already respected traits of zebrafish embryos, this protocol of FISH + IF provides another tool to explore the expression of genes and proteins important to developmental pathway regulation.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work was supported in part by the grant R01DK100237 to R.A.W. and the National Science Foundation Graduate Research Fellowship No. DGE-1313583 to A.N.M. We also thank the College of Science Summer Undergraduate Research Fellowship Program for support to M.U. We would like to thank the Center for Zebrafish Research at the University of Notre Dame for their dedicated care of our zebrafish. We would also like to thank the Department of Biological Sciences as well as the members of our lab for all of their support and valuable insights.
References
- Lieschke GJ, Currie PD. Animal models of human disease: zebrafish swim into view. Nat Rev Genet. 2007;8:353–367. doi: 10.1038/nrg2091. [DOI] [PubMed] [Google Scholar]
- Goldsmith JR, Jobin C. Think small: zebrafish as a model system of human pathology. J Biomed Biotechnol. 2012;2012:817341. doi: 10.1155/2012/817341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe K, et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature. 2013;496:498–503. doi: 10.1038/nature12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond IA. Kidney development and disease in the zebrafish. J Am Soc Nephrol. 2005;16:299–304. doi: 10.1681/ASN.2004090754. [DOI] [PubMed] [Google Scholar]
- Wingert RA, et al. The cdx genes and retinoic acid control the positioning and segmentation of the zebrafish pronephros. PLoS Genet. 2007;3:1922–1938. doi: 10.1371/journal.pgen.0030189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingert RA, Davidson AJ. The zebrafish pronephros: a model to study segmentation. Kindey Int. 2008;73(10):1120–1127. doi: 10.1038/ki.2008.37. [DOI] [PubMed] [Google Scholar]
- Wingert RA, Davidson AJ. Zebrafish nephrogenesis involves dynamic spatiotemporal expression changes in renal progenitors and essential signals from retinoic acid and irx3b. Dev Dyn. 2011;240:2011–2027. doi: 10.1002/dvdy.22691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers BE, Wingert RA. Renal progenitors: roles in kidney disease and regeneration. World J Stem Cells. 2016;8(11):367–375. doi: 10.4252/wjsc.v8.i11.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond BE, Wingert RA. Insights into kidney stem cell development and regeneration using zebrafish. World J Stem Cells. 2016;8(2):22–31. doi: 10.4252/wjsc.v8.i2.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poureetezadi SJ, Wingert RA. Little fish, big catch: zebrafish as a model for kidney disease. Kidney Int. 2016;89(6):1204–1210. doi: 10.1016/j.kint.2016.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer-Zucker AG, Olale F, Haycraft CJ, Yoder BK, Schier AF, Drummond IA. Cilia-driven fluid flow in the zebrafish pronephros, brain and Kupffer's vesicle is required for normal organogenesis. Development. 2005;132:1907–1921. doi: 10.1242/dev.01772. [DOI] [PubMed] [Google Scholar]
- Liu Y, Narendra P, Kramer-Zucker A, Drummond IA. Notch signaling controls the differentiation of transporting epithlelia and multiciliated cells in the zebrafish pronephros. Development. 2007;134:1111–1122. doi: 10.1242/dev.02806. [DOI] [PubMed] [Google Scholar]
- Ma M, Jiang YJ. Jagged2a-Notch signaling mediates cell fate choice in the zebrafish pronephric duct. PLoS Genetics. 2007;3:e18. doi: 10.1371/journal.pgen.0030018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Cheng CN, Verdun VA, Wingert RA. Zebrafish nephrogenesis is regulated by interactions between retinoic acid, mecom, and Notch signaling. Dev Biol. 2014;386(1):111–122. doi: 10.1016/j.ydbio.2013.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, et al. miR-34b regulates multiciliogenesis during organ formation in zebrafish. Development. 2013;140:2755–2764. doi: 10.1242/dev.092825. [DOI] [PubMed] [Google Scholar]
- Marra AN, Wingert RA. Epithelial cell fate in the nephron tubule is mediated by the ETS transcription factors etv5a and etv4 during zebrafish development. Dev Biol. 2016;411(2):231–245. doi: 10.1016/j.ydbio.2016.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marra AN, Li Y, Wingert RA. Antennas of organ morphogenesis: the roles of cilia in vertebrate kidney development. Genesis. 2016;54(9):457–469. doi: 10.1002/dvg.22957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy JL, Suzuki Y. Ciliated human renal proximal tubular cells. Observations in three cases of hypercalcemia. Am J Pathol. 1968;53:609–616. [PMC free article] [PubMed] [Google Scholar]
- Katz SM, Morgan JJ. Cilia in the human kidney. Ultrastruct Pathol. 1984;6:285–294. doi: 10.3109/01913128409018587. [DOI] [PubMed] [Google Scholar]
- Hassan MO, Subramanyan S. Ciliated renal tubular cells in crescentic glomerulonephritis. Ultrastruct Pathol. 1995;19:201–203. doi: 10.3109/01913129509064222. [DOI] [PubMed] [Google Scholar]
- Ong AC, Wagner B. Detection of proximal tubular motile cilia in a patient with renal sarcoidosis associated with hypercalcemia. Am J Kidney Dis. 2005;45:1096–1099. doi: 10.1053/j.ajkd.2005.02.019. [DOI] [PubMed] [Google Scholar]
- Worthington WC, Cathcart RS., III Ependymal cilia: distribution and activity in the adult human brain. Science. 1963;139:221–222. doi: 10.1126/science.139.3551.221. [DOI] [PubMed] [Google Scholar]
- Cowan MJ, Galdwin MT, Shelhamer JH. Disorders of ciliary motility. Am J Med Sci. 2001;321:3–10. doi: 10.1097/00000441-200101000-00002. [DOI] [PubMed] [Google Scholar]
- Jain R, et al. Temporal relationship between primary and motile ciliogenesis in airway epithelial cells. Am J Respir Cell Mol Biol. 2010;43:731–739. doi: 10.1165/rcmb.2009-0328OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbs JL, Vladar EK, Axlerod JD, Kitner C. Multicilin promotes centriole assembly and ciliogenesis during multiciliate cell differentiation. Nat Cell Biol. 2012;14:140–147. doi: 10.1038/ncb2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan FE, et al. Myb promotes centriole amplification and later steps of the multiciliogenesis program. Development. 2013;140:4277–4286. doi: 10.1242/dev.094102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou F, Narasimhan V, Shboul M, Chong YL, Reversade B, Roy S. Gmnc is a master regulator of the multiciliated cell differentiation program. Curr Biol. 2015;25:3267–3273. doi: 10.1016/j.cub.2015.10.062. [DOI] [PubMed] [Google Scholar]
- Jowett T. Analysis of Protein and gene expression. Methods Cell Biol. 1999;59:63–85. doi: 10.1016/s0091-679x(08)61821-x. [DOI] [PubMed] [Google Scholar]
- Schulte-Merker S. Chapter 2: Looking at Embryos. Zebrafish: A practical approach. 2002;261:39–58. [Google Scholar]
- Brend T, Holley SA. Zebrafish whole mount high-resolution double fluorescent in situ hybridization. J Vis Exp. 2009. [DOI] [PMC free article] [PubMed]
- Lauter G, Soll I, Hauptmann G. Two-color fluorescent in situ hybridization in the embryonic zebrafish brain using differential detection systems. BMC Dev Biol. 2011;11 doi: 10.1186/1471-213X-11-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond BE, Li Y, Marra AN, Cheng CN, Wingert RA. The tbx2a/b transcription factors direct pronephros segmentation and corpuscle of Stannius formation in zebrafish. Dev Biol. 2017;421(1):52–66. doi: 10.1016/j.ydbio.2016.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe KM, Thiberge SY, Bisher ME, Burdine RD. Imaging cilia in zebrafish. Methods Cell Biol. 2010;97:415–435. doi: 10.1016/S0091-679X(10)97022-2. [DOI] [PubMed] [Google Scholar]
- Lauter G, Soll I, Hauptmann G. Multicolor fluorescent in situ hybridization to define abutting and overlapping gene expression in the embryonic zebrafish brain. Neural Dev. 2011;6 doi: 10.1186/1749-8104-6-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrells S, Toruno C, Stewart RA, Jette C. Analysis of apoptosis in zebrafish embryos by whole-mount immunofluorescence to detect activated caspase 3. J Vis Exp. 2013. p. e51060. [DOI] [PMC free article] [PubMed]
- Schumacher JA, Zhao EJ, Kofron MJ, Sumanas S. Two-color fluorescent in situ hybridization using chromogenic substrates in zebrafish. Bio Techniques. 2014;57:254–256. doi: 10.2144/000114229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julich D, et al. beamter/deltaC and the role of Notch ligands in the zebrafish somite segmentation, hindbrain neurogenesis and hypochord differentiation. Dev Biol. 2005;286:391–404. doi: 10.1016/j.ydbio.2005.06.040. [DOI] [PubMed] [Google Scholar]
- Gross-Thebing T, Paksa A, Raz E. Simultaneous high-resolution detection of multiple transcripts combined with localization of proteins in whole-mount embryos. BMC Biology. 2014;12 doi: 10.1186/s12915-014-0055-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CN, Li Y, Marra AN, Verdun V, Wingert RA. Flat Mount Preparation for Observation and Analysis of Zebrafish Embryo Specimens Stained by Whole Mount In situ Hybridization. J Vis Exp. 2014. p. e51604. [DOI] [PMC free article] [PubMed]
- Poureetezadi SJ, Donahue EK, Wingert RA. A Manual Small Molecule Screen Approaching High-throughput Using Zebrafish Embryos. J Vis Exp. 2014. p. e52063. [DOI] [PMC free article] [PubMed]